
Studies on the Enantioselective Synthesis of E-Ethylidene-bearing Spiro[indolizidine-1,3′-oxindole] Alkaloids



1
Laboratory of Organic Chemistry, Faculty of Pharmacy and Food Sciences, and Institute of Biomedicine (IBUB), University of Barcelona, 08028 Barcelona, Spain
2
Department of Nutrition, Food Sciences and Gastronomy, Faculty of Pharmacy and Food Sciences, and Institute of Nutrition and Food Safety (INSA-UB), University of Barcelona, 08921 Santa Coloma de Gramanet, Spain
3
Institut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Cerdanyola, Spain
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(2), 428; https://doi.org/10.3390/molecules26020428
Submission received: 29 December 2020
/
Revised: 11 January 2021
/
Accepted: 12 January 2021
/
Published: 15 January 2021
(This article belongs to the Special Issue Application of Organic Synthesis to Bioactive Compounds II)
Abstract
:A synthetic route for the enantioselective construction of the tetracyclic spiro[indolizidine-1,3′-oxindole] framework present in a large number of oxindole alkaloids, with a cis H-3/H-15 stereochemistry, a functionalized two-carbon substituent at C-15, and an E-ethylidene substituent at C-20, is reported. The key steps of the synthesis are the generation of the tetracyclic spirooxindole ring system by stereoselective spirocyclization from a tryptophanol-derived oxazolopiperidone lactam, the removal of the hydroxymethyl group, and the stereoselective introduction of the E-ethylidene substituent by acetylation at the α-position of the lactam carbonyl, followed by hydride reduction and elimination. Following this route, the 21-oxo derivative of the enantiomer of the alkaloid 7(S)-geissoschizol oxindole has been prepared.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The spiro[pyrrolidine-3,3′-oxindole] ring system is a structural moiety found in a large number of natural products, pharmaceuticals, and biologically active compounds. This moiety is present in oxindole alkaloids, which constitute a large group of monoterpenoid alkaloids characterized by a spiro-fusion to a pyrrolidine ring at the 3-position of the indole core [1,2,3]. Oxindole alkaloids possess a variety of pharmacological properties [4,5,6,7,8,9,10,11,12] and have served as an inspiration for the development of new therapeutic agents [13]. One of the most important substructural classes of these alkaloids incorporates a tetracyclic spiro[indolizidine-1,3′-oxindole] framework, in which the pyrrolidine nucleus is embedded in an indolizidine ring. Structurally, these tetracyclic oxindole alkaloids differ in the functionalized substituent at C-15 and the two-carbon substituent at C-20 (usually ethyl, vinyl or E-ethylidene), as well as in the configuration at the C-3, C-7, C-15, and (in some cases) C-20 stereocenters (biogenetic numbering) [14], thus giving rise to a diversified array of relative stereochemical relationships. Some representative examples are depicted in Figure 1.
The members of this group of alkaloids often occur in nature as pairs of C-7 epimers [15,16], interconvertible under acidic or basic conditions by retro-Mannich/Mannich reactions via a ring-opened intermediate [17,18,19,20]. Scheme 1 illustrates this equilibrium for geissoschizol oxindoles [16].
The appealing structure of these alkaloids and their significant biological activities have attracted considerable synthetic attention, resulting in a large number of total syntheses, both in the racemic series and in enantiopure form [21,22,23,24]. However, the access to E-ethylidene-bearing spiro[indolizidine-1,3′-oxindoles] with a cis H-3/H-15 stereochemistry has been little explored and to our knowledge no total syntheses of tetracyclic oxindole alkaloids with this substitution and stereochemical pattern have been reported so far. In fact, although the oxidative rearrangement of tetrahydro-β-carbolines is one of the most frequently used methods to assemble the spiro[pyrrolidine-3,3′-oxindole] system [18,19,21], its application to either cis or trans E-ethylidene-bearing Corynanthe-type indolo[2,3-a]quinolizidine amines (but not amides) leads only to C-7 epimeric mixtures of tetracyclic spirooxindoles with a trans H-3/H-15 stereochemistry due to an equilibration of the C-3 and C-7 stereocenters via a reversible Mannich reaction (Scheme 2) [25,26]. The severe A1,3-interaction between the C-15 side chain and the methyl group on the ethylidene moiety in the cis isomers, which is absent in the trans isomers, could account for the exclusive formation of the latter [26].
2. Results and Discussion
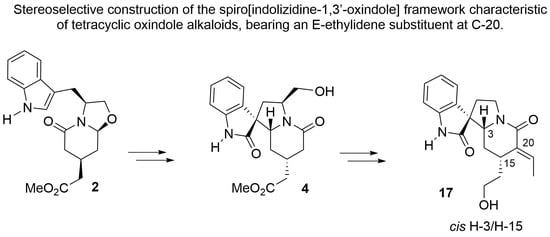
We present herein a procedure for the enantioselective construction of cis H-3/H-15 tetracyclic spiro[indolizidine-1,3′-oxindoles] bearing an E-ethylidene substituent at the C-20 position. The cis H-3/H-15 relationship was secured using the spirocyclization procedure we have recently reported for the direct generation of spirooxindoles from tryptophanol-derived oxazolopiperidone lactams [27], whereas the E-ethylidene substituent was stereoselectively installed, taking advantage of the piperidone carbonyl present in the resulting tetracyclic spirooxindole. Scheme 3 outlines the initial steps of the synthesis.
The required bicyclic lactam 2 was prepared, as previously reported [28], by cyclocondensation of prochiral aldehyde-diester 1 with (S)-tryptophanol in a process that involves the stereoselective desymmetrization of two enantiotopic acetate chains. A subsequent bromination with pyridinium perbromide, with careful control of the reaction time (10 s) and operating under strictly anhydrous conditions to minimize the formation of the corresponding oxindole, afforded 2-bromoindole 3 in an improved yield (92%). The latter underwent smooth spirocyclization on treatment with TFA to provide (71%) tetracyclic spirooxindole 4 as a single stereoisomer, whose absolute configuration was unambiguously confirmed by X-ray crystallographic analysis (Figure 2; see Supplementary Materials).
Removal of the hydroxymethyl substituent, which plays a decisive role as an element of stereocontrol in the spirocyclization reaction, was initially accomplished in four steps: oxidation to carboxylic acid 6 via the corresponding aldehyde 5, subsequent generation of selenoester 7, and finally radical reductive decarbonylation (Scheme 4) [29,30]. Following this procedure, tetracyclic oxindole 8 was obtained in 45% overall yield from aldehyde 5.
Alternatively, 8 was prepared in a more efficient manner by direct rhodium(I)-induced decarbonylation of aldehyde 5. Thus, treatment of 5 with the rhodium(I) complex generated in situ from the chloro(1,5-cyclooctadiene)rhodium(I) dimer {[RhCl(cod)]2} and the bidentate phosphine ligand 1,3-bis(diphenylphosphino)propane (dppp) [31] satisfactorily provided oxindole 8 in 79% yield. The use of the rhodium(I) complex derived from carbonyl(chloro)bis(triphenylphosphine)rhodium(I) [RhCl(CO)(PPh3)2] and dppp [32] gave less satisfactory results (46% yield).
The preparation of 8 in four steps and 40% overall yield from tryptophanol-derived lactam 2 represents a notable improvement with respect to the previously reported eight-step route from the same starting lactam 2, involving the generation of a spiroindoline and the final oxidation of an indoline [27,28], which gave 8 in 13% overall yield (Scheme 5).
Having secured an efficient and reliable sequence to access the tetracyclic scaffold 8, we focused on the introduction of the C-20 two-carbon substituent. Chemoselective LiBH4 reduction of the ester group and subsequent successive protection of the resulting primary alcohol 9 with tert-butydimethylsilyl chloride (TBDMSCl) and the indole nitrogen with either methoxymethyl chloride (MOMCl) or di-tert-butyl dicarbonate (Boc2O) afforded the diprotected intermediates 12 and 13 (Scheme 6). Under the LiBH4 reduction conditions, only minor amounts (5%) of the corresponding indoline 10 were formed.
Initial attempts to directly introduce a 1-hydroxyethyl chain by an aldol-type reaction between 12 or 13 and acetaldehyde (LDA, −78 °C) were unsuccessful, and the corresponding alcohols were only formed in trace amounts. Satisfactorily, the E-ethylidene substituent was installed by acylation at the α-position of the lactam carbonyl of 12, followed by hydride reduction and stereoselective elimination. Thus, acetylation of 12 with methyl acetate in the presence of LDA afforded ketone 14 as a single stereoisomer in 52% yield, with 29% of the starting material being recovered. Although a subsequent reduction with NaBH4 gave a nearly equimolecular mixture of epimeric alcohols 15a and 15b, they could be separated by column chromatography and independently converted to the E-ethylidene derivative 16 in excellent overall yield.
Alcohol 15a was dehydrated by treatment with DCC and CuCl as a catalyst in refluxing toluene [33,34,35] to stereoselectively provide the E-ethylidene derivative 16 in 91% yield. This is a useful method for the syn elimination of β-hydroxycarbonyl compounds that proceeds via an isourea intermediate through a six-membered cyclic transition state [36]. In turn, alcohol 15b was subjected to an anti-elimination sequence [34,35] by treatment of the corresponding mesylate with DBU to give the expected E-ethylidene derivative 16 (60% yield) along with minor amounts of its Z isomer (20% yield). The stereochemistry of both isomers was assigned from their 1H-NMR spectra, in which the olefinic proton of the E isomer appears at a lower field (δ 6.67) than that of the Z isomer (δ 5.94) due to the anisotropic effect of the lactam carbonyl group. A final treatment of 16 under smooth conditions brought about the simultaneous deprotection of the oxindole and alcohol moieties to provide the cis H-3/H-15, E-ethylidene-bearing spiro[indolizidine-1,3′-oxindole] 17.
In summary, we have developed a procedure for the stereoselective construction of the spiro[indolizidine-1,3′-oxindole] framework characteristic of tetracyclic oxindole alkaloids, bearing a cis H-3/H-15 stereochemistry and incorporating a functionalized two-carbon substituent at C-15 and an E-ethylidene substituent at C-20. These studies could open synthetic routes to tetracyclic oxindole alkaloids featuring this substitution and stereochemical pattern, for instance, 7(S)-kopsirensine A or 7(S)-isositsirikine oxindole. In fact, compound 17 can be envisaged as the enantiomer of 21-oxo-7(S)-geissoschizol oxindole. The use of (R)-tryptophanol instead of the S enantiomer in the synthetic sequence outlined in Scheme 6 would provide access to the natural enantiomeric series.
3. Experimental Section
3.1. General Information
All air sensitive manipulations were carried out under a dry argon or nitrogen atmosphere. THF and CH2Cl2 were dried using a column solvent purification system. Analytical thin-layer chromatography was performed on SiO2 (Merck silica gel 60 F254), and the spots were located with 1% aqueous KMnO4. Chromatography refers to flash chromatography and was carried out on SiO2 (SDS silica gel 60 ACC, 35–75 mm, 230–240 mesh ASTM). NMR spectra were recorded at 400 (1H) and 100.6 (13C), and chemical shifts are reported in δ values downfield from TMS or relative to residual chloroform (7.26 ppm, 77.0 ppm) as an internal standard. Data are reported in the following manner: Chemical shift, multiplicity, coupling constant (J) in hertz (Hz), integrated intensity, and assignment (when possible). Assignments and stereochemical determinations are given only when they are derived from definitive two-dimensional NMR experiments (HSQC-COSY). IR spectra were performed in a spectrophotometer Nicolet Avantar 320 FT-IR, and only noteworthy IR absorptions (cm−1) are listed. High resolution mass spectra (HMRS) were performed by Centres Científics i Tecnològics de la Universitat de Barcelona.
3.2. Improved Preparation of Tetracyclic Oxindole 4
(3S,7R,8aS)-3-[(2-Bromo-3-indolyl)methyl]-7-(methoxycarbonylmethyl)-5-oxo-2,3,6,7,8,8a-hexahydro-5H-oxazolo[3,2-a]pyridine (3). A solution of pyridinium tribromide (PyHBr3, 4.20 g, 13.1 mmol) in anhydrous THF (57 mL) was added at 0 °C by a large transfer, under an argon atmosphere, to a stirred solution of lactam 2 [28] (3.21 g, 9.36 mmol) in anhydrous CH2Cl2 (57 mL). Saturated aqueous solutions of Na2S2O3 (19 mL) and NaHCO3 (10 mL) were added 10 s later. Distilled H2O (10 mL) was added, and the mixture was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 7:3 hexane-EtOAc) of the resulting residue gave bromo derivative 3 (3.63 g, 92%) as a white foam: [α]22D = −74.1 (c 1.4, CHCl3); IR (KBr): 3171 (NH), 1630, 1734 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 1.31–1.40 (qm, J = 11.6 Hz, 1H, H-8), 2.17 (dd, J = 17.6, 10.4 Hz, 1H, H-6), 2.32 (dm, J = 11.6 Hz, 1H, H-8), 2.37 (m, 1H, H-7), 2.40 (s, 2H, CH2CO2), 2.65 (dd, J = 17.6, 4.8 Hz, 1H, H-6), 2.69 (dd, J = 14.0, 10.4 Hz, 1H, CH2-Ind), 3.65–3.69 (m, 2H, CH2-Ind, H-2), 3.69 (s, 3H, CH3O), 4.08 (d, J = 8.8 Hz, 1H, H-2), 4.31 (ddd, J = 9.6, 6.8, 3.2 Hz, 1H, H-3), 4.70 (dd, J = 10.0, 3.2 Hz, 1H, H-8a), 7.07 (td, J = 8.0, 0.8 Hz, 1H, HAR), 7.13 (td, J = 8.0, 0.8 Hz, 1H, HAR), 7.28 (dd, J = 8.0, 0.8 Hz, 1H, HAR), 7.76 (dd, J = 8.0, 0.8 Hz, 1H, HAR), 9.28 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 26.4 (CH2-Ind), 27.2 (C-7), 34.4 (C-8), 37.6 (C-6), 40.1 (CH2CO), 51.8 (CH3O), 55.5 (C-3), 70.2 (C-2), 88.3 (C-8a), 108.8 (C-Br), 110.4 (CHAR), 112.0 (CAR), 118.7 (CHAR), 120.3 (CHAR), 122.5 (CHAR), 127.9 (CAR), 136.1 (CAR), 166.8 (CO), 171.8 (CO); HRMS (ESI) calcd for [C19H22BrN2O4 + Na+]: 433.0577, found: 433.0577.
(1’R,3′S,7′R,8a’R)-3′-(Hydroxymethyl)-7′-(methoxycarbonylmethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (4). TFA (3.15 mL, 40.9 mmol) was added at room temperature under an argon atmosphere to a solution of bromo derivative 3 (556 mg, 1.32 mmol) in anhydrous CH2Cl2 (25 mL). The resulting mixture was stirred at room temperature for 16 h and then concentrated under reduced pressure. Flash chromatography (silica previously washed with Et3N; 1:1 to 1:9 hexane-EtOAc) of the resulting residue gave spirooxindole 4 (337 mg, 71%) as a white foam: [α]22D = + 46.1 (c 1.0, CHCl3); IR (KBr): 3401 (NH), 1617, 1724 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.69 (q, J = 12.4 Hz, 1H, H-14), 1.70 (dm, J = 12.4 Hz, 1H, H-14), 1.96 (dd, J = 18.0, 12.0 Hz, 1H, H-20), 2.14–2.26 (m, 4H, H-6, CH2CO), 2.38 (m, 1H, H-15), 2.67 (ddd, J = 18.0, 3.6, 1.2 Hz, 1H, H-20), 3.63 (s, 3H, CH3O), 3.82 (dd, J = 12.0, 2.4 Hz, 1H, CH2OH), 3.89 (dd, J = 12.0, 7.2 Hz, 1H, CH2OH), 4.10 (dd, J = 12.4, 4.4 Hz, 1H, H-3), 4.64 (m, 1H, H-5), 6.92 (d, J = 7.6 Hz, 1H, HAR), 7.00 (d, J = 7.6 Hz, 1H, HAR), 7.07 (td, J = 7.6, 0.8 Hz, 1H, HAR), 7.29 (td, J = 7.6, 1.2 Hz, 1H, HAR), 7.88 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 29.0 (C-15), 29.6 (C-14), 36.7 (C-6), 37.5 (C-20), 39.6 (CH2CO), 51.7 (CH3O), 56.2 (C-7), 60.0 (C-5), 64.5 (C-3), 66.5 (CH2OH), 110.7 (CHAR), 123.2 (CHAR), 123.5 (CHAR), 128.9 (CHAR), 129.4 (CAR), 140.3 (CAR), 170.9 (CO), 171.6 (CO), 177.2 (CO); HRMS (ESI) calcd for [C19H22N2O5 + H+]: 359.1601, found: 359.1601.
3.3. Removal of the Hydroxymethyl Substituent
(1’R,3′S,7′R,8a’R)-3′-Formyl-7′-(methoxycarbonylmethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (5). Dess-Martin periodinane (DMP, 649 mg, 1.53 mmol) and NaHCO3 (257 mg, 3.06 mmol) were added at room temperature under an argon atmosphere to a solution of spirooxindole 4 (367 mg, 1.02 mmol) in anhydrous CH2Cl2 (38 mL). The resulting mixture was stirred at room temperature for 4 h. Saturated aqueous solutions of Na2S2O3 (30 mL) and NaHCO3 (30 mL) were added and the mixture was stirred for 30 min. The aqueous layer was extracted with CH2Cl2. The combined organic extracts were dried, filtered, and concentrated under reduced pressure. Flash chromatography (1:1 hexane-EtOAc to EtOAc) of the resulting residue gave aldehyde 5 (280 mg, 77%) as a white foam: 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.75 (q, J = 12.4 Hz, 1H, H-14), 1.73 (dm, J = 12.4 Hz, 1H, H-14), 1.99 (dd, J = 17.8, 12.0 Hz, 1H, H-20), 2.14–2.33 (m, 3H, CH2CO, H-6), 2.44–2.53 (m, 2H, H-6, H-15), 2.70 (ddd, J = 17.8, 5.4, 1.9 Hz, 1H, H-20), 3.63 (s, 3H, CH3O), 4.15 (dd, J = 11.4, 4.2 Hz, 1H, H-3), 4.88 (t, J = 9.6 Hz, 1H, H-5), 6.90 (d, J = 7.4 Hz, 1H, HAR), 6.97 (d, J = 7.8 Hz, 1H, HAR), 7.07 (td, J = 7.5, 1.0 Hz, 1H, HAR), 7.30 (td, J = 7.6, 0.4 Hz, 1H, HAR), 7.69 (br. s, 1H, NH), 9.73 (d, J = 1.7 Hz, 1H, CHO); 13C-NMR (100.6 MHz, CDCl3): δ = 29.5 (C-15), 29.7 (C-14), 34.6 (C-6), 37.2 (C-20), 39.7 (CH2CO), 51.7 (CH3O), 56.7 (C-7), 62.9 (C-5), 65.0 (C-3), 110.8 (CHAR), 123.3 (CHAR), 123.5 (CHAR), 128.8 (CHAR), 129.1 (CAR), 140.2 (CAR), 169.1 (CO), 171.6 (CO), 176.4 (CO), 197.4 (CHO); HRMS (ESI) calcd for [C19H20N2O5 + H+]: 357.1445, found: 357.1450.
(1′R,3′S,7′R,8a’R)-7′-(Methoxycarbonylmethyl)-2,5′-dioxo-3′-(phenylselenocarbonyl)spi-ro[indoline-3,1′-indolizidine] (7). First step: 2-Methyl-2-butene (2 M in hexane, 6.4 mL) and t-BuOH (25.2 mL) were added at room temperature to a solution of aldehyde 5 (321 mg, 0.9 mmol) in CH3CN (8.1 mL). A solution of NaClO2 (472 mg, 5.22 mmol) and NaH2PO4 (733 mg, 5.31 mmol) in distilled H2O (8.7 mL) was added at 0 °C to the above mixture, which was stirred at room temperature for 1 h. Then, 0.1 M Na2S2O3 and 2 N HCl solutions were added until pH = 1, and the resulting mixture was extracted with EtOAc, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give carboxylic acid 6, which was used in the next step without purification. Second step: Diphenyl diselenide [(PhSe)2, 478 mg, 1.53 mmol] and tri-n-butylphosphine (n-Bu3P, 0.63 mL, 2.52 mmol) were added at room temperature under an argon atmosphere to a solution of crude acid 6 in anhydrous CH2Cl2 (5.4 mL). The resulting mixture was stirred at reflux for 16 h. Distilled H2O was added. The aqueous layer was extracted with CH2Cl2, and the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 7:3 hexane-EtOAc) of the resulting residue gave seleno ester 7 (216 mg, 50% overall yield for the two steps) as a yellow foam: [α]22D = −4.4 (c 1.04, CHCl3); IR (film): 3242 (NH), 1726, 1698, 1660, 1635 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.73 (q, J = 12.8 Hz, 1H, H-14), 1.75–1.82 (dm, J = 12.8 Hz, 1H, H-14), 1.99 (dd, J = 18.0, 12.0 Hz, 1H, H-20), 2.17 (dd, J = 15.6, 7.6 Hz, 1H, CH2CO), 2.30 (dd, J = 15.6, 6.4 Hz, 1H, CH2CO), 2.46 (dd, J = 12.8, 8.4 Hz, 1H, H-6), 2.46–2.58 (m, 1H, H-15), 2.62 (dd, J = 12.8, 8.4 Hz, 1H, H-6), 2.73–2.79 (dm, J = 18.0 Hz, 1H, H-20), 3.64 (s, 3H, CH3O), 4.37 (dd, J = 11.2, 4.4 Hz, H-3), 5.13 (t, J = 9.2 Hz, 1H, H-5), 6.84 (d, J = 7.6 Hz, 1H, HAR), 6.99 (d, J = 7.6 Hz, 1H, H HAR), 7.05 (td, J = 7.6, 1.2 Hz, 1H, HAR), 7.29 (td, J = 8.0, 1.2 Hz, 1H, HAR), 7.36–7.39 (m, 3H, HAR), 7.52–7.55 (m, 2H, HAR), 8.74 (s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 29.3 (C-15), 29.6 (C-14), 37.5 (C-20), 38.0 (C-6), 39.6 (CH2CO), 51.8 (CH3O), 57.1 (C-7), 65.6 (C-3), 66.4 (C-5), 110.8 (CHAR), 123.3 (CHAR), 123.4 (CHAR), 124.8 (CAR), 128.8–129.4 (4CHAR, CAR), 136.1 (2CHAR), 140.2 (CAR), 169.4 (CO), 171.6 (CO), 176.1 (CO), 200.5 (CO); HRMS (ESI) calcd for [C25H24N2O5Se + H+]: 513.0923, found: 513.0927.
(1′R,7′R,8a’R)-7′-(Methoxycarbonylmethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (8). Method A: from seleno derivative 7: Azobisisobutyronitrile (AIBN, 9 mg, 0.05 mmol) was added under an argon atmosphere to a solution of seleno derivative 7 (216 mg, 0.43 mmol) in anhydrous benzene (20 mL). The mixture was heated to reflux, and a solution of tributyltin hydride (TBTH, 180 µL, 0.65 mmol) in anhydrous benzene (4 mL) was added very slowly (over 30 min). The resulting mixture was stirred at reflux for 1 h, and the solvent was evaporated. Flash chromatography (6:4 hexane-EtOAc to 100% EtOAc) of the resulting residue gave spirooxindole 8 (127 mg, 90%). Method B: from aldehyde 5: Argon was bubbled through anhydrous diglyme (3.2 mL) for 30 min. Chloro(1,5-cyclooctadiene)rhodium(I) dimer (3 mg, 0.006 mmol) and 1,3-bis(diphenylphosphino) propane (dppp, 10 mg, 0.023 mmol) were weighed in corning tubes and introduced into the reaction flask under an argon flow using inert glovebox equipment. Anhydrous diglyme (2.2 mL) was transferred into the reaction flask and the bubbling of argon was continued for 15 min. Aldehyde 5 (80 mg, 0.23 mmol) was dissolved in anhydrous diglyme and transferred into the flask. The mixture was stirred at reflux for 24 h. Distilled H2O (2.2 mL) and CH2Cl2 (2.2 mL) were added, the layers were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (1:1 to 1:9 hexane-EtOAc) of the resulting residue gave compound 8 (57 mg, 75%) as a white foam and minor amounts of its 5,6-dehydro derivative, which was hydrogenated (10% Pd/C, absolute EtOH) to give additional compound 8 (3 mg, 4%) after flash chromatography: [α]22D = + 63.0 (c 0.55, CHCl3); IR (film): 3194 (NH), 1727, 1619 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.72 (q, J = 12.4 Hz, 1H, H-14), 1.67 (dm, J = 12.4 Hz, 1H, H-14), 1.95 (dd, J = 17.6, 12.0 Hz, 1H, H-20), 2.09 (dd, J = 12.4, 8.4 Hz, 1H, H-6), 2.14 (dd, J = 15.6, 7.6 Hz, 1H, CH2CO), 2.23 (dd, J = 15.6, 6.4 Hz, 1H, CH2CO), 2.35 (m, 1H, H-15), 2.52 (q, J = 12.4 Hz, 1H, H-6), 2.61 (dd, J = 17.6, 4.8 Hz, 1H, H-20), 3.61 (s, 3H, CH3O), 3.83 (t, J = 11.2 Hz, 1H, H-5), 3.99 (tm, J = 11.2 Hz, 1H, H-5), 4.04 (dd, J = 11.2, 4.4 Hz, 1H, H-3), 6.91 (d, J = 7.2 Hz, 1H, HAR), 6.99 (d, J = 7.6 Hz, 1H, HAR), 7.05 (t, J = 7.6 Hz, 1H, HAR), 7.28 (t, J = 7.6 Hz, 1H, HAR), 8.92 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 29.6 (C-15), 29.7 (C-14), 33.3 (C-6), 37.3 (C-20), 39.9 (CH2CO), 43.9 (C-5), 51.7 (CH3O), 56.9 (C-7), 64.3 (C-3), 110.5 (CHAR), 123.1 (CHAR), 123.7 (CHAR), 128.6 (CHAR), 129.7 (CAR), 140.2 (CAR), 168.4 (CO), 171.7 (CO), 177.5 (CO); HRMS (ESI) calcd for [C18H20N2O4 + H+]: 329.1496, found: 329.1497.
3.4. Introduction of the E-Ethylidene Chain
(1’R,7′S,8a’R)-7′-(2-Hydroxyethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (9): Lithium borohydride (LiBH4, 20 mg, 0.9 mmol) was added at 0 °C under an argon atmosphere to a solution of compound 8 (49 mg, 0.15 mmol) in anhydrous THF (5 mL). The resulting mixture was stirred at room temperature for 72 h. The reaction was quenched at 0 °C by distilled H2O (5 mL), and the mixture was concentrated under reduced pressure using a rotary evaporator with a dry ice condenser. Flash chromatography (95:5 EtOAc-MeOH) of the resulting residue gave oxindole 9 (34 mg, 76%) as a white foam and minor amounts of indoline 10 (3 mg, 5%). Oxindole 9: [α]22D = + 58,0 (c 1.12, MeOH); IR (film): 3100–3600 (NH, OH), 1731, 1714 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.64 (q, J = 12.0 Hz, 1H, H-14), 1.43 (m, 2H, CH2CH2O), 1.64 (dm, J = 12.0 Hz, 1H, H-14), 1.90 (dd, J = 17.6, 12.0 Hz, 1H, H-20), 2.05 (m, 2H, H-6, H-15), 2.49 (dt, J = 10.4 Hz, 1H, H-6), 2.58 (dd, J = 17.6, 5.2 Hz, 1H, H-20), 3.60 (t, J = 6.4 Hz, 2H, CH2O), 3.80 (dd, J = 11.6, 9.2 Hz, 1H, H-5), 3.98 (m, 2H, H-5, H-3), 6.91 (d, J = 7.6 Hz, 1H, HAR), 6.97 (d, J = 8.0 Hz, 1H, HAR), 7.04 (t, J = 7.6 Hz, 1H, HAR), 7.26 (td, J = 7.6, 1.2 Hz, 1H, HAR), 8.83 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 29.5 (C-15), 29.9 (C-14), 33.2 (C-6), 37.9 (C-20), 38.4 (CH2CH2O), 43.8 (C-5), 57.0 (C-7), 59.7 (CH2O), 64.6 (C-3), 110.3 (CHAR), 123.1 (CHAR), 123.8 (CHAR), 128.6 (CHAR), 129.9 (CAR), 140.1 (CAR), 169.2 (CO), 177.5 (CO); HRMS (ESI) calcd for [C17H20N2O3 + Na+]: 323.1366, found: 323.1371. (1′S,7′S,8a’R)-7′-(2-Hydroxyethyl)-5′-oxospiro[indoline-3,1′-indolizidine] (10): IR (film): 3346 (NH, OH), 1606 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 0.69 (q, J = 12.4 Hz, 1H, H-14), 1.48 (m, 2H, CH2CH2O), 1.83–1.92 (m, 2H, H-14, H-20), 1.97–2.08 (m, 2H, H-15, H-6), 2.25 (ddd, J = 12.8, 8.4, 2.0 Hz, 1H, H-6), 2.54 (ddd, J = 17.6, 5.2, 1.6 Hz, 1H, H-20), 3.50–3.66 (m, 2H, H-5, H-3), 3.51 (d, J = 9.2 Hz, 1H, H-2), 3.57 (d, J = 9.2 Hz, 1H, H-2), 3.65 (t, J = 6.4 Hz, 2H, CH2CH2O), 3.83–3.91 (m, 1H, H-5), 6.66 (d, J = 7.6 Hz, 1H, HAR), 6.71 (t, J = 7.6 Hz, 1H, HAR), 6.77 (dd, J = 7.6, 1.6 Hz, 1H, HAR), 7.08 (td, J = 7.6, 1.2 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = 29.7 (C-15), 30.3 (C-14), 35.9 (C-6), 38.2 (C-20), 38.7 (CH2CH2O), 43.5 (C-5), 54.9 (C-2), 55.4 (C-7), 60.0 (CH2CH2O), 66.1 (C-3), 110.1 (CHAR), 119.5 (CHAR), 124.0 (CHAR), 128.5 (CHAR), 131.0 (CAR), 151.2 (CAR), 169.2 (CO); HRMS (ESI) calcd for [C17H22N2O2 + H+]: 287.1754, found: 287.1758.
(1′R,7′S,8a’R)-7′-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-2,5′-dioxospiro[indoline-3,1′-indolizidine] (11). tert-Butyldimethylsilyl chloride (TBDMSCl, 27 mg, 0.18 mmol) and imidazole (25 mg, 0.36 mmol) were added at 0 °C under an argon atmosphere to a solution of alcohol 9 (27 mg, 0.09 mmol) in anhydrous DMF (1 mL). The resulting mixture was stirred at room temperature for 16 h. Brine was added, and the mixture was extracted with EtOAc. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (7:3 hexane-EtOAc to 1:1 hexane-EtOAc) of the resulting residue gave silyl derivative 11 (30 mg, 80%) as a white foam: [α]22D = + 37.09 (c 0.23, CHCl3); IR (film): 3181 (NH), 1725, 1620 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.01 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si), 0.65 (qd, J = 12.4, 3.6 Hz, 1H, H-14), 0.83 [s, 9H, C(CH3)3], 1.39 (m, 2H, CH2CH2O), 1.65 (dm, J = 12.4 Hz, 1H, H-14), 1.89 (dd, J = 16.4, 11.6 Hz, 1H, H-20), 2.03–2.09 (m, 2H, H-15, H-6), 2.50–2.58 (m, 2H, H-6, H-20), 3.55 (m, 2H, CH2O), 3.81 (t, J = 12.4 Hz, 1H, H-5), 3.95–4.03 (m, 2H, H-5, H-3), 6.91–6.95 (m, 2H, HAR), 7.03–7.07 (m, 1H, HAR), 7.27 (m, 1H, HAR), 7.68 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 18.2 [C(CH3)3], 25.8 [C(CH3)3], 29.7 (C-15), 30.1 (C-14), 33.3 (C-6), 37.9 (C-20), 38.5 (CH2CH2O), 43.8 (C-5), 57.0 (C-7), 60.1 (CH2O), 64.8 (C-3), 110.2 (CHAR), 123.1 (CHAR), 123.9 (CHAR), 128.5 (CHAR), 129.9 (CAR), 140.0 (CAR), 169.2 (CO), 177.5 (CO); HRMS (ESI) calcd for [C23H34N2O3Si + H+]: 415.2411, found: 415.2419.
(1′R,7′S,8a’R)-7′-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-1-(methoxymethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (12). A solution of compound 11 (386 mg, 0.93 mmol) in anhydrous THF (2.5 mL) was transferred at 0 °C under an argon atmosphere to a suspension of NaH (95%, 36 mg, 1.4 mmol) in anhydrous DMF (2.5 mL). The resulting mixture was stirred at 0 °C for 30 min. Methoxymethyl chloride (MOMCl, 0.12 mL, 1.4 mmol) was added, and the resulting mixture was stirred at room temperature for 1.5 h. The mixture was cooled to 0 °C and saturated aqueous NaHCO3 (11.2 mL) was added. The mixture was extracted with EtOAc and the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 7:3 hexane-EtOAc) of the resulting residue gave the N-MOM derivative 12 (283 mg, 67%) as a white foam: [α]22D = + 50.6 (c 2.26, CHCl3); IR (film): 1725, 1651 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.02 (s, 3H, CH3Si), −0.03 (s, 3H, CH3Si), 0.64 (q, J = 12.4 Hz, 1H, H-14), 0.82 [s, 9H, C(CH3)3], 1.37 (dd, J = 12.8, 6.4 Hz, 2H, CH2CH2O), 1.55 (dm, J = 12.4 Hz, 1H, H-14), 1.89 (dd, J = 17.6, 12.0 Hz, 1H, H-20), 2.00–2.07 (m, 2H, H-15, H-6), 2.50–2.57 (m, 2H, H-20, H-6), 3.32 (s, 3H, CH3O), 3.54 (t, J = 6.0 Hz, 2H, CH2CH2O), 3.81 (t, J = 11.6 Hz, 1H, H-5), 3.96–4.03 (m, 2H, H-5, H-3), 5.15 (s, 2H, NCH2O), 6.94 (d, J = 7.6 Hz, 1H, HAR), 7.08–7.11 (m, 2H, HAR), 7.32 (t, J = 7.6 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 18.2 [C(CH3)3], 25.8 [C(CH3)3], 29.6 (C-15), 30.2 (C-14), 33.7 (C-6), 37.8 (C-20), 38.6 (CH2CH2O), 43.8 (C-5), 56.3, 56.9 (C-7, CH3O), 60.0 (CH2CH2O), 64.8 (C-3), 71.5 (NCH2O), 110.0 (CHAR), 123.6 (CHAR), 123.7 (CHAR), 128.6 (CHAR), 129.1 (CAR), 141.2 (CAR), 169.1 (CO), 176.3 (CO); HRMS (ESI) calcd for [C25H38N2O4Si + H+]: 459.2674, found: 459.2675.
(1′R,7′S,8a’R)-7′-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-1-(tert-butoxycarbonyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (13). NaH (95%, 25 mg, 0.64 mmol) and (Boc)2O (70 mg, 0.32 mmol) were added at 0 °C under an argon atmosphere to a solution of compound 11 (33 mg, 0.08 mmol) in anhydrous DMF (1 mL). The mixture was stirred at room temperature for 16 h. The reaction was quenched by the addition of a few drops of distilled H2O, and the resulting residue was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 4:1 EtOAc-MeOH) gave N-Boc derivative 13 (26 mg, 64%): [α]22D = + 56.7 (c 0.92, CHCl3); IR (film): 1794, 1766, 1733 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.28 (s, 3H, CH3Si), −0.21 (s, 3H, CH3Si), 0.60 (q, J = 12.0 Hz, 1H, H-14), 0.82 [s, 9H, SiC(CH3)3], 1.37–1.45 (m, 2H, CH2CH2O), 1.65 [s, 9H, C(CH3)3], 1.65 (masked, 1H, H-14), 1.88 (dd, J = 17.6, 12.0 Hz, 1H, H-20), 2.01 (m, 1H, H-15), 2.08 (dd, J = 12.8, 6.8 Hz, 1H, H-6), 2.52 (m, 2H, H-6, H-20), 3.50–3.57 (m, 2H, CH2CH2O), 3.79 (t, J = 12.0 Hz, 1H, H-5), 3.94–4.03 (m, 2H, H-3, H-5), 6.90 (d, J = 6.8 Hz, 1H, HAR), 7.16 (t, J = 7.6 Hz, 1H, HAR), 7.34 (td, J = 7.2, 1.2 Hz, 1H, HAR), 7.90 (d, J = 8.0 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 18.1 [SiC(CH3)3], 25.8 [SiC(CH3)3], 28.1 [C(CH3)3], 29.9 (C-15), 30.2 (C-14), 34.5 (C-6), 37.8 (C-20), 38.5 (CH2CH2O), 43.7 (C-5), 56.8 (C-7), 60.2 (CH2CH2O), 65.5 (C-3), 84.9 [C(CH3)3], 115.3 (CHAR), 123.3 (CHAR), 125.1 (CHAR), 128.5 (CAR), 128.7 (CHAR), 138.9 (CAR), 148.9 (CO), 169.1 (CO), 174.6 (CO); HRMS (ESI) calcd for [C28H42N2O5Si + H+]: 515.2936, found: 515.2941.
(1′R,6′R,7′S,8a’R)-6′-Acetyl-7′-{2-[(tert-butyldimethylsilyl)oxy]ethyl}-1-(methoxymtehyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (14). Lithium diisopropylamide (LDA, 0.26 mL of a 2.0 M solution in THF/heptane/ethylbenzene, 0.42 mmol) was added at −78 °C under an argon atmosphere to a solution of spiro compound 12 (62 mg, 0.14 mmol) in anhydrous THF (0.7 mL), and the mixture was stirred at −78 °C for 1 h. Methyl acetate (0.05 mL, 0.56 mmol) was added at −78 °C, and the resulting mixture was stirred at room temperature for 4 h. Saturated aqueous NH4Cl was added, and the mixture was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 7:3 hexane-EtOAc) of the resulting residue gave starting material 12 (18 mg, 29%) and acetyl derivative 14 (35 mg, 52%) as a white foam: [α]22D = + 26.2 (c 0.82, CHCl3); IR (film): 1723, 1642, 1613 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.07 (s, 3H, CH3Si), −0.06 (s, 3H, CH3Si), 0.70–0.84 (m, 1H, H-14), 0.79 [s, 9H, C(CH3)3], 1.19–1.25 (m, 1H, CH2CH2O), 1.41–1.49 (m, 1H, CH2CH2O), 1.64–1.70 (m, 1H, H-14), 2.05 (dd, J = 12.8, 2.8 Hz, 1H, H-6), 2.37 (s, 3H, COCH3), 2.40–2.46 (m, 1H, H-15), 2.55 (dt, J = 12.8, 10.8 Hz, 1H, H-6), 3.21 (d, J = 10.8 Hz, 1H, H-20), 3.32 (s, 3H, CH3O), 3.47 (t, J = 6.4 Hz, 2H, CH2CH2O), 3.81 (dd, J = 12.8, 10.8 Hz, 1H, H-5), 3.96–4.02 (m, 1H, H-5), 4.09 (dd, J = 11.6, 4.0 Hz, 1H, H-3), 5.13 (d, J = 10.8 Hz, 1H, NCH2O), 5.15 (d, J = 10.8 Hz, 1H, NCH2O), 6.92 (d, J = 7.6 Hz, 1H, HAR), 7.08–7.12 (m, 2H, HAR), 7.33 (td, J = 9.2, 1.2 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.6 (CH3Si), −5.5 (CH3Si), 18.1 [C(CH3)3], 25.8 [C(CH3)3], 28.8 (C-14), 31.1 (CH3CO), 32.7 (C-15), 33.7 (C-6), 36.6 (CH2CH2O), 44.3 (C-5), 56.3 (CH3O), 56.9 (C-7), 60.2 (CH2CH2O), 61.7 (C-20), 64.3 (C-3), 71.5 (NCH2O), 110.1 (CHAR), 123.6 (CHAR), 123.7 (CHAR), 128.7 (CAR), 128.8 (CHAR), 141.2 (CAR), 165.8 (CO), 176.0 (CO), 205.2 (CO); HRMS (ESI) calcd for [C27H40N2O5Si + H+]: 501.2779, found: 501.2791.
(1′R,6′R,7′S,8a’R)-7′-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-6′-(1R- and 1S-hydroxyethyl)-1-(methoxymethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (15a and 15b). NaBH4 (10 mg, 0.24 mmol) was added at −10 °C under an argon atmosphere to a solution of ketone 14 (60 mg, 0.12 mmol) in anhydrous MeOH (2 mL). The resulting mixture was stirred at −10 °C for 1 h. Saturated aqueous NaHCO3 (1.3 mL) and CH2Cl2 were added, and the mixture was stirred for 5 min. The organic solvent was evaporated, and the resulting aqueous mixture was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (7:3 hexane-EtOAc to 1:1 hexane-EtOAc) of the resulting residue gave alcohols 15a (28 mg, 46%) and 15b (27 mg, 46%) as white foams. 15a: [α]22D = + 30.3 (c 1.16, CHCl3); IR (film): 3427 (OH), 1725, 1614 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.05 (s, 3H, CH3Si), −0.04 (s, 3H, CH3Si), 0.65–0.81 (m, 1H, H-14), 0.81 [s, 9H, C(CH3)3], 1.24–1.32 (m, 1H, CH2CH2O), 1.39 (d, J = 6.4 Hz, 3H, CH3CHOH), 1.58 (dt, J = 13.2, 3.9 Hz, 1H, H-14), 1.76–1.85 (m, 1H, CH2CH2O), 1.92–1.95 (m, 1H, H-15), 2.00–2.05 (m, 1H, H-6), 2.11 (dd, J = 9.2, 4.8 Hz, 1H, H-20), 2.54 (td, J = 12.4, 9.6 Hz, 1H, H-6), 3.32 (s, 3H, CH3O), 3.45–3.56 (m, 2H, CH2CH2O), 3.81 (dd, J = 12.4, 9.6 Hz, 1H, H-5), 3.96–4.03 (m, 3H, H-3, H-5, CHOH), 5.15 (s, 2H, NCH2O), 6.96 (dm, J = 8.0 Hz, 1H, HAR), 7.08–7.12 (m, 2H, HAR), 7.33 (td, J = 8.0, 1.2 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 18.1 [C(CH3)3], 22.1 (CH3CHOH), 25.8 [C(CH3)3], 30.4 (C-14), 32.4 (C-15), 33.9 (C-6), 38.0 (CH2CH2O), 44.7 (C-5), 53.3 (C-20), 56.3 (CH3O), 57.2 (C-7), 60.7 (CH2CH2O), 63.6 (C-3), 69.7 (CHOH), 71.5 (NCH2O), 110.1 (CHAR), 123.6 (CHAR), 123.8 (CHAR), 128.7 (CHAR, CAR), 141.2 (CAR), 171.2 (CO), 176.2 (CO); HRMS (ESI) calcd for [C27H42N2O5Si + H+]: 503.2936, found: 503.2937. 15b: [α]22D = + 14.0 (c 1.13, CHCl3); IR (film): 3418 (OH), 1725, 1614 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.07 (s, 3H, CH3Si), −0.05 (s, 3H, CH3Si), 0.70–0.84 (m, 1H, H-14), 0.80 [s, 9H, C(CH3)3], 1.18–1.20 (m, 1H, CH2CH2O), 1.19 (d, J = 6.4 Hz, 3H, CH3CHOH), 1.61–1.75 (m, 3H, H-14, H-15, CH2CH2O), 2.06 (dd, J = 12.8, 6.4 Hz, 1H, H-6), 2.34 (dd, J = 10.8, 3.6 Hz, 1H, H-20), 2.55 (tm, J = 12.8 Hz, 1H, H-6), 3.32 (s, 3H, CH3O), 3.44–3.54 (m, 2H, CH2CH2O), 3.84 (dd, J = 12.8, 10.8 Hz, 1H, H-5), 3.96–4.01 (m, 3H, H-3, H-5, CHOH), 5.15 (s, 2H, NCH2O), 6.94 (d, J = 7.2 Hz, 1H, HAR), 7.09–7.13 (m, 2H, HAR), 7.34 (t, J = 7.6 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 18.0 [C(CH3)3], 18.6 (CH3CHOH), 25.8 [C(CH3)3], 29.3 (C-14), 32.2 (C-15), 33.8 (C-6), 36.3 (CH2CH2O), 44.1 (C-5), 52.3 (C-20), 56.3 (C-7), 57.0 (CH3O), 59.9 (CH2CH2O), 64.1 (C-3), 67.1 (CHOH), 71.5 (NCH2O), 110.2 (CHAR), 123.7 (CHAR), 123.8 (CHAR), 128.7 (CAR), 128.8 (CHAR), 141.2 (CAR), 172.2 (CO), 176.1 (CO); HRMS (ESI) calcd for [C27H42N2O5Si + H+]: 503.2936, found: 503.2933.
(1′R,6′E,7′S,8a’R)-7′-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-6′-ethylidene-1-(methoxymethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (16). From alcohol 15a: N,N′-Dicyclohexylcarbodiimide (DCC, 54 mg, 0.26 mmol) and copper(I) chloride (CuCl, 52 mg, 0.52 mmol) were added under an argon atmosphere to a solution of alcohol 15a (26 mg, 0.05 mmol) in anhydrous toluene (1.6 mL), and the resulting mixture was stirred at reflux for 5 h. The suspension was filtered through Celite®, and the residue was washed with CH3CN. The resulting filtrate was kept in the freezer overnight and filtered again through Celite®, washing with minimal amounts of cold CH3CN. The organic filtrate was concentrated under reduced pressure. Flash chromatography (1:9 hexane-EtOAc) of the resulting residue gave the E-ethylidene derivative 16 (22 mg, 91%). From alcohol 15b: First step: Et3N (18 μL, 0.13 mmol) and mesyl chloride (MsCl, 9 μL, 0.11 mmol) were added at 0 °C under an argon atmosphere to a solution of alcohol 15b (21 mg, 0.04 mmol) in anhydrous CH2Cl2 (0.6 mL). The resulting mixture was stirred at 0 °C for 4 h. Saturated aqueous NH4Cl (1.2 mL) was added, and the mixture was extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give the corresponding mesylate, which was used in the next step without further purification. Second step: Diazabicycloundecene (DBU, 27 μL, 0.18 mmol) was added under an argon atmosphere to a solution of the above mesylate in anhydrous THF (0.6 mL), and the resulting mixture was stirred at reflux overnight. Distilled H2O was added, and the mixture was extracted with EtOAc. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 7:3 hexane-EtOAc) of the residue gave the Z isomer of compound 16 (4 mg, 20%) and the E-ethylidene derivative 16 (12 mg, 60%) as white foams. 16: [α]22D = −2.9 (c 0.76, CHCl3); IR (film): 1725, 1613 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = −0.1 (s, 3H, CH3Si), −0.07 (s, 3H, CH3Si), 0.78 [s, 9H, C(CH3)3], 0.94–1.05 (q, J = 12.0 Hz, 1H, H-14), 1.29–1.36 (m, 1H, CH2CH2O), 1.65–1.70 (m, 1H, H-14), 1.77–1.85 (m, 4H, CH2CH2O, =CHCH3), 1.87–1.95 (dm, J = 12.4 Hz, 1H, H-6), 2.51 (m, 1H, H-6), 2.87–2.96 (m, 1H, H-15), 3.32 (s, 3H, CH3O), 3.36–3.52 (m, 2H, CH2CH2O), 3.88–4.30 (m, 3H, H-5, H-3), 5.14 (d, J = 10.8 Hz, 1H, NCH2O), 5.17 (d, J = 10.8 Hz, 1H, NCH2O), 6.67 (m, 1H, =CHCH3), 7.09–7.16 (m, 3H, HAR), 7.33 (td, J = 7.2, 1.6 Hz, HAR); 13C-NMR (100.6 MHz, CDCl3): δ = −5.5 (2CH3Si), 14.4 (=CHCH3), 18.1 [C(CH3)3], 25.8 [C(CH3)3], 30.8 (C-15), 31.1 (C-14), 34.8 (C-6), 39.4 (CH2CH2O), 44.6 (C-5), 56.3 (CH3O), 57.4 (C-7), 60.4 (CH2CH2O), 61.6 (C-3), 71.5 (NCH2O), 110.1 (CHAR), 123.5 (CHAR), 124.3 (CHAR), 128.7 (CHAR), 128.8 (CAR), 133.5 (=CHCH3), 135.7 (C-20), 141.2 (CAR), 167.6 (CO), 176.6 (CO); HRMS (ESI) calcd for [C27H40N2O4Si + H+]: 485.2830, found: 485.2825. Z-isomer of 16: IR (film): 1726, 1614 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC, selected resonances): δ = −0.05 (s, 3H, CH3Si), −0.04 (s, 3H, CH3Si), 0.81 [s, 9H, C(CH3)3], 1.54–1.64 (m, 1H, H-14), 1.83 (m, 1H, CH2CH2O), 2.04 (ddd, J = 12.4, 7.6, 1.6 Hz, 1H, H-6), 2.20 (dd, J = 7.2, 2.0 Hz, 3H, =CHCH3), 2.54–2.60 (m, 1H, H-6), 3.32 (s, 3H, CH3O), 3.48–3.60 (m, 2H, CH2CH2O), 3.83–3.89 (m, 1H, H-5), 4.02–4.08 (m, 2H, H-3, H-5), 5.15 (s, 2H, NCH2O), 5.94 (qd, J = 7.2, 2.0 Hz, 1H, =CHCH3), 6.99 (dd, J = 8.0, 1.2 Hz, 1H, HAR), 7.07–7.11 (m, 2H, HAR), 7.31 (td, J = 7.6, 1.2 Hz, 1H, HAR); 13C-NMR (100.6 MHz, CDCl3, selected resonances): δ = −5.5 (CH3Si), −5.4 (CH3Si), 15.8 (=CHCH3), 18.1 [C(CH3)3], 25.8 [C(CH3)3], 29.8 (C-14), 34.1 (C-6), 36.2 (CH2CH2O), 44.0 (C-5), 56.3 (CH3O), 57.0 (C-7), 60.3 (CH2CH2O), 63.9 (C-3), 71.4 (NCH2O), 109.9 (CHAR), 123.6 (CHAR), 123.9 (CHAR), 128.6 (CHAR), 132.1 (CAR), 134.2 (=CHCH3), 141.2 (CAR), 165.5 (CO), 176.4 (CO); HRMS (ESI) calcd for [C27H40N2O4Si + H+]: 485.2830, found: 485.2825.
(1′R,6′E,7′S,8a’R)-6′-Ethylidene-7′-(2-hydroxyethyl)-2,5′-dioxospiro[indoline-3,1′-indolizidine] (17). TMSCl (30 μL, 0.23 mmol) and sodium iodide (35 mg, 0.23 mmol) were added at 0 °C under an argon atmosphere to a solution of compound 16 (25 mg, 0.05 mg) in anhydrous CH3CN (0.9 mL). The resulting mixture was stirred at 0 °C for 2 h. Saturated aqueous NaHCO3 was added and the mixture was extracted with EtOAc. The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The resulting residue was taken in MeOH (4.7 mL), Et3N (22 μL, 0.15 mmol) was added to the solution, and the mixture was stirred at 55 °C for 1 h. Saturated aqueous NH4Cl (1.7 mL) was added, the organic solvent was evaporated, and the aqueous mixture was extracted with EtOAc. The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography (hexane to 1:1 hexane-EtOAc) of the residue gave compound 17 (11 mg, 68%) as a white foam: [α]22D = −4.3 (c 0.44, CHCl3); IR (film): 3500–3000 (NH, OH), 1721, 1651 (CO) cm−1; 1H-NMR (400 MHz, CDCl3, COSY, g-HSQC): δ = 1.06 (q, J = 12.0 Hz, 1H, H-14), 1.39–1.48 (m, 1H, CH2CH2O), 1.70–1.76 (m, 1H, H-14), 1.80 (dd, J = 7.2, 1.2 Hz, 1H, =CHCH3), 1.83–1.91 (m, 1H, CH2CH2O), 2.01–2.06 (m, 1H, H-6), 2.46–2.54 (m, 1H, H-6), 2.93–3.00 (m, 1H, H-15), 3.43–3.49 (m, 1H, CH2CH2O), 3.53–3.59 (m, 1H, CH2CH2O), 3.88–4.02 (m, 3H, H-5, H-3), 6.71 (qd, J = 7.2, 2.0 Hz, 1H, =CHCH3), 6.97 (d, J = 7.6 Hz, 1H, HAR), 7.05–7.13 (m, 2H, HAR), 7.26–7.30 (m, 1H, HAR), 8.01 (br. s, 1H, NH); 13C-NMR (100.6 MHz, CDCl3): δ = 14.5 (=CHCH3), 30.5 (C-14), 30.9 (C-15), 34.4 (C-6), 38.5 (CH2CH2O), 44.8 (C-5), 57.4 (C-7), 59.9 (CH2CH2O), 61.5 (C-3), 110.5 (CHAR), 122.9 (CHAR), 124.4 (CHAR), 128.6 (CHAR), 129.7 (CAR), 133.6 (=CHCH3), 134.7 (C-20), 140.2 (CAR), 167.1 (CO), 177.7 (CO); HRMS (ESI) calcd for [C19H22N2O3 + H+]: 327.1703, found: 327.1702.
Supplementary Materials
The following are available online. Copies of 1H and 13C NMR spectra and crystallographic data for spirooxindole 4 (CCDC 2052158). CCDC 2052158 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
Author Contributions
M.A. designed and planned the research; M.P. supervised the experimental work; N.Y. performed the experimental work and characterized the compounds; E.M. carried out the X-ray analysis; J.B. discussed the results and prepared the manuscript for publication. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICIU/FEDER, Spain (RTI2018-093974-B-I00).
Data Availability Statement
The data presented in this study are available in the article and the Supplementary Materials section.
Acknowledgments
Financial support from the MICIU/FEDER, Spain (RTI2018-093974-B-I00) is gratefully acknowledged. Thanks are also due to the AGAUR (Generalitat de Catalunya) for a fellowship to N.Y. (AP2010-1663). We also acknowledge the networking contribution from the COST Action CM1407.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Saxton, J.E. Alkaloids of Mitragyna and Ourouparia species. In The Alkaloids; Manske, R.H.F., Ed.; Academic Press: New York, NY, USA, 1965; Volume 8, pp. 59–91. [Google Scholar]
- Bindra, J.S. Oxindole alkaloids. In The Alkaloids; Manske, R.H.F., Ed.; Academic Press: New York, NY, USA, 1973; Volume 14, pp. 83–121. [Google Scholar]
- Brown, R.T. Indoles, The Monoterpenoid Indole Alkaloids. In The Chemistry of Heterocyclic Compounds; Saxton, J.E., Weissberger, A., Taylor, E.C., Eds.; Wiley: New York, NY, USA, 1983; Volume 25, pp. 85–97. [Google Scholar]
- Shi, J.-S.; Yu, J.-X.; Chen, X.-P.; Xu, R.-X. Pharmacological actions of Uncaria alkaloids, rhynchophylline and isorhynchophylline. Acta Pharmacol. Sin. 2003, 24, 97–101. [Google Scholar] [PubMed]
- Heitzman, M.E.; Neto, C.C.; Winiarz, E.; Vaisberg, A.J.; Hammond, G.B. Ethnobotany, phytochemistry and pharmacology of Uncaria (Rubiaceae). Phytochemistry 2005, 66, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, S. Antihypertensive and neuroprotective activities of rhynchophylline: The role of rhynchophylline in neurotransmission and ion channel activity. J. Ethnopharmacol. 2010, 132, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Eisenbrand, G. Handbook of Chinese Medicinal Plants; Wiley: Weinheim, Germany, 2011; Volume 2, pp. 1213–1221. [Google Scholar]
- Zhou, J.-Y.; Zhou, S.-W. Isorhynchophylline: A plant alkaloid with therapeutic potential for cardiovascular and central nervous system diseases. Fitoterapia 2012, 83, 617–626. [Google Scholar] [CrossRef]
- Ndagijimana, A.; Wang, X.; Pan, G.; Zhang, F.; Feng, H.; Olaleye, O. A review on indole alkaloids isolated from Uncaria rhynchophylla and their pharmacological studies. Fitoterapia 2013, 86, 35–47. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, J.J.; Xu, J.; Feng, F.; Qu, W. Medicinal uses, phytochemistry and pharmacology of the genus Uncaria. J. Ethnopharmacol. 2015, 173, 48–80. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.-Q.; Liu, H.-M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.-Y.; Zhou, J. Therapeutic potential of spirooxindoles as antiviral agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef]
- Le Men, J.; Taylor, W.I. A uniform numbering system for indole alkaloids. Experientia 1965, 21, 508–510. [Google Scholar] [CrossRef]
- Ban, Y.; Seto, M.; Oishi, T. The synthesis of 3-spirooxindole derivatives. VII. Total synthesis of alkaloids (±)-rhynchophylline and (±)-isorhynchophylline. Chem. Pharm. Bull. 1975, 23, 2605–2613. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; Sim, K.-M.; Tan, G.-H.; Kam, T.-S. Four tetracyclic oxindole alkaloids and a taberpsychine derivative from a Malayan Tabernaemontana. Phytochemistry 2009, 70, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Seaton, J.C.; Nair, M.D.; Edwards, O.E.; Marion, L. The structure and stereoisomerism of three mitragyna alkaloids. Can. J. Chem. 1960, 38, 1035–1042. [Google Scholar] [CrossRef]
- Finch, N.; Taylor, W.I. Oxidative transformations of indole alkaloids. I. The preparation of oxindoles from yohimbine; the structures and partial syntheses of mitraphylline, rhyncophylline and corynoxeine. J. Am. Chem. Soc. 1962, 84, 3871–3877. [Google Scholar] [CrossRef]
- Deiters, A.; Pettersson, M.; Martin, S.F. General strategy for the syntheses of corynanthe, tacaman, and oxindole alkaloids. J. Org. Chem. 2006, 71, 6547–6561. [Google Scholar] [CrossRef]
- Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Total synthesis of the spirocyclic oxindole alkaloids Corynoxine, Corynoxine B, Corynoxeine, and Rhynchophylline. Eur. J. Org. Chem. 2013, 1100–1106. [Google Scholar] [CrossRef]
- Marti, C.; Carreira, E.M. Construction of spiro[pyrrolidine-3,3′-oxindoles]—Recent applications to the synthesis of oxindole alkaloids. Eur. J. Org. Chem. 2003, 2209–2219. [Google Scholar] [CrossRef]
- Trost, B.M.; Brennan, M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 3003–3025. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.-L.; Zhou, J. Catalytic asymmetric synthesis of oxindoles bearing a tetrasubstituted stereocenter at the C-3 position. Adv. Synth. Catal. 2010, 325, 1381–1407. [Google Scholar] [CrossRef]
- Santos, M.M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Martin, S.F.; Mortimore, M. New methods for the synthesis of oxindole alkaloids. Total syntheses of isopteropodine and pteropodine. Tetrahedron Lett. 1990, 31, 4557–4560. [Google Scholar] [CrossRef]
- Ito, M.; Clark, C.W.; Mortimore, M.; Goh, J.B.; Martin, S.F. Biogenetic approach to the Strychnos alkaloids. Concise syntheses of (±)-Akuammicine and (±)-Strychnine. J. Am. Chem. Soc. 2001, 123, 8003–8010. [Google Scholar] [CrossRef]
- Pérez, M.; Ramos, C.; Massi, L.; Gazzola, S.; Taglienti, C.; Yayik, N.; Molins, E.; Viayna, A.; Luque, F.J.; Bosch, J.; et al. Enantioselective synthesis of spiro[indolizidine-1,3′-oxindoles]. Org. Lett. 2017, 19, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Amat, M.; Ramos, C.; Pérez, M.; Molins, E.; Florindo, P.; Santos, M.M.M.; Bosch, J. Enantioselective formal synthesis of ent-rhynchophylline and ent-isorhynchophylline. Chem. Commun. 2013, 49, 1954–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allin, S.M.; Thomas, C.I.; Allard, J.E.; Doyle, K.; Elsegood, M.R.J. Highly stereoselective synthesis of the indolo[2,3-a]quinolizine ring system and application to indole natural product synthesis. Tetrahedron Lett. 2004, 45, 7103–7105. [Google Scholar] [CrossRef]
- Amat, M.; Santos, M.M.M.; Bassas, O.; Llor, N.; Escolano, C.; Gómez-Esqué, A.; Molins, E.; Allin, S.M.; McKee, V.; Bosch, J. Straightforward methodology for the enantioselective synthesis of benzo[a]- and indolo[2,3-a]quinolizidines. J. Org. Chem. 2007, 72, 5193–5201. [Google Scholar] [CrossRef]
- Breman, A.C.; van der Heijden, G.; van Maarseveen, J.H.; Ingemann, S.; Hiemstra, H. Synthetic and organocatalytic studies of quinidine analogues with ring-size modifications in the quinuclidine moiety. Chem. Eur. J. 2016, 22, 14247–14256. [Google Scholar] [CrossRef]
- Kikukawa, K.; Westcott, S.A.; Croatt, M.P.; Williams, T.J.; Wender, P.A.; Li, Y.; Jiang, X. Carbonyl(chloro)bis(triphenylphosphine)rhodium(I). Encycl. Reag. Org. Synth. 2015, 1–22. [Google Scholar] [CrossRef]
- Corey, E.J.; Andersen, N.H.; Carlson, R.M.; Paust, J.; Vedejs, E.; Vlattas, I.; Winter, R.E.K. Total synthesis of prostaglandins. Synthesis of the pure dl-E1, -F1α, -F1β, -A1, and -B1 hormones. J. Am. Chem. Soc. 1968, 90, 3245–3247. [Google Scholar] [CrossRef]
- Fox, D.N.A.; Lathbury, D.; Mahon, M.F.; Molloy, K.C.; Gallagher, T. Enantioselective synthesis of pumiliotoxin 25ID. A strategy employing an allene-based electrophile-mediated cyclization. J. Am. Chem. Soc. 1991, 113, 2652–2656. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.; Zhang, Y.; Huang, J.; Nie, L.; Chen, J.; Cao, W.; Wu, X. Enantioselective synthesis of indoloquinolizidines via asymmetric catalytic hydrogenation/lactamization of imino diesters. J. Org. Chem. 2013, 78, 12009–12017. [Google Scholar] [CrossRef] [PubMed]
- Sai, H.; Ogikub, T.; Hiroshi Ohmizu, H. Facile stereoselective synthesis of (E)- and (Z)-α-substituted cinnamates: Stereospecific dehydration reaction with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and copper(II) chloride. Tetrahedron 2007, 63, 10345–10353. [Google Scholar] [CrossRef]
Figure 1.
Representative tetracyclic spiro[indolizidine-1,3′-oxindole] alkaloids.

Scheme 1.
Equilibrating a C-7 epimeric pair of geissoschizol oxindoles.

Scheme 2.
Oxidative rearrangements of indolo[2,3-a]quinolizidines reported by S. F. Martin [26].
Scheme 2.
Oxidative rearrangements of indolo[2,3-a]quinolizidines reported by S. F. Martin [26].

Scheme 3.
Initial steps of the synthesis.

Figure 2.
ORTEP plot of the X-ray structure of spiro[indolizidine-1,3′-oxindole] 4.

Scheme 4.
Removal of the hydroxymethyl substituent.

Scheme 5.
Previous synthesis of oxindole 8 via oxidation of the indoline moiety.

Scheme 6.
Stereoselective introduction of the E-ethylidene chain.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yayik, N.; Pérez, M.; Molins, E.; Bosch, J.; Amat, M. Studies on the Enantioselective Synthesis of E-Ethylidene-bearing Spiro[indolizidine-1,3′-oxindole] Alkaloids. Molecules 2021, 26, 428. https://doi.org/10.3390/molecules26020428
AMA Style
Yayik N, Pérez M, Molins E, Bosch J, Amat M. Studies on the Enantioselective Synthesis of E-Ethylidene-bearing Spiro[indolizidine-1,3′-oxindole] Alkaloids. Molecules. 2021; 26(2):428. https://doi.org/10.3390/molecules26020428
Chicago/Turabian StyleYayik, Nihan, Maria Pérez, Elies Molins, Joan Bosch, and Mercedes Amat. 2021. "Studies on the Enantioselective Synthesis of E-Ethylidene-bearing Spiro[indolizidine-1,3′-oxindole] Alkaloids" Molecules 26, no. 2: 428. https://doi.org/10.3390/molecules26020428