
Autophagic Activation and Decrease of Plasma Membrane Cholesterol Contribute to Anticancer Activities in Non-Small Cell Lung Cancer


Abstract
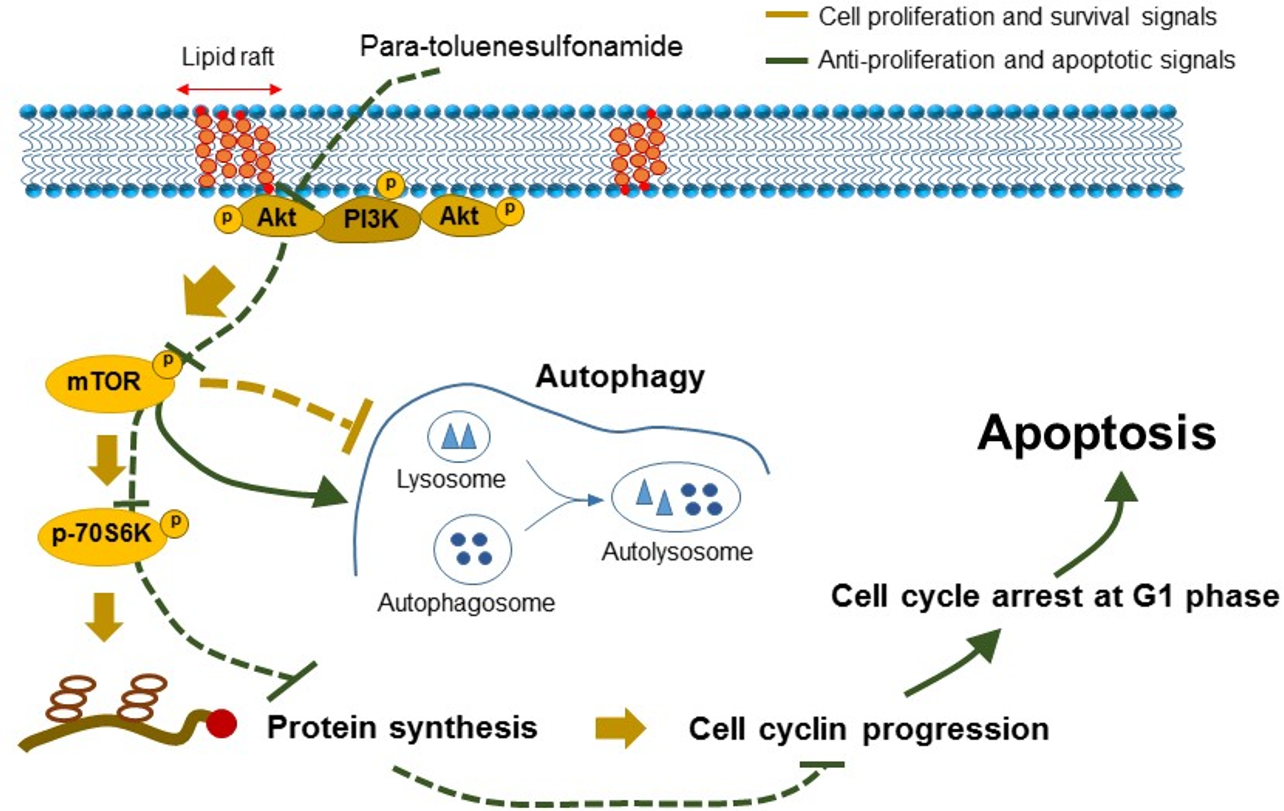
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
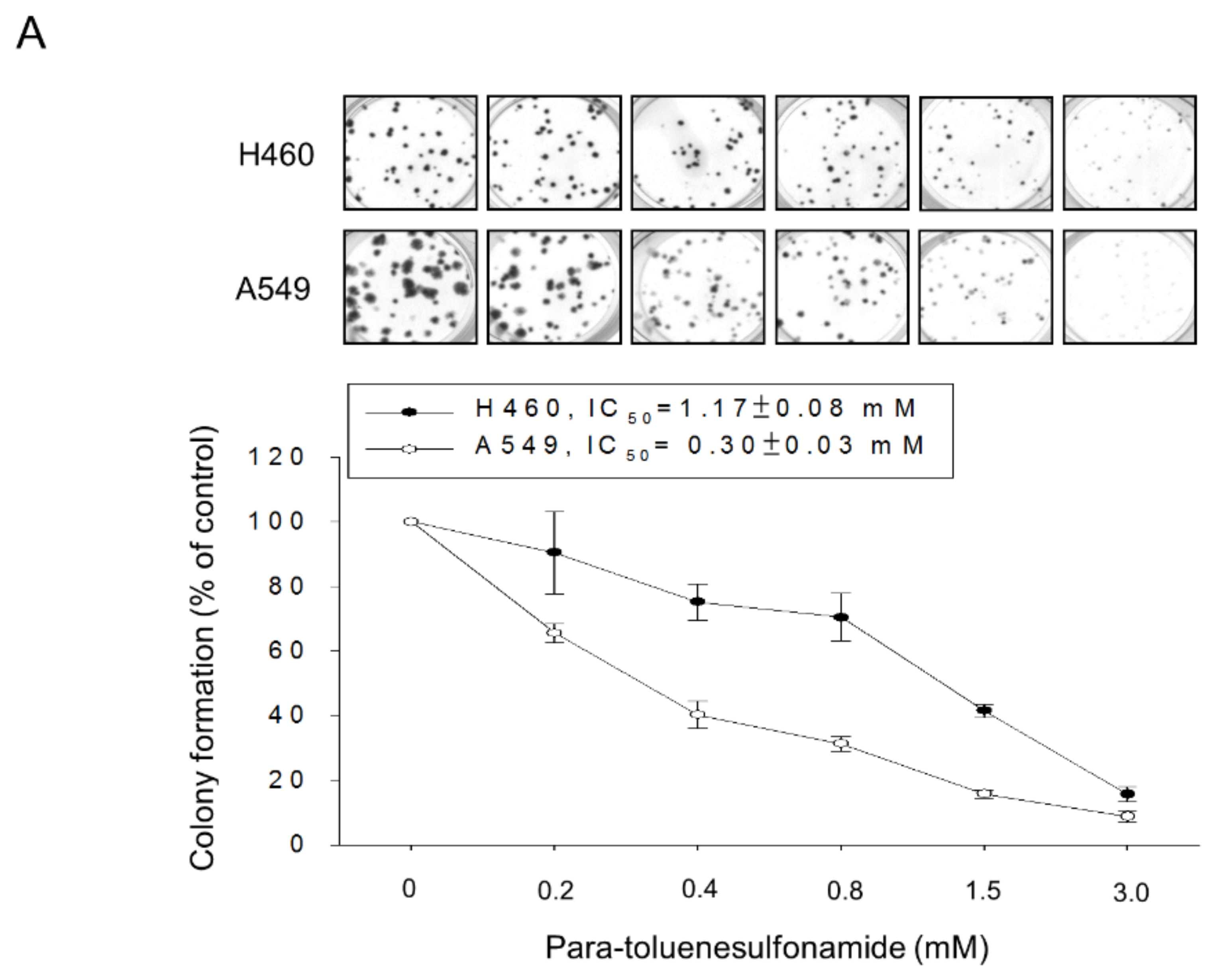
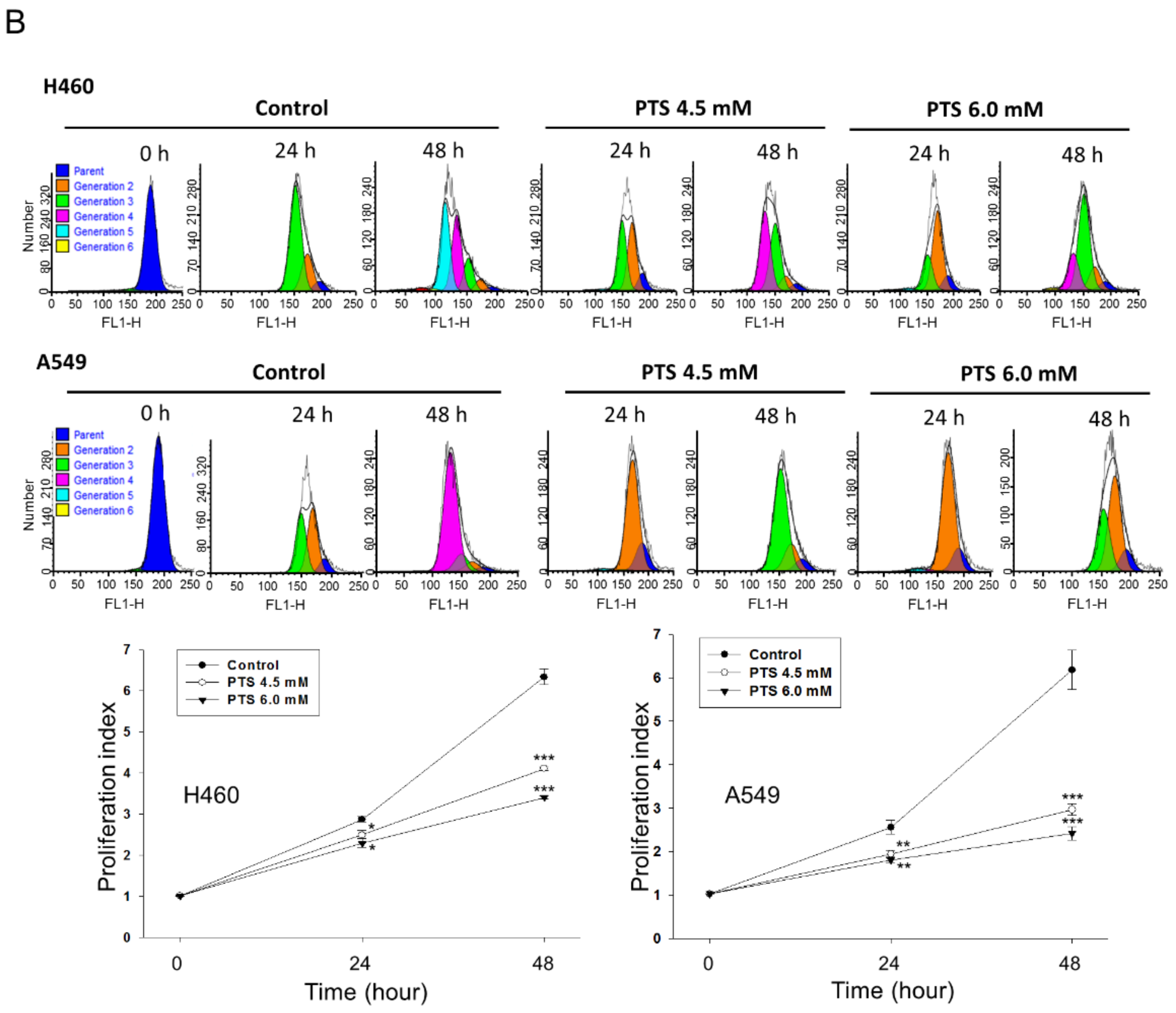
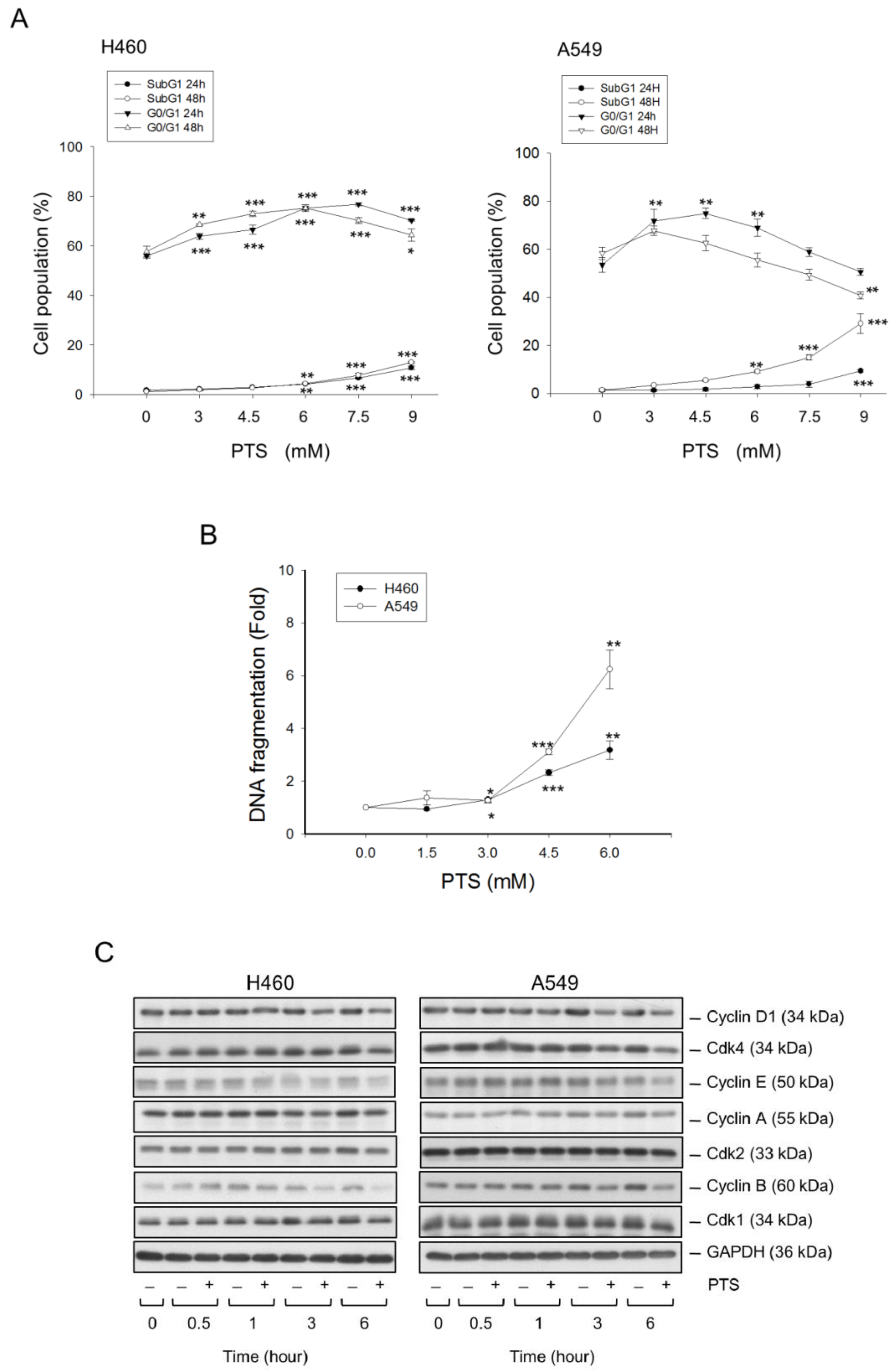
2.1. Para-Toluenesulfonamide Induces Anti-NSCLC Effects through Inhibition of Akt/mTOR/p70S6K Pathway
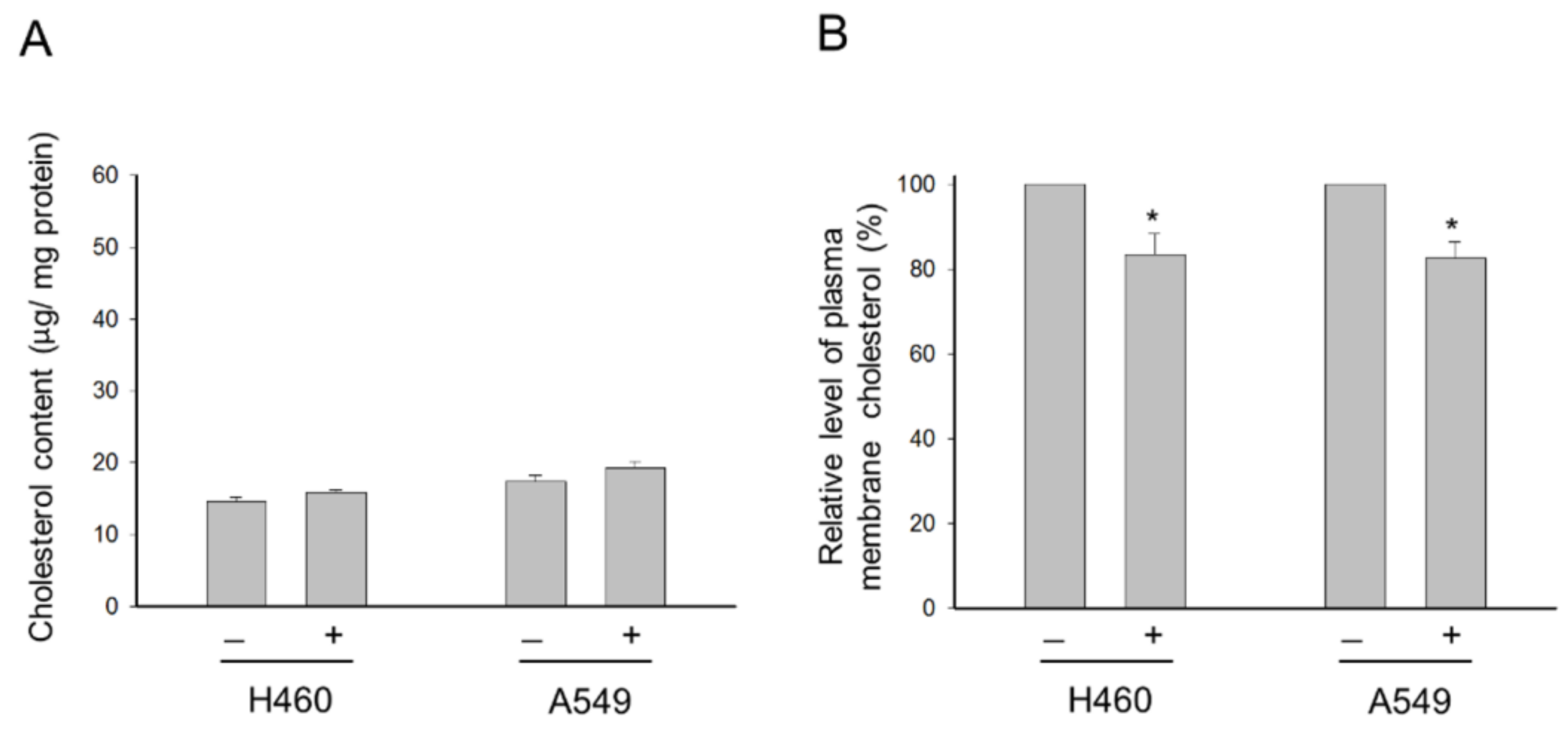
2.2. Para-Toluenesulfonamide Suppresses Cholesterol Levels of the Plasma Membrane
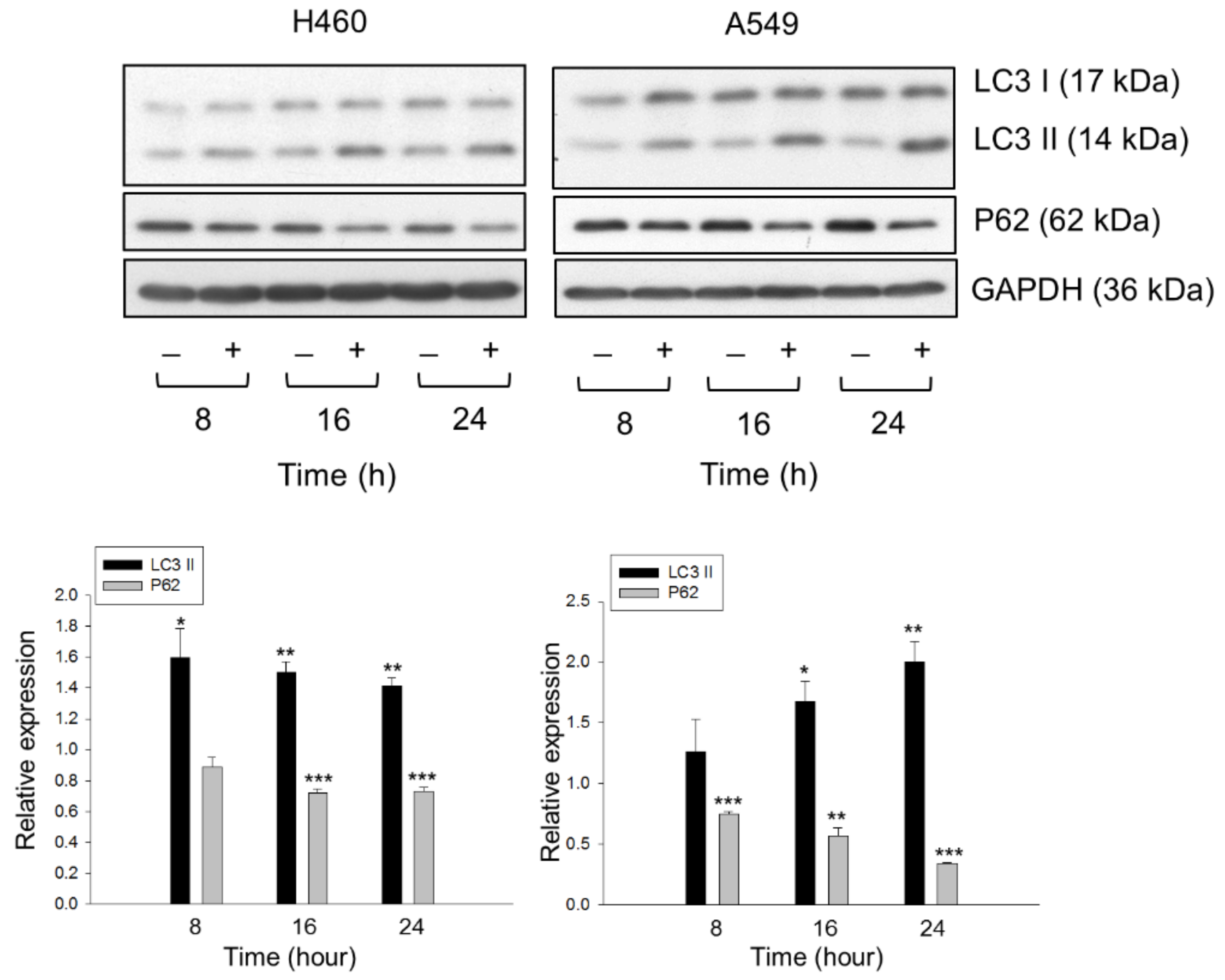
2.3. Lysosome Plays a Role in Determining the Fate of Cholesterol through Autophagic Activation
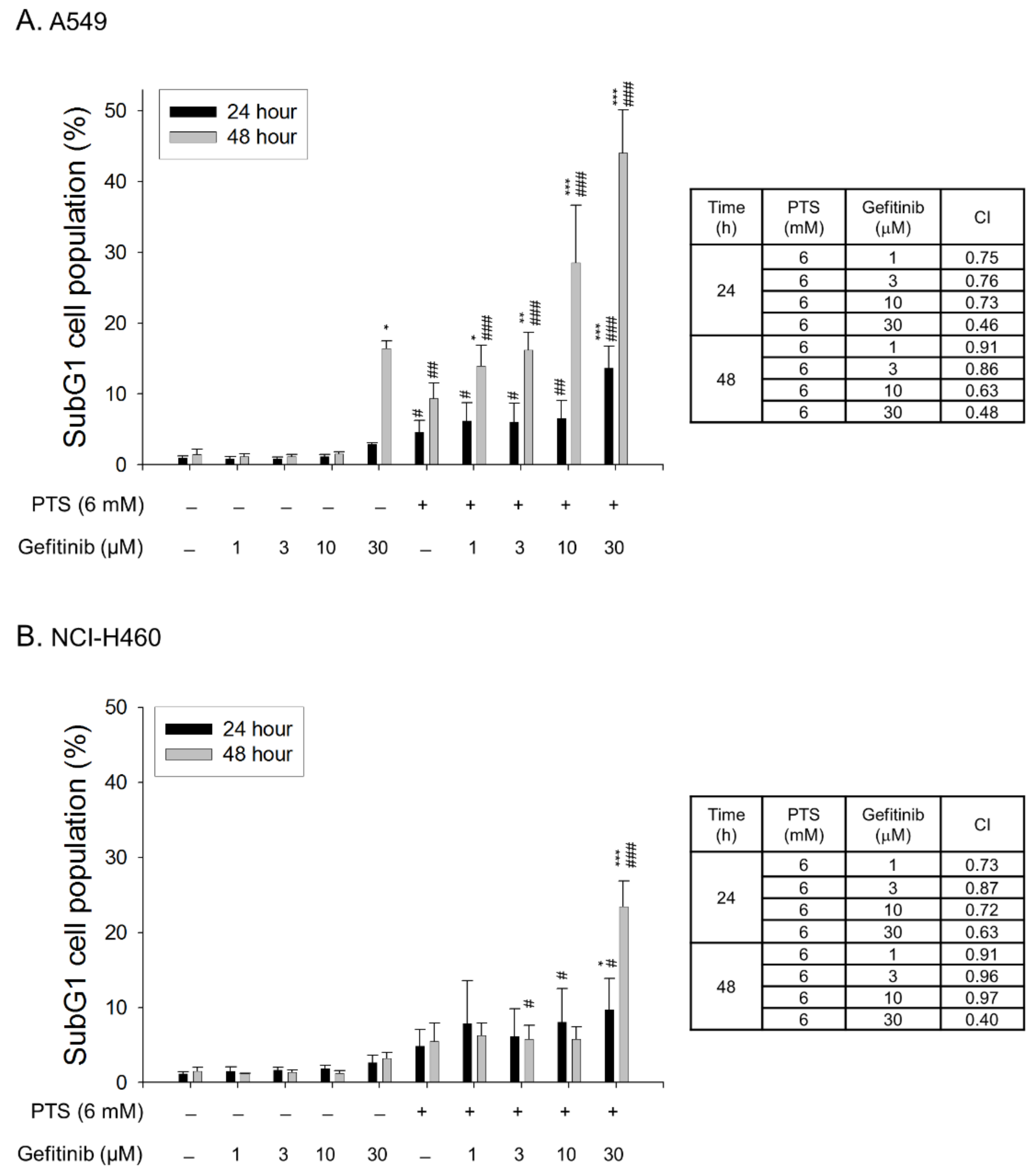
2.4. Para-Toluenesulfonamide Synergistically Potentiates Gefitinib-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. SRB Assay
4.4. Colony Formation Assay
4.5. CFSE Staining Assay
4.6. Detection of Cell Population in Cell Cycle Progression
4.7. Detection of Nucleosomal DNA Cleavage
4.8. Western Blot Analysis
4.9. Immunofluorescence Staining of Lysosomes and Cholesterol
4.10. Detection of Total Cellular Cholesterol Levels
4.11. Purification of Plasma Membrane
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| Cdk | Cyclin-dependent kinase |
| CFSE | Carboxyfluorescein succinimidyl ester |
| 4E-BP1 | eIF4E-binding protein 1 |
| EGFR | Epidermal growth factor receptor |
| eIF4E | Eukaryotic translation initiation factor 4E |
| FBS | Fetal bovine serum |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| NSCLC | Non-small-cell lung cancer |
| p70S6K | p70S6 kinase |
| PBS | Phosphate-buffered saline |
| PI | Propidium iodide |
| PI3K | Phosphatidylinositol 3-kinase |
| PMSF | Phenylmethylsulfonylfluoride |
| SRB | Sulforhodamine B |
| TCA | Trichloroacetic acid |
| TCGA | The Cancer Genome Atlas |
References
- Testa, U.; Castelli, G.; Pelosi, E. Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, H.; Dally, H.; Popanda, O.; Risch, A.; Schmezer, P. Genetic risk profiles for cancer susceptibility and therapy response. Recent Results Cancer Res. 2007, 174, 19–36. [Google Scholar]
- Di Maio, M.; Gridelli, C.; Gallo, C.; Shepherd, F.; Piantedosi, F.V.; Cigolari, S.; Manzione, L.; Illiano, A.; Barbera, S.; Robbiati, S.F.; et al. Chemotherapy-induced neutropenia and treatment efficacy in advanced non-small-cell lung cancer: A pooled analysis of three randomized trials. Lancet Oncol. 2005, 6, 669–677. [Google Scholar] [CrossRef]
- Ribaudo, G.; Zanforlin, E.; Zagotto, G. Overcoming resistance in non-small-cell lung cancer: A practical lesson for the medicinal chemist. Arch. Pharm. 2018, 351, e1800037. [Google Scholar] [CrossRef]
- Pakkala, S.; Ramalingam, S.S. Personalized therapy for lung cancer: Striking a moving target. JCI Insight 2018, 3, 120858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L.; Canadian Cancer Registries Epidemiology Research Group. Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500. [Google Scholar] [PubMed]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol metabolism: A potential therapeutic target in glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusselmans, K.; Timmermans, L.; Van de Sande, T.; Van Veldhoven, P.P.; Guan, G.; Shechter, I.; Claessens, F.; Verhoeven, G.; Swinnen, J.V. Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J. Biol. Chem. 2007, 282, 18777–18785. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.F.; Jan, Y.H.; Liu, Y.P.; Yang, C.J.; Su, C.Y.; Chang, Y.C.; Lai, T.C.; Chiou, J.; Tsai, H.Y.; Lu, J.; et al. Squalene synthase induces tumor necrosis factor receptor 1 enrichment in lipid rafts to promote lung cancer metastasis. Am. J. Respir. Crit. Care Med. 2014, 190, 675–687. [Google Scholar] [CrossRef]
- Hanai, J.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J. Cell Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Gao, Y.; Guan, W.; Huang, L.; Xu, X.; Zhang, C.; Chen, X.; Wu, Y.; Zeng, G.; Zhong, N. Antitumor effect of para-toluenesulfonamide against lung cancer xenograft in a mouse model. J. Thorac. Dis. 2013, 5, 472–483. [Google Scholar] [PubMed]
- Liu, Z.; Liang, C.; Zhang, Z.; Pan, J.; Xia, H.; Zhong, N.; Li, L. Para-toluenesulfonamide induces tongue squamous cell carcinoma cell death through disturbing lysosomal stability. Anticancer Drugs 2015, 26, 1026–1033. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.L.; Leu, W.J.; Hsu, L.C.; Liu, S.P.; Zhong, N.S.; Guh, J.H. Para-toluenesulfonamide induces anti-tumor activity through Akt-dependent and –independent mTOR/p70S6K pathway: Roles of lipid raft and cholesterol contents. Front. Pharmacol. 2018, 9, 1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Kuang, A.R.; Guan, Y.S.; Liu, Y.Q. Puncture injection of para-toluenesulfonamide combined with chemoembolization for advanced hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 6861–6864. [Google Scholar] [CrossRef]
- He, J.; Ying, W.; Yang, H.; Xu, X.; Shao, W.; Guan, Y.; Jiang, M.; Wu, Y.; Zhong, B.; Wang, D.; et al. Gemcitabine plus cisplatin chemotherapy with concurrent para-toluenesulfonamide localinjection therapy for peripherally advanced nonsmall cell lung cancer larger than 3 cm in the greatest dimension. Anticancer Drugs 2009, 20, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Li, S.Y.; Zhong, N.S. Effects of para-toluenesulfonamide intratumoral injection on pulmonary adenoid cystic carcinoma complicating with severe central airway obstruction: A 5-year follow-up study. J. Thorac. Dis. 2018, 10, 2448–2455. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Li, Q.; Guan, W.J.; Huang, J.; Yang, H.P.; Wu, G.M.; Jin, F.G.; Hu, C.P.; Chen, L.A.; Xu, G.L.; et al. Effects of para-toluenesulfonamide intratumoral injection on non-small cell lung carcinoma with severe central airway obstruction: A multi-center, non-randomized, single-arm, open-label trial. Lung Cancer 2016, 98, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Hu, W.; Han, W.B.; Liu, Y.L.; Tu, Y.; Yang, H.M.; Fang, Q.J.; Zhou, M.Y.; Wan, Z.Y.; Tang, R.M.; et al. Inhibition of Akt/mTOR/p70S6K signaling activity with Huangkui capsule alleviates the early glomerular pathological changes in diabetic nephropathy. Front. Pharmacol. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Roux, P.P. Translational control by oncogenic signaling pathways. Biochim. Biophys. Acta. 2015, 1849, 753–765. [Google Scholar] [CrossRef]
- Ghayad, S.E.; Cohen, P.A. Inhibitors of the PI3K/Akt/mTOR pathway: New hope for breast cancer patients. Recent Pat. Anticancer Drug Discov. 2010, 5, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Silvius, J.R. Role of cholesterol in lipid raft formation: Lessons from lipid model systems. Biochim. Biophys. Acta 2003, 1610, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Lin, J.; Lu, M.L.; Solomon, K.R.; Freeman, M.R. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar]
- Calay, D.; Vind-Kezunovic, D.; Frankart, A.; Lambert, S.; Poumay, Y.; Gniadecki, R.J. Inhibition of Akt signaling by exclusion from lipid rafts in normal and transformed epidermal keratinocytes. J. Investig. Dermatol. 2010, 130, 1136–1145. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Ohsaki, Y.; Tauchi-Sato, K.; Fujita, A.; Fujimoto, T. Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 2006, 351, 246–252. [Google Scholar] [CrossRef]
- Motoyama, K.; Kameyama, K.; Onodera, R.; Araki, N.; Hirayama, F.; Uekama, K.; Arima, H. Involvement of PI3K-Akt-Bad pathway in apoptosis induced by 2,6-di-O-methyl-beta-cyclodextrin, not 2,6-di-O-methyl-alpha-cyclodextrin, through cholesterol depletion from lipid rafts on plasma membranes in cells. Eur. J. Pharm. Sci. 2009, 38, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liu, L.; Fu, Y.; Gao, J.; He, Y.; Wu, Y.; Lian, X. Dietary cholesterol intake and risk of lung cancer: A meta-analysis. Nutrients 2018, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Henslee, A.B.; Steele, T.A. Combination statin and chemotherapy inhibits proliferation and cytotoxicity of an aggressive natural killer cell leukemia. Biomark. Res. 2018, 6, 26. [Google Scholar] [CrossRef]
- Mandal, C.C.; Rahman, M.M. Targeting intracellular cholesterol is a novel therapeutic strategy for cancer treatment. J. Cancer Sci. Ther. 2014, 6, 510–513. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Qiu, Z.X.; Zhang, K.; Qiu, X.S.; Zhou, M.; Li, W.M. The prognostic value of phosphorylated AKT expression in non-small cell lung cancer: A meta-analysis. PLoS ONE 2013, 8, e81451. [Google Scholar] [CrossRef]
- Gately, K.; Al-Alao, B.; Dhillon, T.; Mauri, F.; Cuffe, S.; Seckl, M.; O’Byrne, K. Overexpression of the mammalian target of rapamycin (mTOR) and angioinvasion are poor prognostic factors in early stage NSCLC: A verification study. Lung Cancer 2012, 75, 217–222. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Y.; Chen, B.; Zeng, J.; Guo, N.; Zhang, S.; Liu, L.; Xu, H.; Mo, X.; Li, W. Activation of mammalian target of rapamycin pathway confers adverse outcome in nonsmall cell lung carcinoma. Cancer 2011, 117, 3763–3773. [Google Scholar]
- Ayuso, M.I.; Hernández-Jiménez, M.; Martín, M.E.; Salinas, M.; Alcázar, A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-bindingprotein 1 (4E-BP1) in ischemia-reperfusion stress. J. Biol. Chem. 2010, 285, 34355–34363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinnebusch, A.G. Translational homeostasis via eIF4E and 4E-BP1. Mol. Cell. 2012, 46, 717–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Boyle, D.W.; Liechty, E.A. Changes in 4E-BP1 and p70S6K phosphorylation in skeletal muscle of the ovine fetus after prolonged maternal fasting: Effects of insulin and IGF-I. Pediatr. Res. 2005, 58, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Zhang, H.; Tan, Y.; Sun, C.; Liang, Y.; Yu, J.; Zou, H. Aggregation of lipid rafts activates c-met and c-Src in non-small cell lung cancer cells. BMC Cancer 2018, 18, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, R.T.; Sousa, D.; Gomes, A.S.; Mendes, N.; Matthiesen, R.; Pedro, M.; Marques, F.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. The antitumor activity of a lead thioxanthone is associated with alterations in cholesterol localization. Molecules 2018, 23, 3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell. Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Eid, W.; Dauner, K.; Courtney, K.C.; Gagnon, A.; Parks, R.J.; Sorisky, A.; Zha, X. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. USA 2017, 114, 7999–8004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Duarte, A.; Poderoso, C.; Cooke, M.; Soria, G.; Cornejo Maciel, F.; Gottifredi, V.; Podestá, E.J. Mitochondrial fusion is essential for steroid biosynthesis. PLoS ONE 2012, 7, e45829. [Google Scholar] [CrossRef] [Green Version]
- Hall, P.F.; Almahbobi, G. Roles of microfilaments and intermediate filaments in adrenal steroidogenesis. Microsc. Res. Tech. 1997, 36, 463–479. [Google Scholar] [CrossRef]
- Martin, L.A.; Kennedy, B.E.; Karten, B.J. Mitochondrial cholesterol: Mechanisms of import and effects on mitochondrial function. Bioenerg. Biomembr. 2016, 48, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basañez, G.; Antonsson, B.; Prieto, J.; García-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccone, G.; Herbst, R.S.; Manegold, C.; Scagliotti, G.; Rosell, R.; Miller, V.; Natale, R.B.; Schiller, J.H.; Von Pawel, J.; Pluzanska, A.; et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: A phase III trial--INTACT 1. J. Clin. Oncol. 2004, 22, 777–784. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, J.-L.; Leu, W.-J.; Zhong, N.-S.; Guh, J.-H. Autophagic Activation and Decrease of Plasma Membrane Cholesterol Contribute to Anticancer Activities in Non-Small Cell Lung Cancer. Molecules 2021, 26, 5967. https://doi.org/10.3390/molecules26195967
Hsu J-L, Leu W-J, Zhong N-S, Guh J-H. Autophagic Activation and Decrease of Plasma Membrane Cholesterol Contribute to Anticancer Activities in Non-Small Cell Lung Cancer. Molecules. 2021; 26(19):5967. https://doi.org/10.3390/molecules26195967
Chicago/Turabian StyleHsu, Jui-Ling, Wohn-Jenn Leu, Nan-Shan Zhong, and Jih-Hwa Guh. 2021. "Autophagic Activation and Decrease of Plasma Membrane Cholesterol Contribute to Anticancer Activities in Non-Small Cell Lung Cancer" Molecules 26, no. 19: 5967. https://doi.org/10.3390/molecules26195967