
Proteasomal Degradation of Zn-Dependent Hdacs: The E3-Ligases Implicated and the Designed Protacs That Enable Degradation


Abstract
: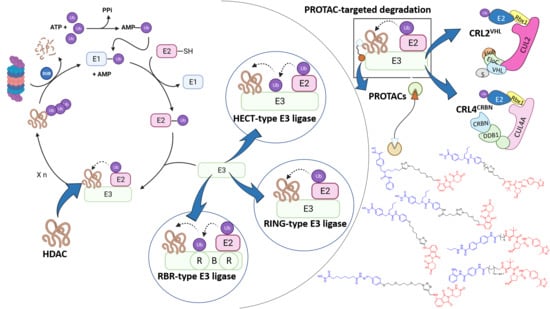
1. Introduction
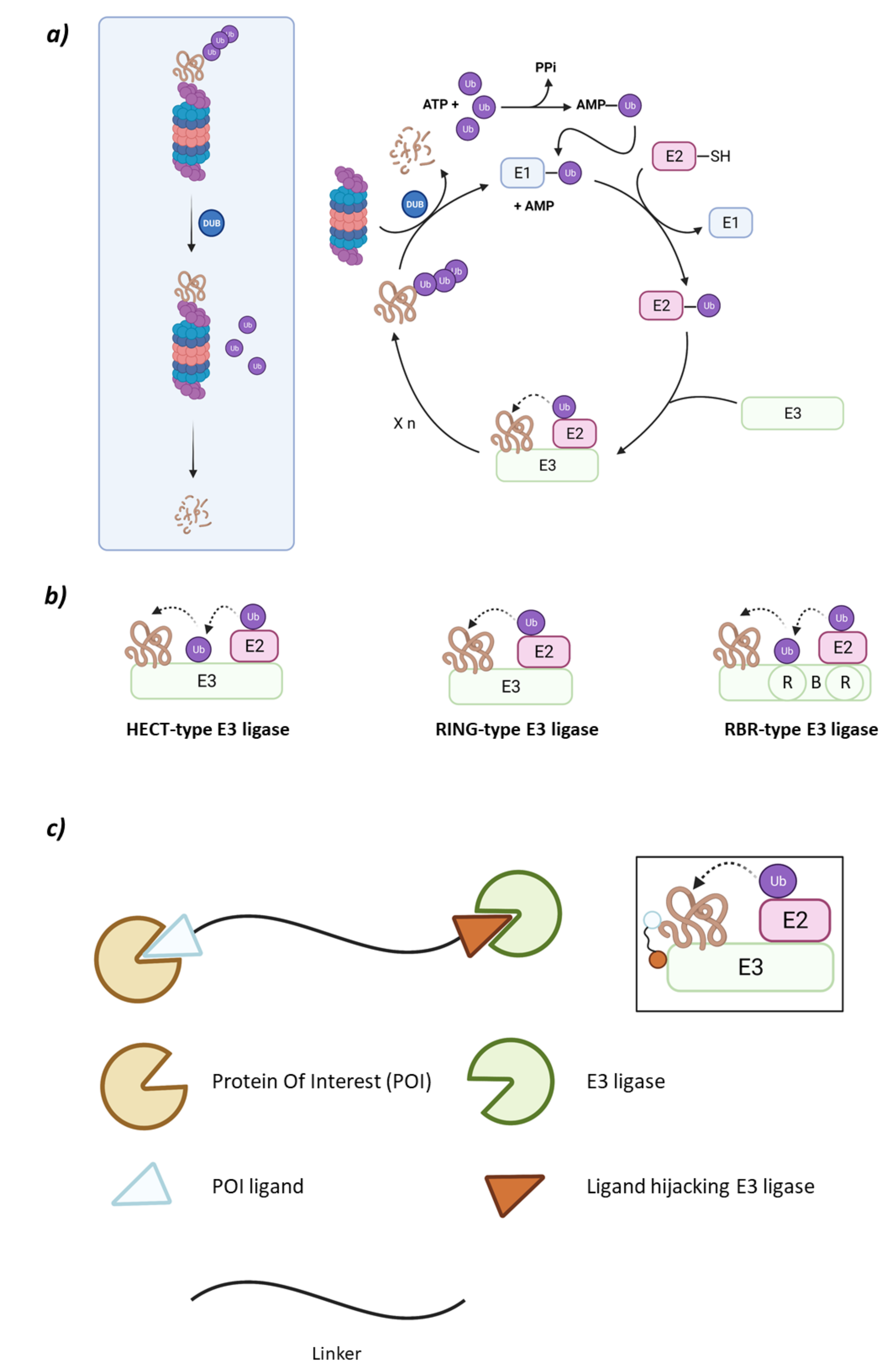
1.1. The Ubiquitin-Proteasome System
1.2. The HDAC Protein Family
1.3. PROteolysis TArgeting Chimaeras (PROTACs)
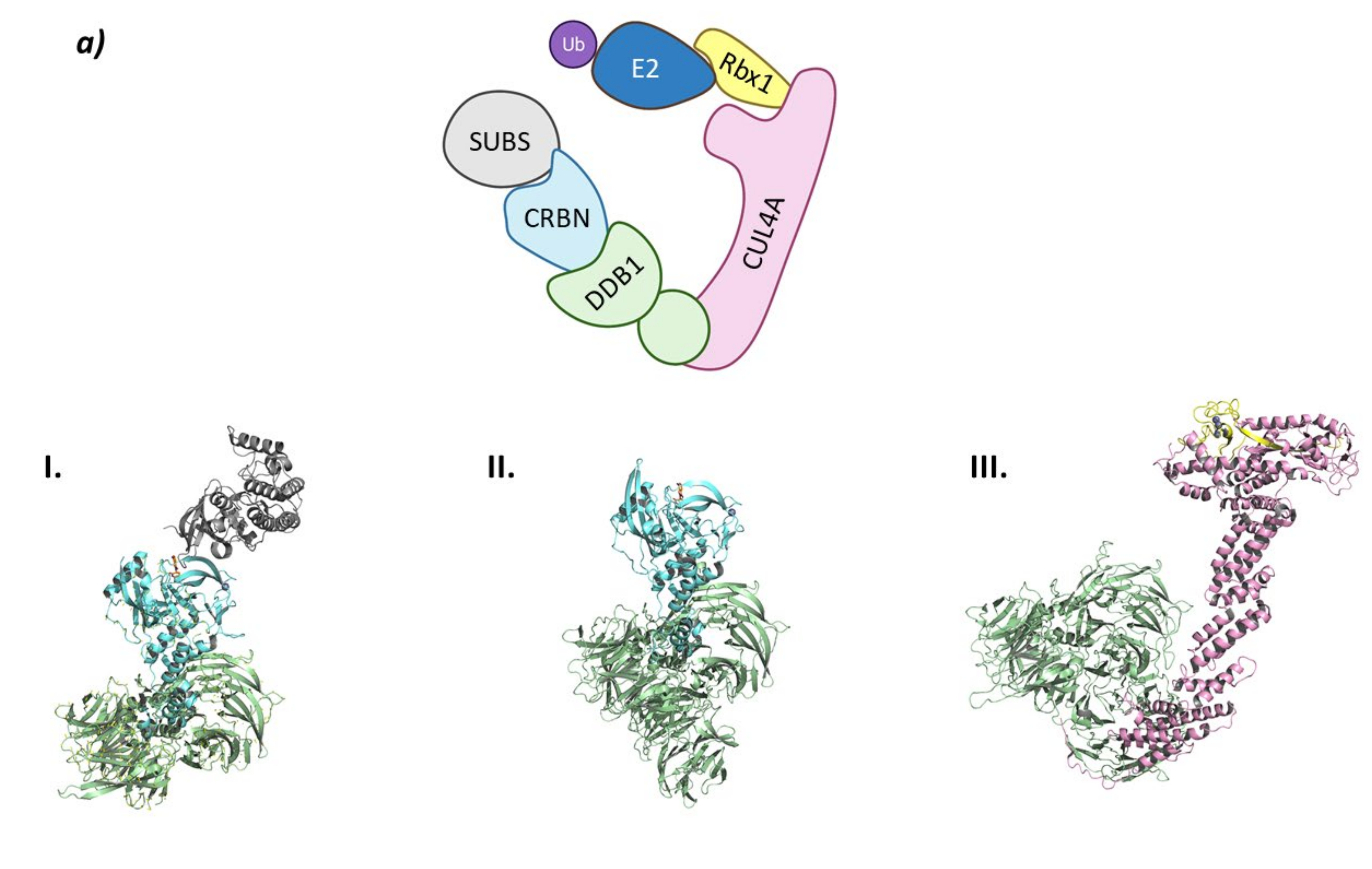
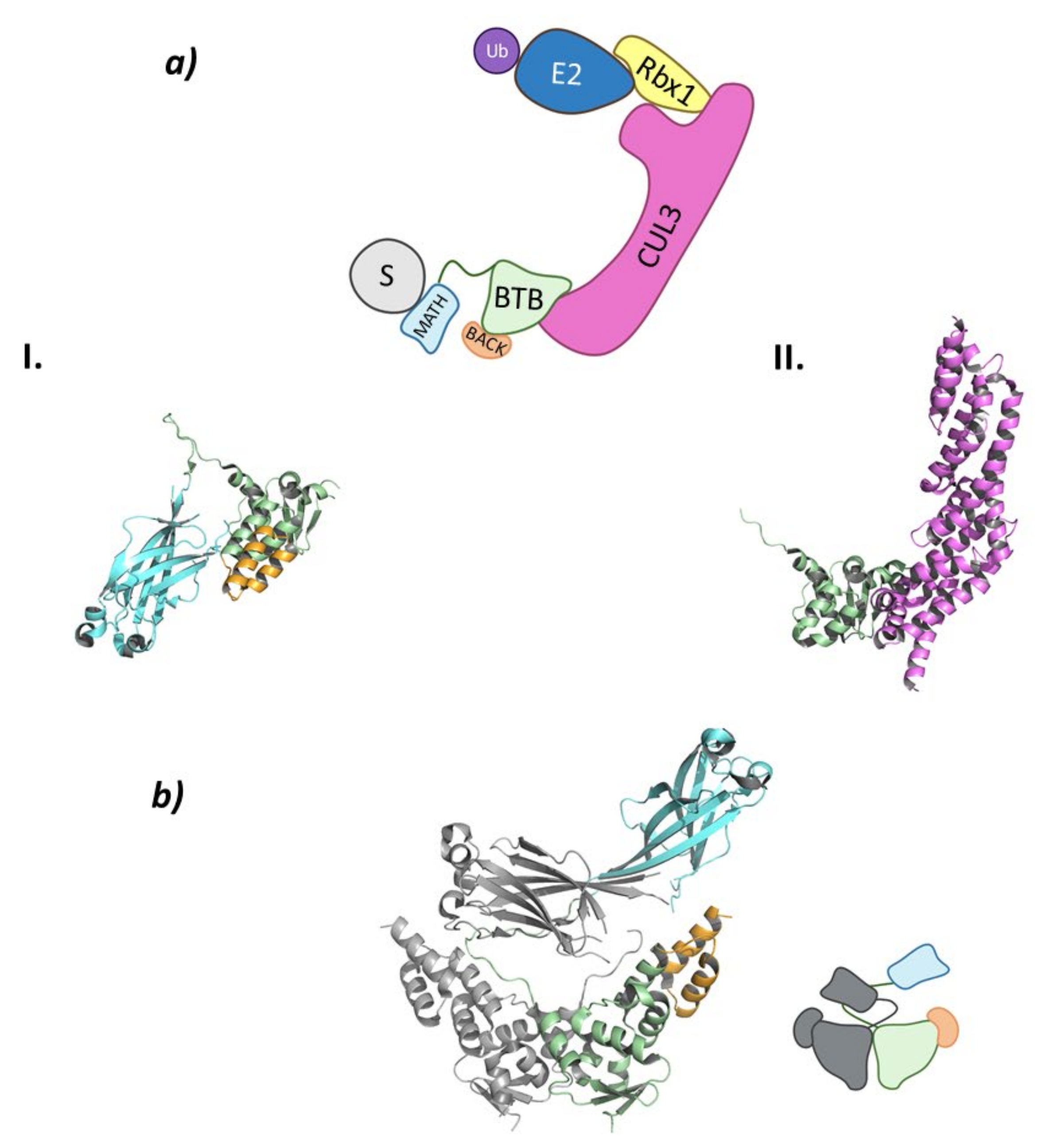
2. RING E3 Ligases
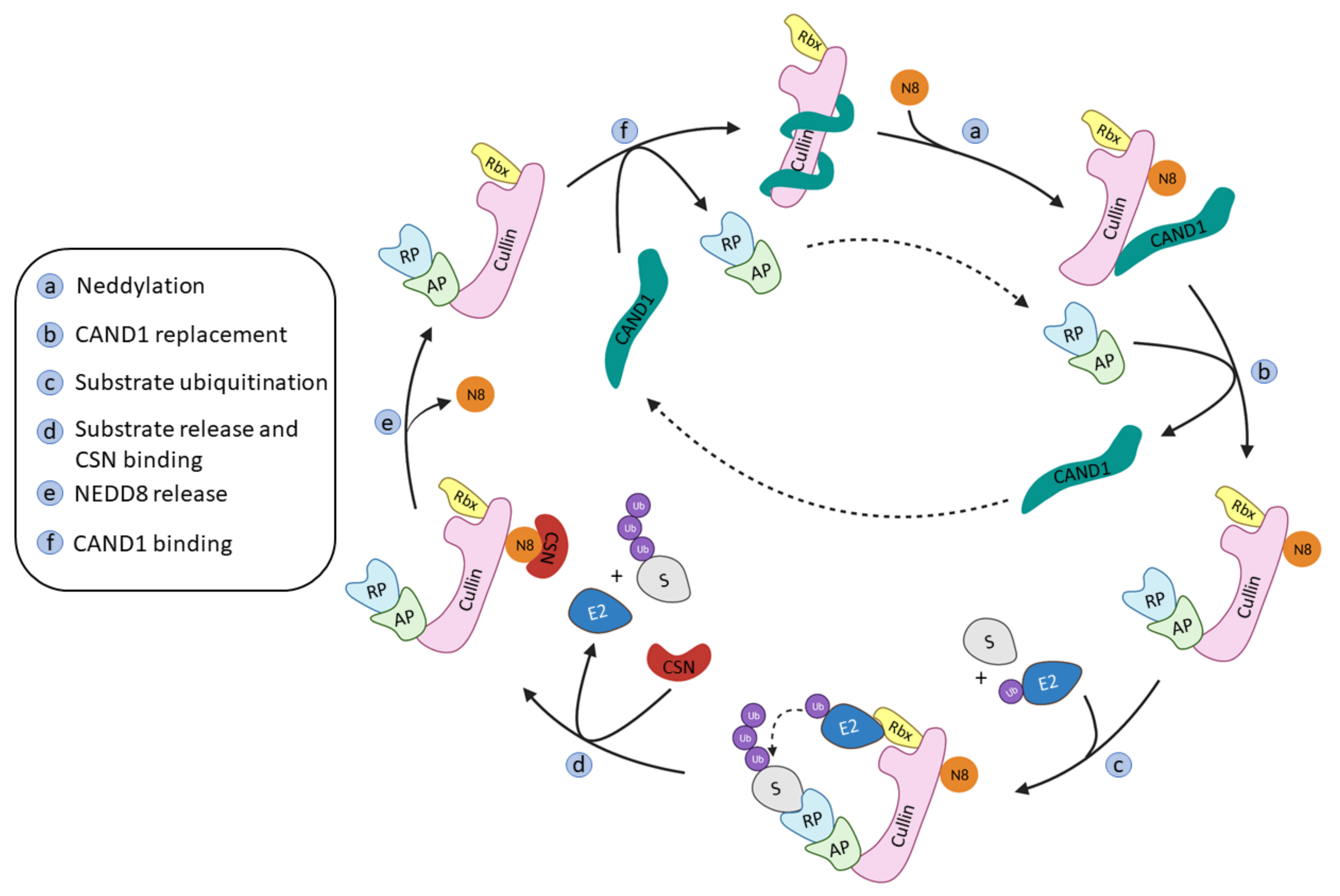
2.1. Cullin-RING Ligases
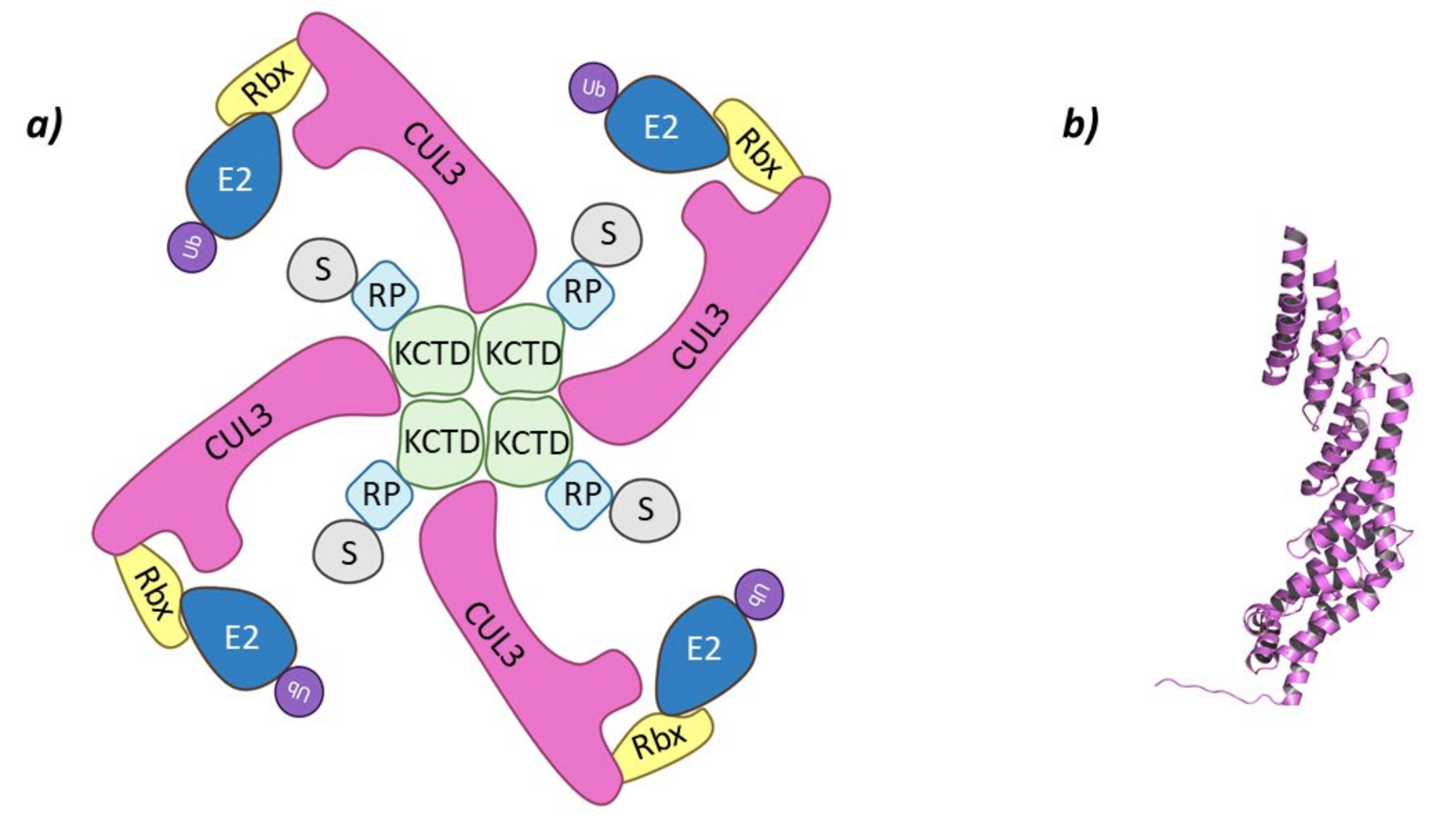
2.1.1. Cul3-KCTD
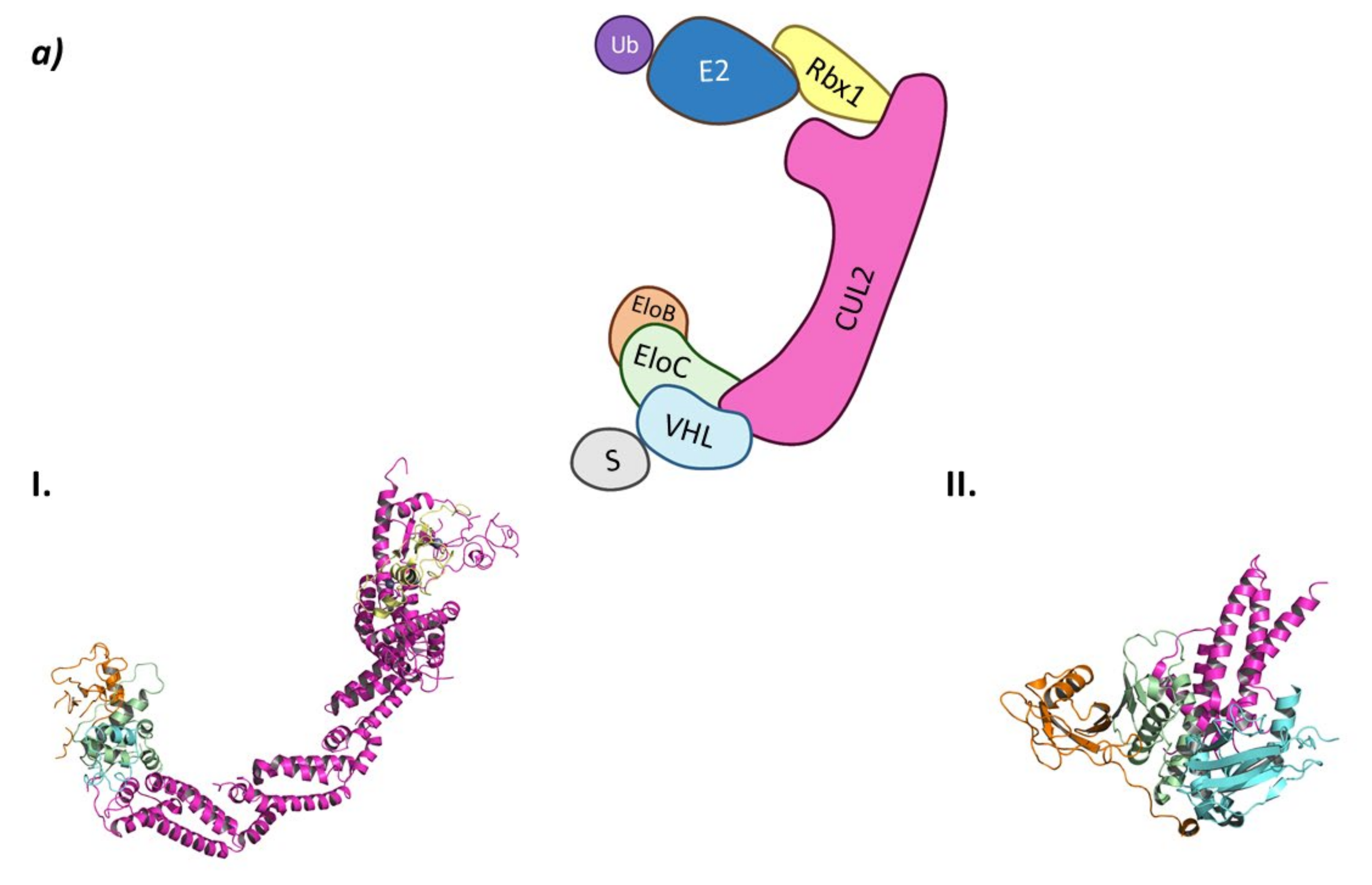
2.1.2. CRL2VHL
2.1.3. CRL4CRBN
2.1.4. Cul3SPOP
2.2. Monomeric RING
2.2.1. CHFR
2.2.2. PIRH2 (RCHY1)
2.2.3. RLIM (RNF12)
2.3. Homodimeric RING
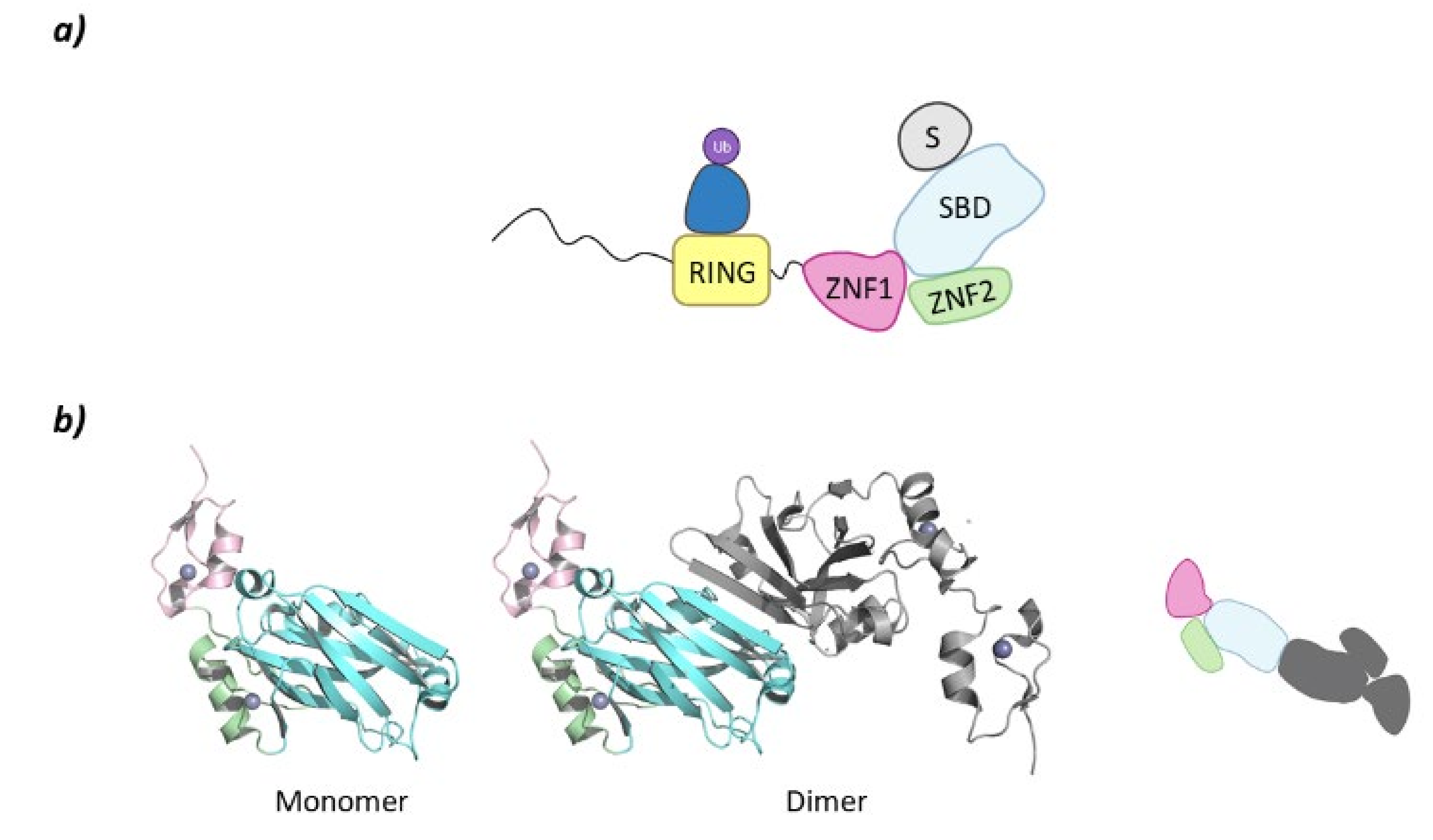
SIAH2
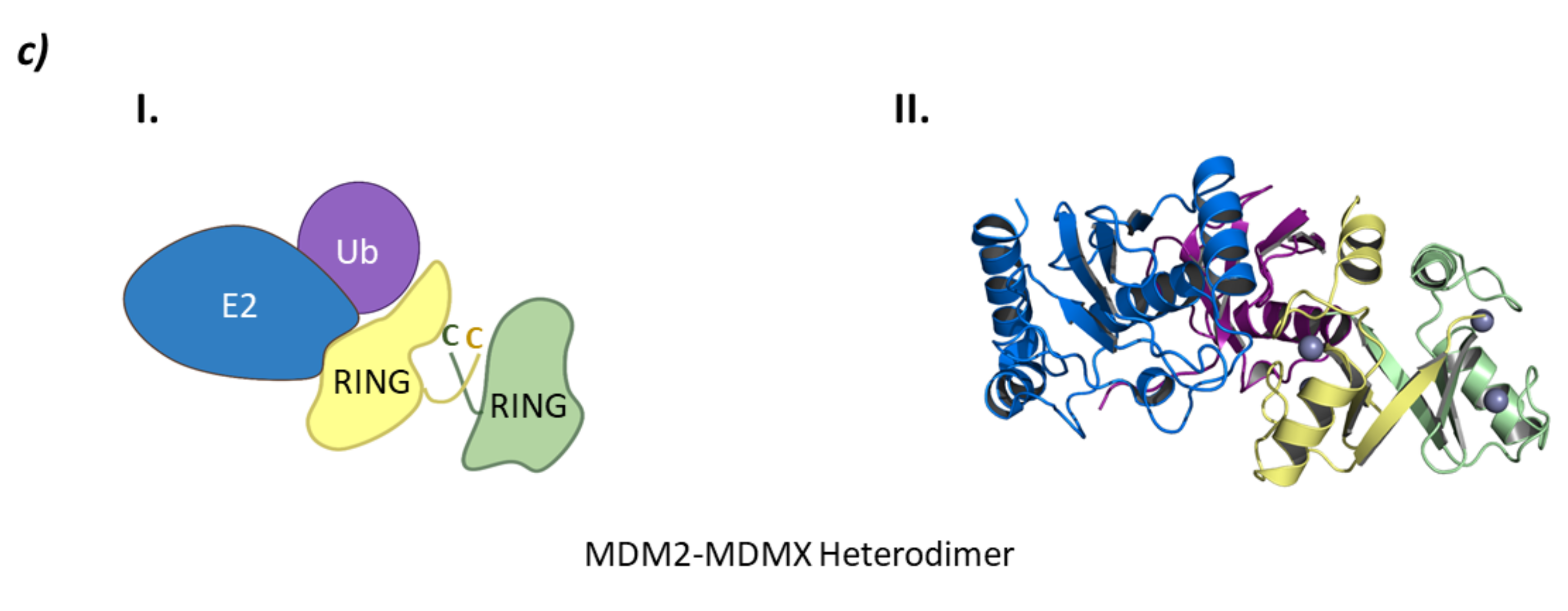
2.4. Heterodimeric RING Ligases
MDM
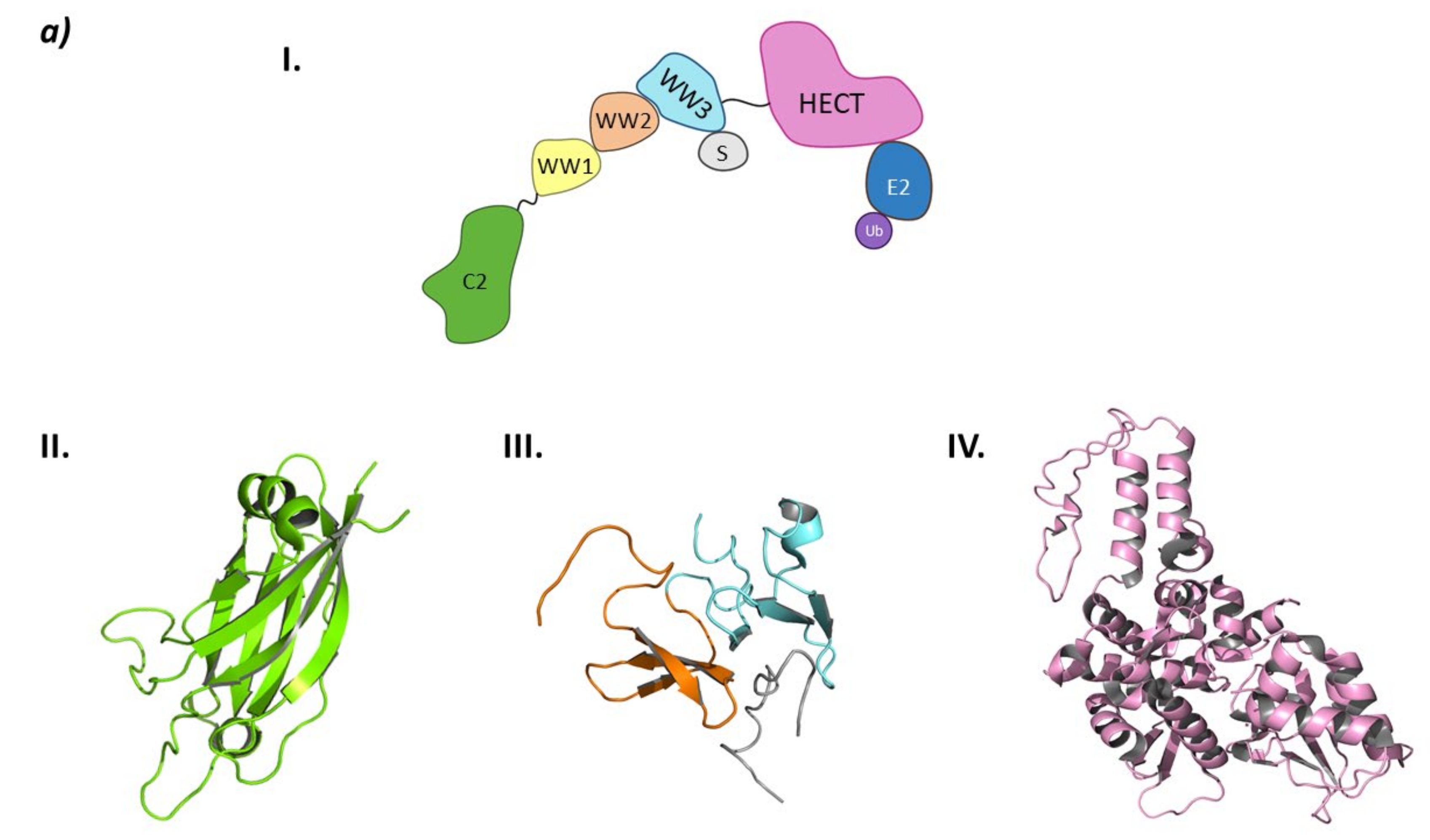
3. Hect Ligases
3.1. Smurf2
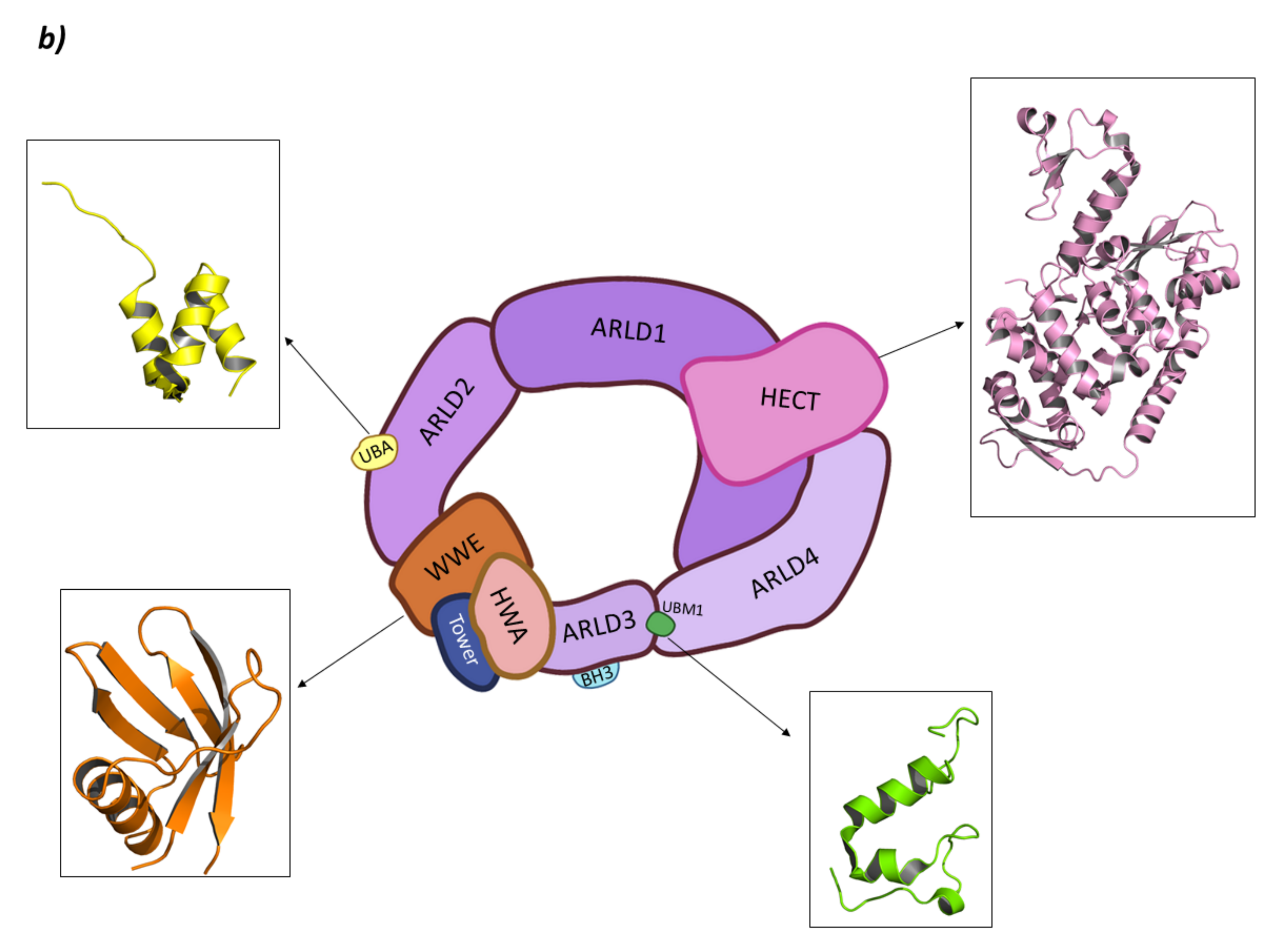
3.2. HUWE1 (Mule)
4. Known PROTACs for HDACs
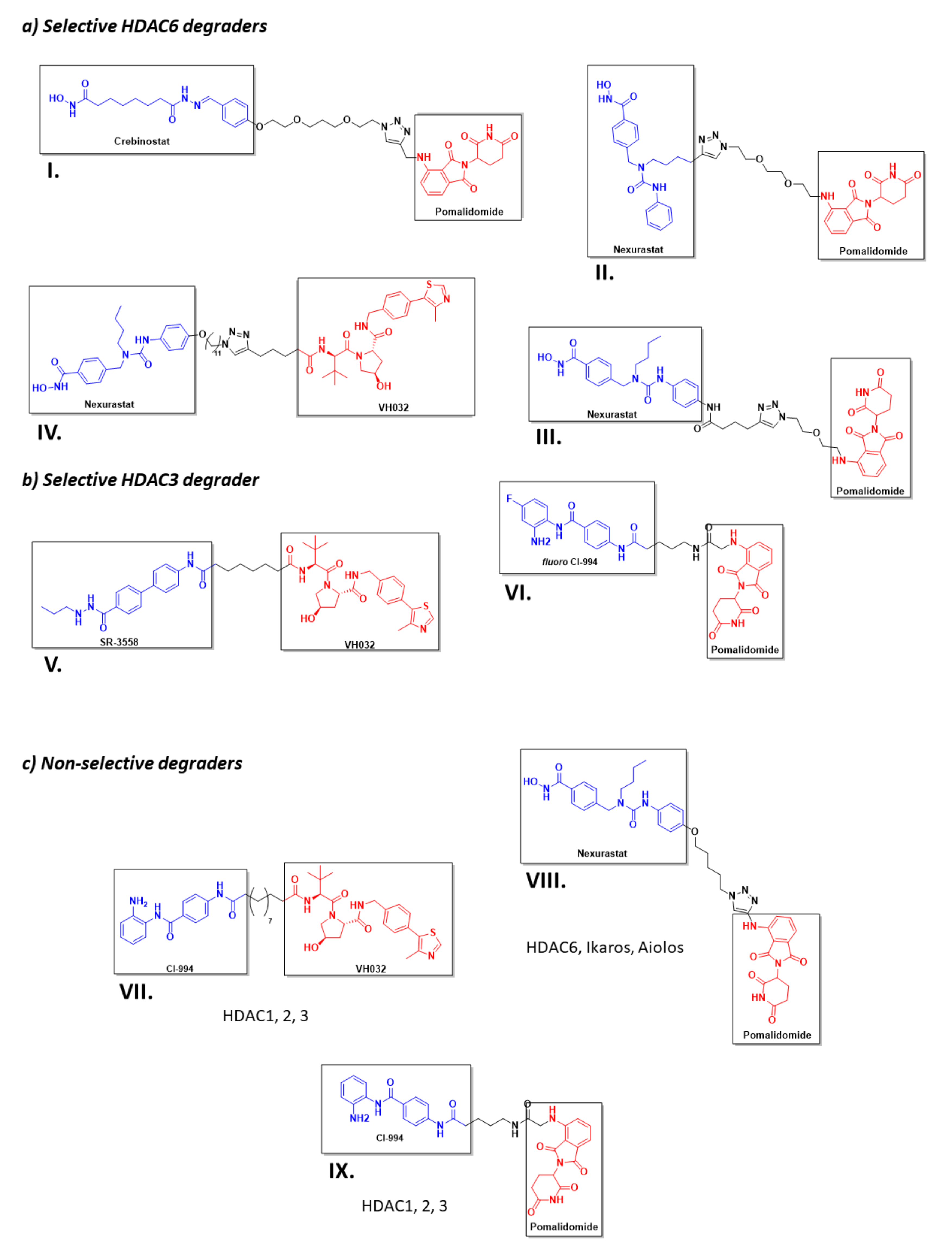
5. Conclusions and Future Work
6. Materials and Methods
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef]
- Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules 2020, 10, 796. [Google Scholar] [CrossRef]
- Dwane, L.; Gallagher, W.M.; Ní Chonghaile, T.; O’Connor, D.P. The Emerging Role of Non-traditional Ubiquitination in Oncogenic Pathways*. J. Biol. Chem. 2017, 292, 3543–3551. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, E.; Richard, F. The-N-End Rule: The Beginning Determines the End. Protein Pept. Lett. 2016, 23, 343–348. [Google Scholar] [CrossRef]
- Ottis, P.; Toure, M.; Cromm, P.M.; Ko, E.; Gustafson, J.L.; Crews, C.M. Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol. 2017, 12, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Bielskienė, K.; Bagdonienė, L.; Mozūraitienė, J.; Kazbarienė, B.; Janulionis, E. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina 2015, 51, 1–9. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Busino, L. E3 ubiquitin ligases in B-cell malignancies. Cell. Immunol. 2019, 340, 103905. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Benirschke, R.C.; Thompson, J.R.; Nominé, Y.; Wasielewski, E.; Juranić, N.; Macura, S.; Hatakeyama, S.; Nakayama, K.I.; Botuyan, M.V.; Mer, G. Molecular Basis for the Association of Human E4B U Box Ubiquitin Ligase with E2-Conjugating Enzymes UbcH5c and Ubc4. Structure 2010, 18, 955–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardley, H.C.; Robinson, P.A. E3 ubiquitin ligases. Essays Biochem. 2005, 41, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Zagidullin, A.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Small Molecule Modulators of RING-Type E3 Ligases: MDM and Cullin Families as Targets. Front. Pharmacol. 2018, 9, 450. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e241. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, L.; Yang, Y.; Zhang, X.; Gang, Y.; Bai, L. The Role of HDACs and HDACi in Cartilage and Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 966. [Google Scholar] [CrossRef]
- Sixto-López, Y.; Gómez-Vidal, J.A.; de Pedro, N.; Bello, M.; Rosales-Hernández, M.C.; Correa-Basurto, J. Hydroxamic acid derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with antiproliferative activity in cancer cell lines. Sci. Rep. 2020, 10, 10462. [Google Scholar] [CrossRef]
- Núñez-Álvarez, Y.; Suelves, M. HDAC11: A multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2021. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [Green Version]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorganic Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef]
- Wu, D.; Qiu, Y.; Jiao, Y.; Qiu, Z.; Liu, D. Small Molecules Targeting HATs, HDACs, and BRDs in Cancer Therapy. Front. Oncol. 2020, 10, 2474. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Pinheiro, P.d.S.M.; Sagrillo, F.S.; Bolognesi, M.L.; Fraga, C.A.M. Histone deacetylases as targets for the treatment of neurodegenerative disorders: Challenges and future opportunities. Med. Res. Rev. 2020, 40, 2177–2211. [Google Scholar] [CrossRef]
- Lee, J.O.; Byun, W.S.; Kang, M.J.; Han, J.A.; Moon, J.; Shin, M.-J.; Lee, H.J.; Chung, J.H.; Lee, J.-S.; Son, C.-G.; et al. The myokine meteorin-like (metrnl) improves glucose tolerance in both skeletal muscle cells and mice by targeting AMPKα2. FEBS J. 2020, 287, 2087–2104. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Zwinderman, M.R.H.; van Merkerk, R.; Ettema, P.E.; Quax, W.J.; Dekker, F.J. Inhibitory selectivity among class I HDACs has a major impact on inflammatory gene expression in macrophages. Eur. J. Med. Chem. 2019, 177, 457–466. [Google Scholar] [CrossRef]
- Schiedel, M.; Conway, S.J. Small molecules as tools to study the chemical epigenetics of lysine acetylation. Curr. Opin. Chem. Biol. 2018, 45, 166–178. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation*. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Puppala, D.; Choi, E.-Y.; Swanson, H.; Kim, K.-B. Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool. Chembiochem A Eur. J. Chem. Biol. 2007, 8, 2058–2062. [Google Scholar] [CrossRef]
- Vassilev, L.; Vu, B.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Vogelmann, A.; Robaa, D.; Sippl, W.; Jung, M. Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr. Opin. Chem. Biol. 2020, 57, 8–16. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e75. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidou, M.; Li, J.; Zhang, B.; Wang, Z.; Shaabani, S.; Ter Brake, F.; Essa, K.; Dömling, A. PROTACs—A game-changing technology. Expert Opin. Drug Discov. 2019, 14, 1255–1268. [Google Scholar] [CrossRef]
- Daniels, D.L.; Riching, K.M.; Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discov. Today Technol. 2019, 31, 61–68. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Cheng, J.; Guo, J.; North, B.J.; Tao, K.; Zhou, P.; Wei, W. The emerging role for Cullin 4 family of E3 ligases in tumorigenesis. Biochim. Biophys. Acta—Rev. Cancer 2019, 1871, 138–159. [Google Scholar] [CrossRef]
- Sarikas, A.; Hartmann, T.; Pan, Z.-Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Xiong, X.; Zhao, Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smaldone, G.; Pirone, L.; Balasco, N.; Di Gaetano, S.; Pedone, E.M.; Vitagliano, L. Cullin 3 Recognition Is Not a Universal Property among KCTD Proteins. PLoS ONE 2015, 10, e0126808. [Google Scholar] [CrossRef]
- Teng, X.; Aouacheria, A.; Lionnard, L.; Metz, K.A.; Soane, L.; Kamiya, A.; Hardwick, J.M. KCTD: A new gene family involved in neurodevelopmental and neuropsychiatric disorders. CNS Neurosci. Ther. 2019, 25, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Ji, A.X.; Chu, A.; Nielsen, T.K.; Benlekbir, S.; Rubinstein, J.L.; Privé, G.G. Structural Insights into KCTD Protein Assembly and Cullin3 Recognition. J. Mol. Biol. 2016, 428, 92–107. [Google Scholar] [CrossRef] [Green Version]
- De Smaele, E.; Di Marcotullio, L.; Moretti, M.; Pelloni, M.; Occhione, M.A.; Infante, P.; Cucchi, D.; Greco, A.; Pietrosanti, L.; Todorovic, J.; et al. Identification and Characterization of KCASH2 and KCASH3, 2 Novel Cullin3 Adaptors Suppressing Histone Deacetylase and Hedgehog Activity in Medulloblastoma. Neoplasia 2011, 13, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Correale, S.; Pirone, L.; Di Marcotullio, L.; De Smaele, E.; Greco, A.; Mazzà, D.; Moretti, M.; Alterio, V.; Vitagliano, L.; Di Gaetano, S.; et al. Molecular organization of the cullin E3 ligase adaptor KCTD11. Biochimie 2011, 93, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Mancarelli, M.M.; Zazzeroni, F.; Ciccocioppo, L.; Capece, D.; Po, A.; Murgo, S.; Di Camillo, R.; Rinaldi, C.; Ferretti, E.; Gulino, A.; et al. The tumor suppressor gene KCTD11RENis regulated by Sp1 and methylation and its expression is reduced in tumors. Mol. Cancer 2010, 9, 172. [Google Scholar] [CrossRef] [Green Version]
- Di Marcotullio, L.; Ferretti, E.; De Smaele, E.; Argenti, B.; Mincione, C.; Zazzeroni, F.; Gallo, R.; Masuelli, L.; Napolitano, M.; Maroder, M.; et al. RENKCTD11 is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc. Natl. Acad. Sci. USA 2004, 101, 10833–10838. [Google Scholar] [CrossRef] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef]
- Zazzeroni, F.; Nicosia, D.; Tessitore, A.; Gallo, R.; Verzella, D.; Fischietti, M.; Vecchiotti, D.; Ventura, L.; Capece, D.; Gulino, A.; et al. KCTD11 Tumor Suppressor Gene Expression Is Reduced in Prostate Adenocarcinoma. Biomed Res. Int. 2014, 2014, 380398. [Google Scholar] [CrossRef]
- Lange, S.; Perera, S.; Teh, P.; Chen, J. Obscurin and KCTD6 regulate cullin-dependent small ankyrin-1 (sAnk1.5) protein turnover. Mol. Biol. Cell 2012, 23, 2490–2504. [Google Scholar] [CrossRef]
- Smaldone, G.; Pirone, L.; Pedone, E.; Marlovits, T.; Vitagliano, L.; Ciccarelli, L. The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett. 2016, 590, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xia, W.; Qiu, M.; Wang, X.; Jiang, F.; Yin, R.; Xu, L. Atlas on substrate recognition subunits of CRL2 E3 ligases. Oncotarget 2016, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Maeda, Y.; Suzuki, T.; Pan, X.; Chen, G.; Pan, S.; Bartman, T.; Whitsett, J.A. CUL2 Is Required for the Activity of Hypoxia-inducible Factor and Vasculogenesis*. J. Biol. Chem. 2008, 283, 16084–16092. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yang, H. The von Hippel-Lindau Tumor Suppressor Protein Promotes c-Cbl-Independent Poly-Ubiquitylation and Degradation of the Activated EGFR. PLoS ONE 2011, 6, e23936. [Google Scholar] [CrossRef]
- Anderson, K.; Nordquist, K.A.; Gao, X.; Hicks, K.C.; Zhai, B.; Gygi, S.P.; Patel, T.B. Regulation of Cellular Levels of Sprouty2 Protein by Prolyl Hydroxylase Domain and von Hippel-Lindau Proteins*. J. Biol. Chem. 2011, 286, 42027–42036. [Google Scholar] [CrossRef] [Green Version]
- Alexandru, G.; Graumann, J.; Smith, G.T.; Kolawa, N.J.; Fang, R.; Deshaies, R.J. UBXD7 Binds Multiple Ubiquitin Ligases and Implicates p97 in HIF1α Turnover. Cell 2008, 134, 804–816. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Cardote, T.A.F.; Gadd, M.S.; Ciulli, A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017, 25, 901–911.e903. [Google Scholar] [CrossRef] [Green Version]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
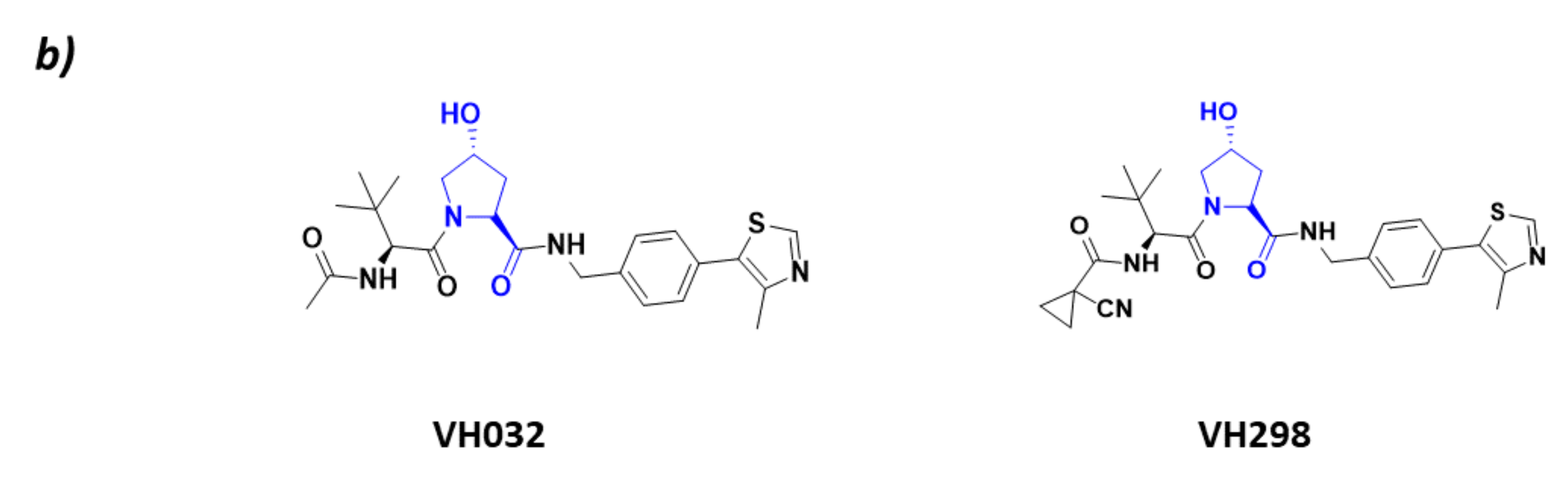
- Soares, P.; Gadd, M.S.; Frost, J.; Galdeano, C.; Ellis, L.; Epemolu, O.; Rocha, S.; Read, K.D.; Ciulli, A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61, 599–618. [Google Scholar] [CrossRef]
- Dias, D.M.; Van Molle, I.; Baud, M.G.J.; Galdeano, C.; Geraldes, C.F.G.C.; Ciulli, A. Is NMR Fragment Screening Fine-Tuned to Assess Druggability of Protein–Protein Interactions? ACS Med. Chem. Lett. 2014, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
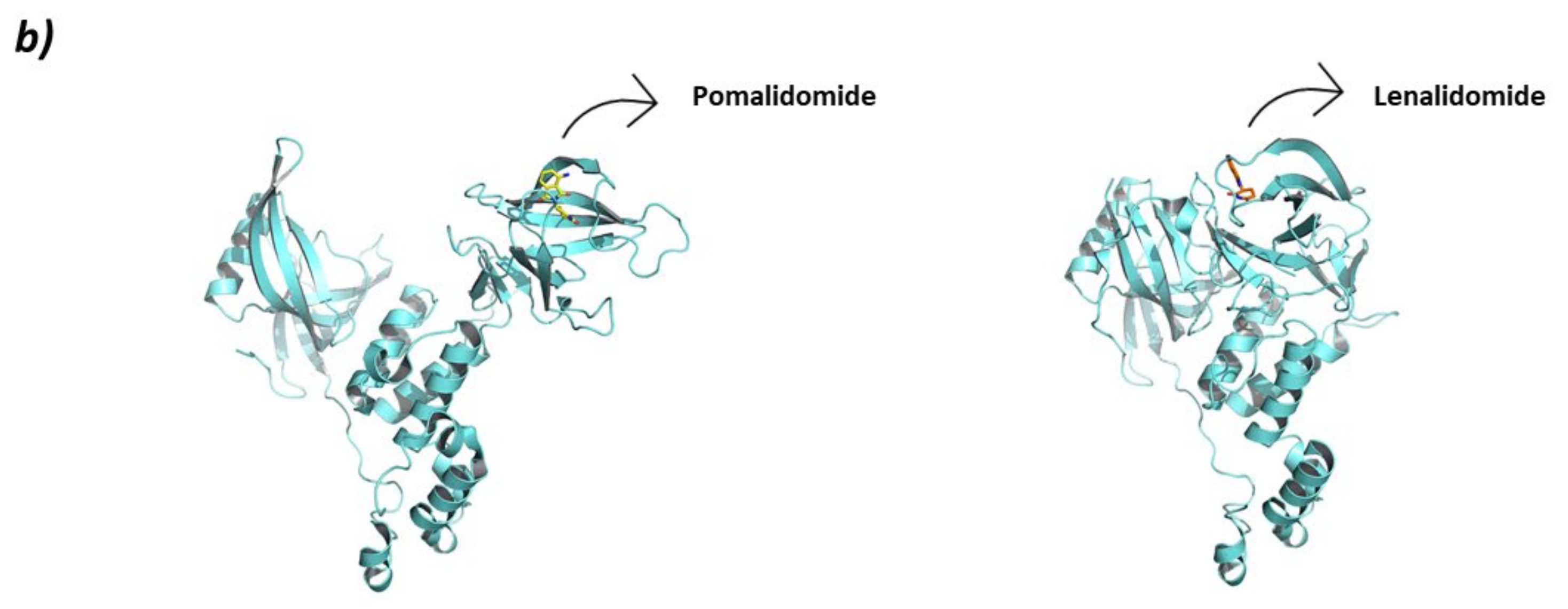
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef]
- Dijkhuizen, T.; van Essen, T.; van der Vlies, P.; Verheij, J.B.G.M.; Sikkema-Raddatz, B.; van der Veen, A.Y.; Gerssen-Schoorl, K.B.J.; Buys, C.H.C.M.; Kok, K. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am. J. Med Genet. Part. A 2006, 140A, 2482–2487. [Google Scholar] [CrossRef]
- Jo, S.; Lee, K.-H.; Song, S.; Jung, Y.-K.; Park, C.-S. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 2005, 94, 1212–1224. [Google Scholar] [CrossRef]
- Kim, H.K.; Ko, T.H.; Nyamaa, B.; Lee, S.R.; Kim, N.; Ko, K.S.; Rhee, B.D.; Park, C.-S.; Nilius, B.; Han, J. Cereblon in health and disease. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1299–1309. [Google Scholar] [CrossRef]
- Mori, T.; Ito, T.; Liu, S.; Ando, H.; Sakamoto, S.; Yamaguchi, Y.; Tokunaga, E.; Shibata, N.; Handa, H.; Hakoshima, T. Structural basis of thalidomide enantiomer binding to cereblon. Sci. Rep. 2018, 8, 1294. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Słabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thomä, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Chen, L. Cereblon: A Protein Crucial to the Multiple Functions of Immunomodulatory Drugs as well as Cell Metabolism and Disease Generation. J. Immunol. Res. 2017, 2017, 9130608. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Chen, H.; Chen, F.; Liu, N.; Wang, X.; Gou, S. Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin-proteasome pathway. Bioorganic Chem. 2018, 81, 536–544. [Google Scholar] [CrossRef]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef]
- Li, L.; Xue, W.; Shen, Z.; Liu, J.; Hu, M.; Cheng, Z.; Wang, Y.; Chen, Y.; Chang, H.; Liu, Y.; et al. A Cereblon Modulator CC-885 Induces CRBN- and p97-Dependent PLK1 Degradation and Synergizes with Volasertib to Suppress Lung Cancer. Mol. Ther. Oncolytics 2020, 18, 215–225. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.-C.; Miller, K.; Fang, W.; Wang, N.-Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef]
- Surka, C.; Jin, L.; Mbong, N.; Lu, C.-C.; Jang, I.S.; Rychak, E.; Mendy, D.; Clayton, T.; Tindall, E.; Hsu, C.; et al. CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells. Blood 2021, 137, 661–677. [Google Scholar] [CrossRef]
- Gao, S.; Wang, S.; Song, Y. Novel immunomodulatory drugs and neo-substrates. Biomark. Res. 2020, 8, 2. [Google Scholar] [CrossRef]
- Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular Mechanisms of the Teratogenic Effects of Thalidomide. Pharmaceuticals 2020, 13, 95. [Google Scholar] [CrossRef]
- Yamshon, S.; Ruan, J. IMiDs New and Old. Curr. Hematol. Malig. Rep. 2019, 14, 414–425. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, P.P.; Cathers, B.E. Cereblon modulators: Low molecular weight inducers of protein degradation. Drug Discov. Today Technol. 2019, 31, 29–34. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thomä, N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, M.J.; Mittag, T. The ubiquitin ligase adaptor SPOP in cancer. FEBS J. 2019, 286, 3946–3958. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-Substrate Complexes: Insights into Molecular Architectures of BTB-Cul3 Ubiquitin Ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.-Q.; Zheng, T.; Chen, B.; Luo, C.; Ouyang, S.; Gong, S.; Li, J.; Mao, L.-L.; Lian, F.; Yang, Y.; et al. Small-Molecule Targeting of E3 Ligase Adaptor SPOP in Kidney Cancer. Cancer Cell 2016, 30, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Ci, Y.; Dai, X.; Wu, F.; Guo, J.; Liu, D.; North, B.J.; Huo, J.; Zhang, J. Cullin 3 SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget 2017, 8, 47890–47901. [Google Scholar] [CrossRef] [Green Version]
- Van Geersdaele, L.K.; Stead, M.A.; Harrison, C.M.; Carr, S.B.; Close, H.J.; Rosbrook, G.O.; Connell, S.D.; Wright, S.C. Structural basis of high-order oligomerization of the cullin-3 adaptor SPOP. Acta Crystallogr. Sect. D 2013, 69, 1677–1684. [Google Scholar] [CrossRef]
- Marzahn, M.R.; Marada, S.; Lee, J.; Nourse, A.; Kenrick, S.; Zhao, H.; Ben-Nissan, G.; Kolaitis, R.-M.; Peters, J.L.; Pounds, S.; et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016, 35, 1254–1275. [Google Scholar] [CrossRef]
- Errington, W.J.; Khan, M.Q.; Bueler, S.A.; Rubinstein, J.L.; Chakrabartty, A.; Privé, G.G. Adaptor Protein Self-Assembly Drives the Control of a Cullin-RING Ubiquitin Ligase. Structure 2012, 20, 1141–1153. [Google Scholar] [CrossRef] [Green Version]
- Scolnick, D.M.; Halazonetis, T.D. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 2000, 406, 430–435. [Google Scholar] [CrossRef]
- Yu, X.; Minter-Dykhouse, K.; Malureanu, L.; Zhao, W.-M.; Zhang, D.; Merkle, C.J.; Ward, I.M.; Saya, H.; Fang, G.; van Deursen, J.; et al. Chfr is required for tumor suppression and Aurora A regulation. Nat. Genet. 2005, 37, 401–406. [Google Scholar] [CrossRef]
- Kim, M.; Kwon, Y.E.; Song, J.O.; Bae, S.J.; Seol, J.H. CHFR negatively regulates SIRT1 activity upon oxidative stress. Sci. Rep. 2016, 6, 37578. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.M.; Kwon, Y.E.; Kim, J.M.; Bae, S.J.; Lee, B.K.; Yoo, S.J.; Chung, C.H.; Deshaies, R.J.; Seol, J.H. Chfr is linked to tumour metastasis through the downregulation of HDAC1. Nat. Cell Biol. 2009, 11, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.; Heimsath, E.G.; Loring, G.L.; Brenner, C. FHA-RING ubiquitin ligases in cell division cycle control. Cell. Mol. Life Sci. 2008, 65, 3458–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavridi, E.S.; Huyen, Y.; Loreto, I.R.; Scolnick, D.M.; Halazonetis, T.D.; Pavletich, N.P.; Jeffrey, P.D. Crystal Structure of the FHA Domain of the Chfr Mitotic Checkpoint Protein and Its Complex with Tungstate. Structure 2002, 10, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-D. FHA: A Signal Transduction Domain with Diverse Specificity and Function. Structure 2002, 10, 887–888. [Google Scholar] [CrossRef] [Green Version]
- Oberoi, J.; Richards, M.W.; Crumpler, S.; Brown, N.; Blagg, J.; Bayliss, R. Structural Basis of Poly(ADP-ribose) Recognition by the Multizinc Binding Domain of Checkpoint with Forkhead-associated and RING Domains (CHFR)*. J. Biol. Chem. 2010, 285, 39348–39358. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Qian, Y.; Chen, X. Pirh2 RING-finger E3 ubiquitin ligase: Its role in tumorigenesis and cancer therapy. FEBS Lett. 2012, 586, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.; Sunnerhagen, M.; Srisailam, S.; Karra, M.; et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.-j.; Hakem, R.; Hakem, A. Pirh2. Cell Cycle 2013, 12, 2733–2737. [Google Scholar] [CrossRef] [Green Version]
- Logan, I.R.; Gaughan, L.; McCracken, S.R.C.; Sapountzi, V.; Leung, H.Y.; Robson, C.N. Human PIRH2 Enhances Androgen Receptor Signaling through Inhibition of Histone Deacetylase 1 and Is Overexpressed in Prostate Cancer. Mol. Cell. Biol. 2006, 26, 6502–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Choi, Y.M.; An, I.-S.; Bae, S.; Jung, J.H.; An, S. E3 ligase RCHY1 negatively regulates HDAC2. Biochem. Biophys. Res. Commun. 2020, 521, 37–41. [Google Scholar] [CrossRef]
- Shimada, M.; Kitagawa, K.; Dobashi, Y.; Isobe, T.; Hattori, T.; Uchida, C.; Abe, K.; Kotake, Y.; Oda, T.; Suzuki, H.; et al. High expression of Pirh2, an E3 ligase for p27, is associated with low expression of p27 and poor prognosis in head and neck cancers. Cancer Sci. 2009, 100, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohgaki, M.; Hakem, A.; Halaby, M.J.; Bohgaki, T.; Li, Q.; Bissey, P.A.; Shloush, J.; Kislinger, T.; Sanchez, O.; Sheng, Y.; et al. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ. 2013, 20, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chen, J.; Han, X.; Zhang, E.; Huang, X.; Guo, X.; Chen, Q.; Wu, W.; Zheng, G.; He, D.; et al. Pirh2 mediates the sensitivity of myeloma cells to bortezomib via canonical NF-κB signaling pathway. Protein Cell 2018, 9, 770–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Bach, I.; Rodriguez-Esteban, C.; Carrière, C.; Bhushan, A.; Krones, A.; Rose, D.W.; Glass, C.K.; Andersen, B.; Belmonte, J.C.I.; Rosenfeld, M.G. RLIM inhibits functional activity of LIM homeodomain transcription factors via recruitment of the histone deacetylase complex. Nat. Genet. 1999, 22, 394–399. [Google Scholar] [CrossRef]
- Wang, F.; Bach, I. Rlim/Rnf12, Rex1, and X Chromosome Inactivation. Front. Cell Dev. Biol. 2019, 7, 258. [Google Scholar] [CrossRef]
- Frints, S.G.M.; Ozanturk, A.; Rodríguez Criado, G.; Grasshoff, U.; de Hoon, B.; Field, M.; Manouvrier-Hanu, S.E.; Hickey, S.; Kammoun, M.; Gripp, K.W.; et al. Pathogenic variants in E3 ubiquitin ligase RLIM/RNF12 lead to a syndromic X-linked intellectual disability and behavior disorder. Mol. Psychiatry 2019, 24, 1748–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, F.; Segarra-Fas, A.; Chaugule, V.K.; Brandenburg, L.; Branigan, E.; Toth, R.; Macartney, T.; Knebel, A.; Hay, R.T.; Walden, H.; et al. RNF12 X-Linked Intellectual Disability Mutations Disrupt E3 Ligase Activity and Neural Differentiation. Cell Rep. 2018, 23, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, H.; Zhou, F.; Schimmel, J.; Pardo, C.G.; Zhang, T.; Barakat, T.S.; Sheppard, K.-A.; Mickanin, C.; Porter, J.A.; et al. RNF12 Controls Embryonic Stem Cell Fate and Morphogenesis in Zebrafish Embryos by Targeting Smad7 for Degradation. Mol. Cell 2012, 46, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Wang, L.; Cai, H.; Zhu, J.; Yu, L. E3 Ubiquitin Ligase RLIM Negatively Regulates c-Myc Transcriptional Activity and Restrains Cell Proliferation. PLoS ONE 2016, 11, e0164086. [Google Scholar] [CrossRef]
- Wagner, T.; Brand, P.; Heinzel, T.; Krämer, O.H. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 524–538. [Google Scholar] [CrossRef]
- Jin, J.-O.; Lee, G.D.; Nam, S.H.; Lee, T.H.; Kang, D.H.; Yun, J.K.; Lee, P.C.-W. Sequential ubiquitination of p53 by TRIM28, RLIM, and MDM2 in lung tumorigenesis. Cell Death Differ. 2021, 28, 1790–1803. [Google Scholar] [CrossRef] [PubMed]
- Middleton, A.J.; Zhu, J.; Day, C.L. The RING Domain of RING Finger 12 Efficiently Builds Degradative Ubiquitin Chains. J. Mol. Biol. 2020, 432, 3790–3801. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, C. Regulation of the SIAH2-HIF-1 Axis by Protein Kinases and Its Implication in Cancer Therapy. Front. Cell Dev. Biol. 2021, 9, 687. [Google Scholar]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Chillappagari, S.; Belapurkar, R.; Möller, A.; Molenda, N.; Kracht, M.; Rohrbach, S.; Schmitz, M.L. SIAH2-mediated and organ-specific restriction of HO-1 expression by a dual mechanism. Sci. Rep. 2020, 10, 2268. [Google Scholar] [CrossRef] [Green Version]
- Scortegagna, M.; Hockemeyer, K.; Dolgalev, I.; Poźniak, J.; Rambow, F.; Li, Y.; Feng, Y.; Tinoco, R.; Otero, D.C.; Zhang, T.; et al. Siah2 control of T-regulatory cells limits anti-tumor immunity. Nat. Commun. 2020, 11, 99. [Google Scholar] [CrossRef]
- Zhao, H.-L.; Ueki, N.; Hayman, M.J. The Ski protein negatively regulates Siah2-mediated HDAC3 degradation. Biochem. Biophys. Res. Commun. 2010, 399, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, Z.; Hou, F.; Harding, R.; Huang, X.; Dong, A.; Walker, J.R.; Tong, Y. The substrate binding domains of human SIAH E3 ubiquitin ligases are now crystal clear. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3095–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, V.; Xiong, S.; Iwakuma, T.; Quintás-Cardama, A.; Lozano, G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. USA 2011, 108, 11995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnussen, H.M.; Ahmed, S.F.; Sibbet, G.J.; Hristova, V.A.; Nomura, K.; Hock, A.K.; Archibald, L.J.; Jamieson, A.G.; Fushman, D.; Vousden, K.H.; et al. Structural basis for DNA damage-induced phosphoregulation of MDM2 RING domain. Nat. Commun. 2020, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Frenk, R.; Stad, R.; de Graaf, P.; Marine, J.-C.; Vousden Karen, H.; Jochemsen Aart, G. Critical Role for a Central Part of Mdm2 in the Ubiquitylation of p53. Mol. Cell. Biol. 2003, 23, 4929–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uldrijan, S.; Pannekoek, W.-J.; Vousden, K.H. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007, 26, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Klejnot, M.; Kowalczyk, D.; Hock, A.K.; Sibbet, G.J.; Vousden, K.H.; Huang, D.T. Structural analysis of MDM2 RING separates degradation from regulation of p53 transcription activity. Nat. Struct. Mol. Biol. 2017, 24, 578–587. [Google Scholar] [CrossRef]
- Poyurovsky, M.V.; Priest, C.; Kentsis, A.; Borden, K.L.B.; Pan, Z.-Q.; Pavletich, N.; Prives, C. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007, 26, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Liao, G.; Yu, B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: Advances and perspectives. Acta Pharm. Sin. B 2020, 10, 1253–1278. [Google Scholar] [CrossRef] [PubMed]
- Kallen, J.; Izaac, A.; Chau, S.; Wirth, E.; Schoepfer, J.; Mah, R.; Schlapbach, A.; Stutz, S.; Vaupel, A.; Guagnano, V.; et al. Structural States of Hdm2 and HdmX: X-ray Elucidation of Adaptations and Binding Interactions for Different Chemical Compound Classes. ChemMedChem 2019, 14, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Twarda-Clapa, A.; Neochoritis, C.G.; Surmiak, E.; Machula, M.; Wisniewska, A.; Labuzek, B.; Ali, A.M.; Krzanik, S.; Dubin, G.; et al. A fluorinated indole-based MDM2 antagonist selectively inhibits the growth of p53wt osteosarcoma cells. FEBS J. 2019, 286, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Surmiak, E.; Twarda-Clapa, A.; Zak, K.M.; Musielak, B.; Tomala, M.D.; Kubica, K.; Grudnik, P.; Madej, M.; Jablonski, M.; Potempa, J.; et al. A Unique Mdm2-Binding Mode of the 3-Pyrrolin-2-one- and 2-Furanone-Based Antagonists of the p53-Mdm2 Interaction. Acs Chem. Biol. 2016, 11, 3310–3318. [Google Scholar] [CrossRef] [PubMed]
- Bogen, S.L.; Pan, W.; Gibeau, C.R.; Lahue, B.R.; Ma, Y.; Nair, L.G.; Seigel, E.; Shipps, G.W.; Tian, Y.; Wang, Y.; et al. Discovery of Novel 3,3-Disubstituted Piperidines as Orally Bioavailable, Potent, and Efficacious HDM2-p53 Inhibitors. Acs Med. Chem. Lett. 2016, 7, 324–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollner, A.; Rudolph, D.; Arnhof, H.; Bauer, M.; Blake, S.M.; Boehmelt, G.; Cockroft, X.-L.; Dahmann, G.; Ettmayer, P.; Gerstberger, T.; et al. Discovery of Novel Spiro[3H-indole-3,2′-pyrrolidin]-2(1H)-one Compounds as Chemically Stable and Orally Active Inhibitors of the MDM2–p53 Interaction. J. Med. Chem. 2016, 59, 10147–10162. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollner, A.; Weinstabl, H.; Fuchs, J.E.; Rudolph, D.; Garavel, G.; Hofbauer, K.S.; Karolyi-Oezguer, J.; Gmaschitz, G.; Hela, W.; Kerres, N.; et al. Targeted Synthesis of Complex Spiro[3H-indole-3,2′-pyrrolidin]-2(1H)-ones by Intramolecular Cyclization of Azomethine Ylides: Highly Potent MDM2–p53 Inhibitors. ChemMedChem 2019, 14, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.-H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.-K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2-E3 Enzyme Cascade. Science 1999, 286, 1321. [Google Scholar] [CrossRef]
- Weber, J.; Polo, S.; Maspero, E. HECT E3 Ligases: A Tale With Multiple Facets. Front. Physiol 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, P.; Levy-Cohen, G.; Blank, M. Smurfs in Protein Homeostasis, Signaling, and Cancer. Front. Oncol. 2018, 8, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Ying, Y. The Post-translational Modifications of Smurf2 in TGF-β Signaling. Front. Mol. Biosci. 2020, 7, 128. [Google Scholar] [CrossRef]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. An Expanded WW Domain Recognition Motif Revealed by the Interaction between Smad7 and the E3 Ubiquitin Ligase Smurf2*. J. Biol. Chem. 2006, 281, 17069–17075. [Google Scholar] [CrossRef] [Green Version]
- David, D.; Nair, S.A.; Pillai, M.R. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 119–128. [Google Scholar] [CrossRef]
- Obri, A.; Makinistoglu, M.P.; Zhang, H.; Karsenty, G. HDAC4 integrates PTH and sympathetic signaling in osteoblasts. J. Cell Biol. 2014, 205, 771–780. [Google Scholar] [CrossRef]
- Fu, L.; Cui, C.-P.; Zhang, X.; Zhang, L. The functions and regulation of Smurfs in cancers. Semin. Cancer Biol. 2020, 67, 102–116. [Google Scholar] [CrossRef]
- Ogunjimi, A.A.; Briant, D.J.; Pece-Barbara, N.; Le Roy, C.; Di Guglielmo, G.M.; Kavsak, P.; Rasmussen, R.K.; Seet, B.T.; Sicheri, F.; Wrana, J.L. Regulation of Smurf2 Ubiquitin Ligase Activity by Anchoring the E2 to the HECT Domain. Mol. Cell 2005, 19, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, S.; Ogunjimi, A.A.; Wang, H.-R.; Rotin, D.; Sicheri, F.; Wrana, J.L.; Forman-Kay, J.D. Autoinhibition of the HECT-Type Ubiquitin Ligase Smurf2 through Its C2 Domain. Cell 2007, 130, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529. [Google Scholar] [CrossRef]
- Zhang, J.; Kan, S.; Huang, B.; Hao, Z.; Mak, T.W.; Zhong, Q. Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 2011, 25, 2610–2618. [Google Scholar] [CrossRef] [Green Version]
- Giles, A.C.; Grill, B. Roles of the HUWE1 ubiquitin ligase in nervous system development, function and disease. Neural Dev. 2020, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Heng, J.I.-T.; Guardavaccaro, D.; Jiang, R.; Pagano, M.; Guillemot, F.; Iavarone, A.; Lasorella, A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 2008, 10, 643–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.-H.; Wu, H.-T.; Wu, K.-J. Ubiquitination by HUWE1 in tumorigenesis and beyond. J. Biomed. Sci. 2018, 25, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myant, K.B.; Cammareri, P.; Hodder, M.C.; Wills, J.; Von Kriegsheim, A.; Győrffy, B.; Rashid, M.; Polo, S.; Maspero, E.; Vaughan, L.; et al. HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 2017, 9, 181–197. [Google Scholar] [CrossRef]
- Peter, S.; Bultinck, J.; Myant, K.; Jaenicke, L.A.; Walz, S.; Müller, J.; Gmachl, M.; Treu, M.; Boehmelt, G.; Ade, C.P.; et al. Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol. Med. 2014, 6, 1525–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, L.J.; Campbell, D.C.; Morgan, J.J.; Lawson, M.A.; Down, J.M.; Chauhan, D.; McAvera, R.M.; Morris, T.C.; Hamilton, C.; Krishnan, A.; et al. The E3 ligase HUWE1 inhibition as a therapeutic strategy to target MYC in multiple myeloma. Oncogene 2020, 39, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Striegl, H.; Andrade-Navarro, M.A.; Heinemann, U. Armadillo Motifs Involved in Vesicular Transport. PLoS ONE 2010, 5, e8991. [Google Scholar] [CrossRef]
- Hunkeler, M.; Jin, C.Y.; Ma, M.W.; Overwijn, D.; Monda, J.K.; Bennett, E.J.; Fischer, E.S. Modular HUWE1 architecture serves as hub for degradation of cell-fate decision factors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sander, B.; Xu, W.; Eilers, M.; Popov, N.; Lorenz, S. A conformational switch regulates the ubiquitin ligase HUWE1. eLife 2017, 6, e21036. [Google Scholar] [CrossRef]
- Bricelj, A.; Steinebach, C.; Kuchta, R.; Gütschow, M.; Sosič, I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 2021, 9, 463. [Google Scholar] [CrossRef]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bübül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. Chem. Med. Chem. 2021, 16, 1336–1359. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhao, W.; Zhao, C.; Liu, Q.; Li, S.; Zhang, G.; Chou, C.J.; Zhang, Y. Development of a Bestatin-SAHA Hybrid with Dual Inhibitory Activity against APN and HDAC. Molecules 2020, 25, 4991. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C. Drugging the undruggable: Targeting challenging E3 ligases for personalized medicine. Future Med. Chem. 2017, 9, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.-P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E3 Family | E3 Subfamily | Implicated Ligase | HDAC | PROTAC |
|---|---|---|---|---|
| RING | Cullin RING | Cul3-RENKCTD11 | 1 | |
| CRL2VHL | 1 †, 2 †, 3 †, and 6 † | YES | ||
| CRL4CRBN | 1 †, 2 †, 3 †, and 6 † | YES | ||
| Cul3SPOP | 6 | |||
| Monomeric RING | CHFR * | 1 and 2 | ||
| PIRH2 (RCHY1) * | 1 and 2 | |||
| RLIM (RNF12) * | 2 | |||
| Homodimeric RING | SIAH2 | 3 | ||
| Heterodimeric RING | MDM | 2 | ||
| HECT | NEDD4 | Smurf2 | 4 | |
| Others | HUWE1 (Mule) * | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márquez-Cantudo, L.; Ramos, A.; Coderch, C.; de Pascual-Teresa, B. Proteasomal Degradation of Zn-Dependent Hdacs: The E3-Ligases Implicated and the Designed Protacs That Enable Degradation. Molecules 2021, 26, 5606. https://doi.org/10.3390/molecules26185606
Márquez-Cantudo L, Ramos A, Coderch C, de Pascual-Teresa B. Proteasomal Degradation of Zn-Dependent Hdacs: The E3-Ligases Implicated and the Designed Protacs That Enable Degradation. Molecules. 2021; 26(18):5606. https://doi.org/10.3390/molecules26185606
Chicago/Turabian StyleMárquez-Cantudo, Laura, Ana Ramos, Claire Coderch, and Beatriz de Pascual-Teresa. 2021. "Proteasomal Degradation of Zn-Dependent Hdacs: The E3-Ligases Implicated and the Designed Protacs That Enable Degradation" Molecules 26, no. 18: 5606. https://doi.org/10.3390/molecules26185606