
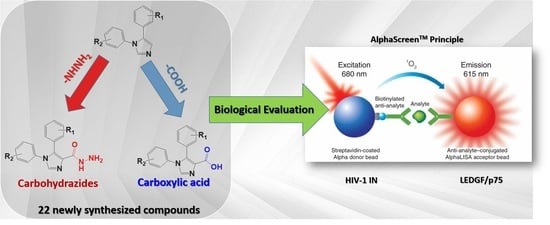
Studies towards the Design and Synthesis of Novel 1,5-Diaryl-1H-imidazole-4-carboxylic Acids and 1,5-Diaryl-1H-imidazole-4-carbohydrazides as Host LEDGF/p75 and HIV-1 Integrase Interaction Inhibitors


Abstract
:
1. Introduction
2. Results and Discussion
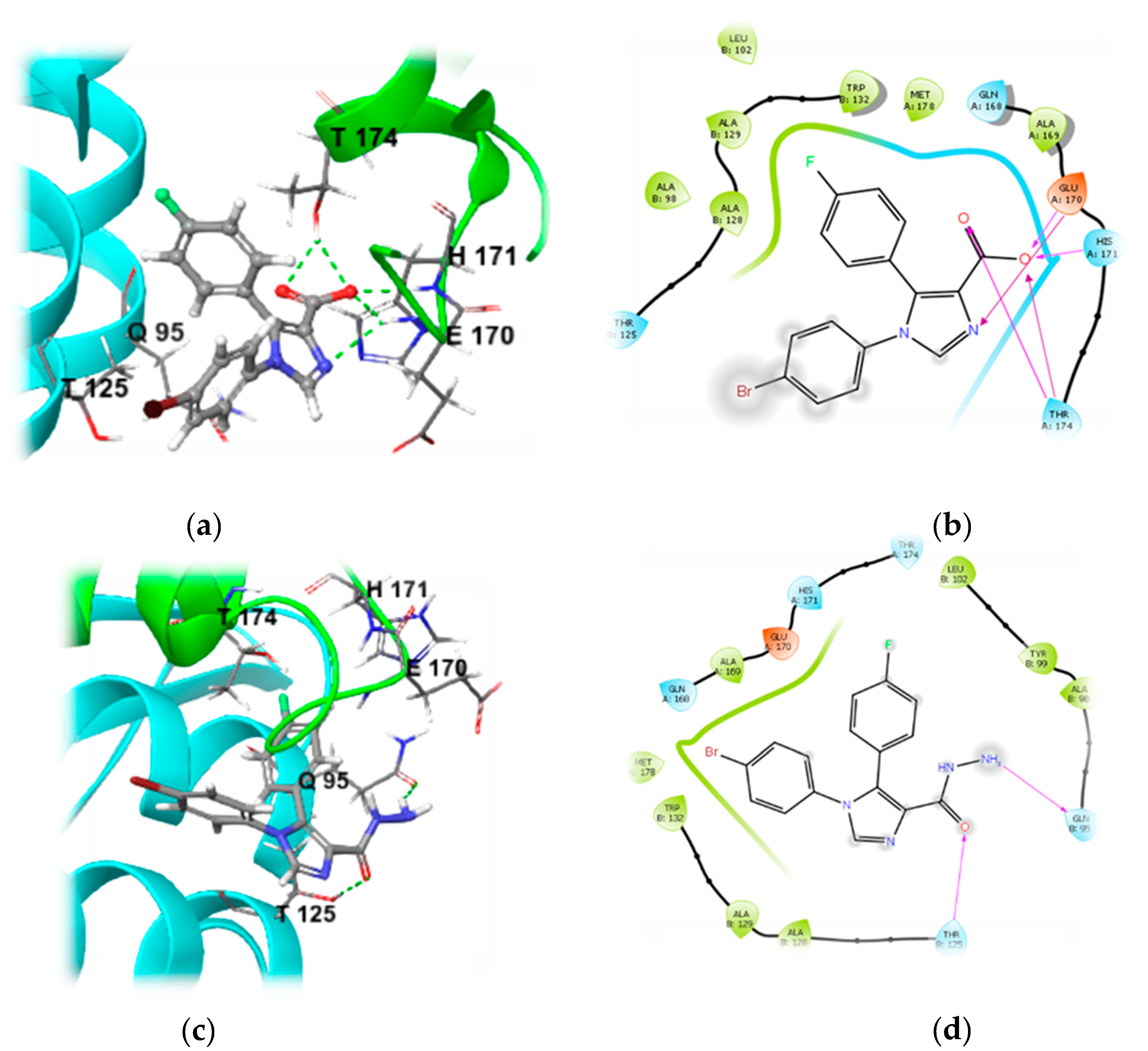
2.1. Testing the Hypothesis In Silico
2.2. Chemistry
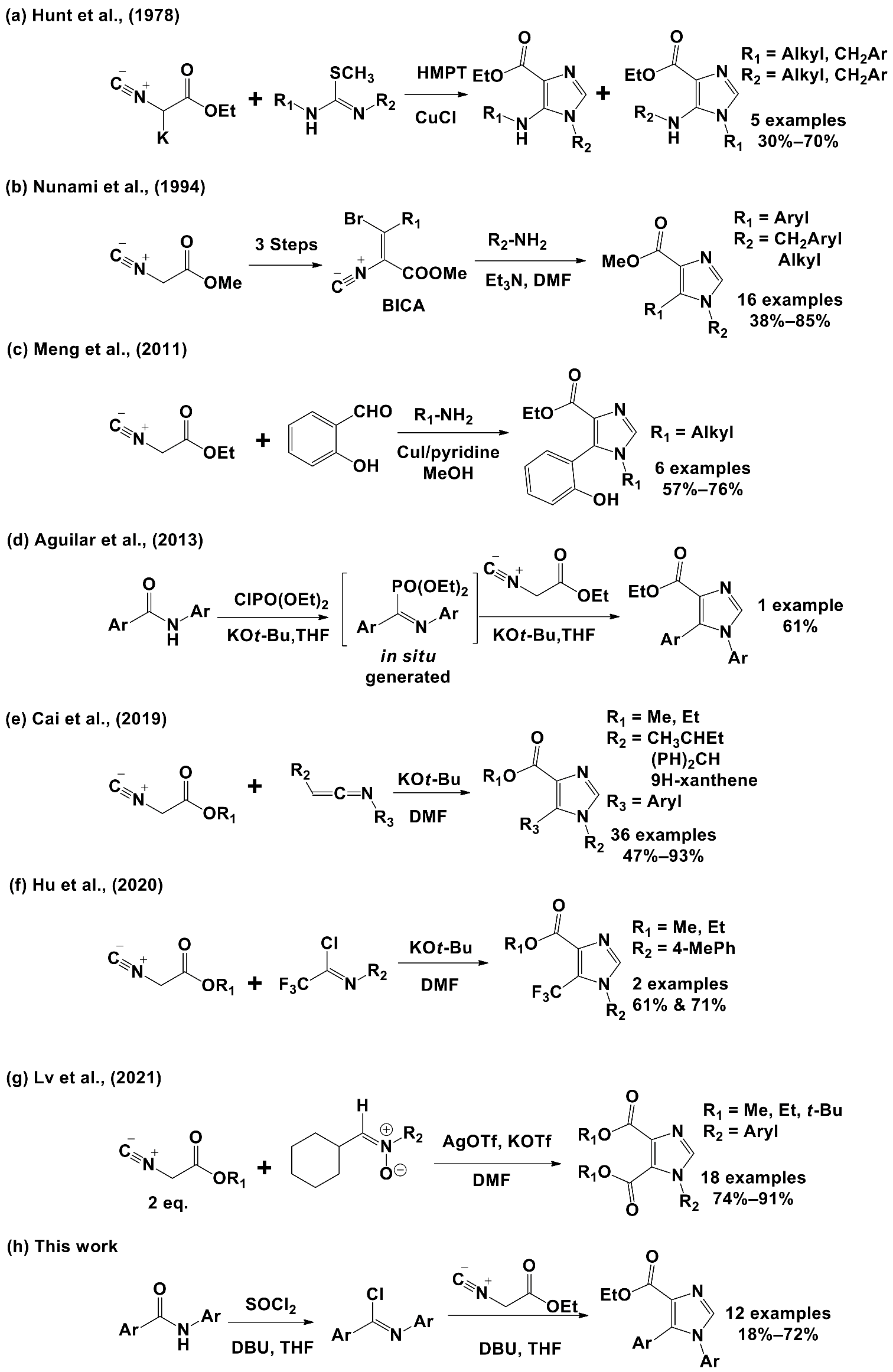
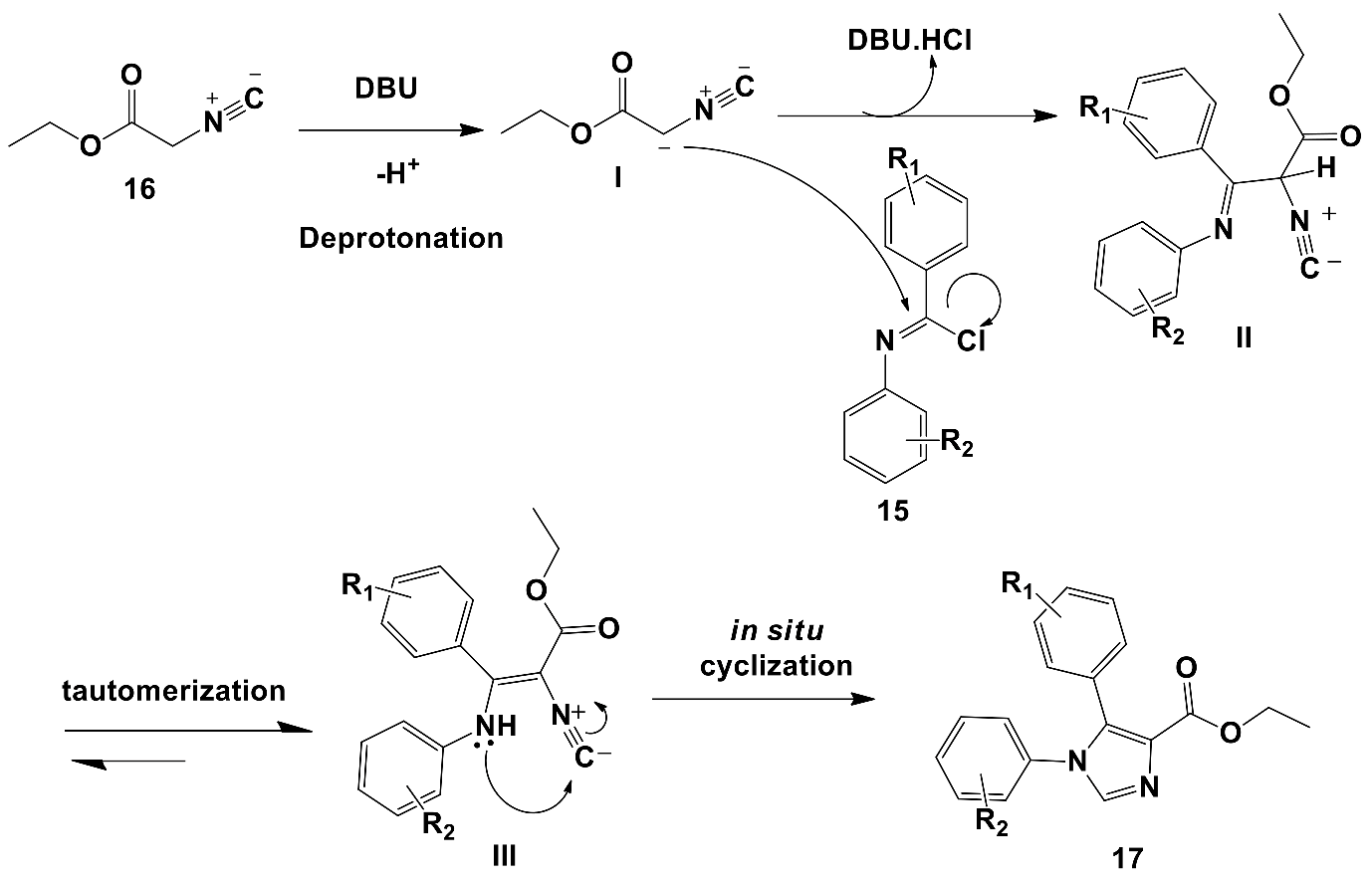
2.2.1. Synthesis of Key 1,5-Diaryl-1H-imidazole-4-carboxylate Esters
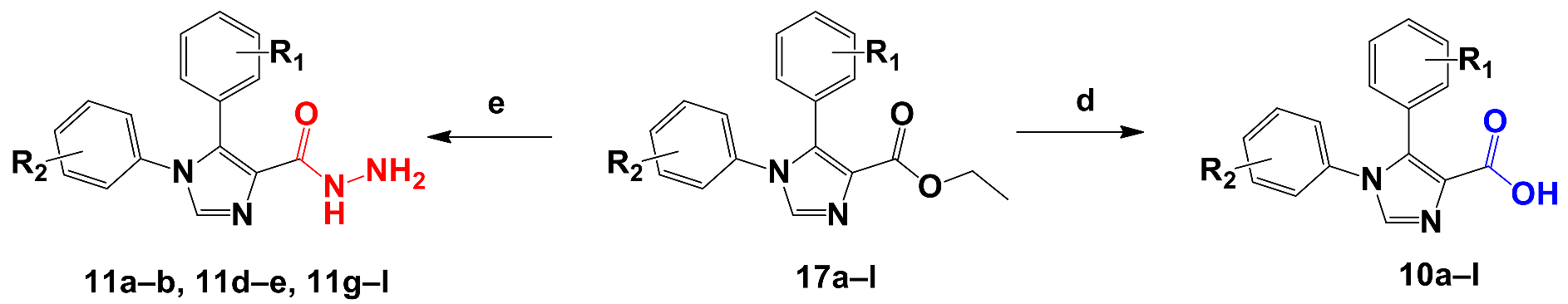
2.2.2. Synthesis of Novel 5-Diaryl-1H-imidazole-4-carboxylic Acids and 1,5-Diaryl-1H-imidazole-4-carbohydrazides
2.3. Biological Evaluation
2.3.1. HIV-1 IN-LEDGF AlphaScreenTM Assays
2.3.2. Direct ELISA Assay
2.3.3. Cytotoxicity and Antiviral Assays
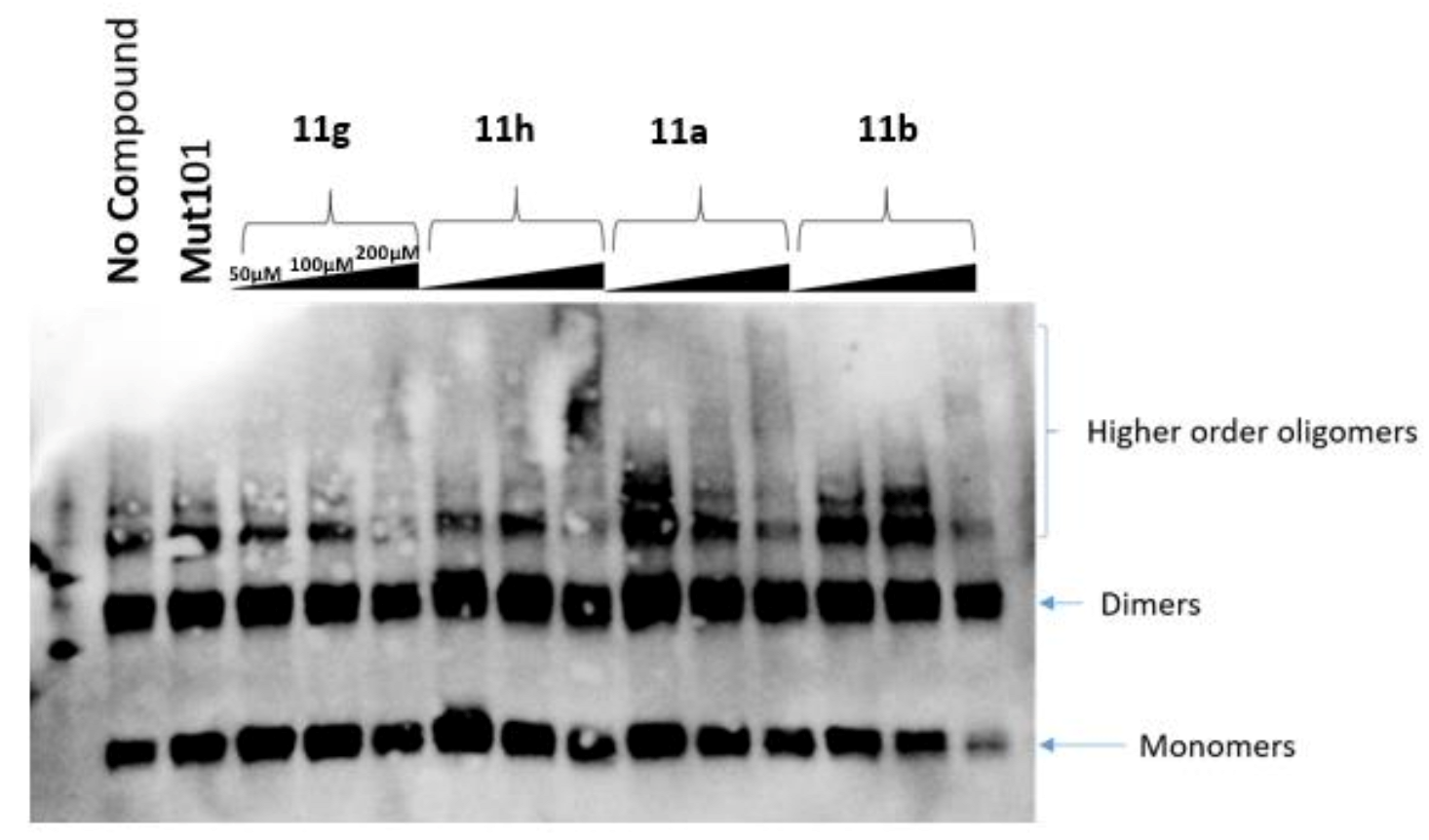
2.4. Multimerization Gel Assay as a Secondary Screening Assay
3. Experimental Protocols
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. General Procedure for the Synthesis of N-Aryl-benzamides 14a–p
3.1.3. General Procedure for the Synthesis of N-Aryl-benzimidoyl Chloride Intermediates 15a–n
N-(4-Bromophenyl)-4-fluorobenzimidoyl Chloride 15a
N-(4-Bromophenyl)-3-fluorobenzimidoyl Chloride 15b
N-(4-Bromophenyl)-3-methoxybenzimidoyl Chloride 15c
N-(4-Bromophenyl)-3-methylbenzimidoyl Chloride 15d
N-(4-Bromophenyl)-4-methylbenzimidoyl Chloride 15e
N-(4-Bromophenyl)benzimidoyl Chloride 15f
N-(4-Chlorophenyl)-4-fluorobenzimidoyl Chloride 15g
N-(4-Chlorophenyl)-3-fluorobenzimidoyl Chloride 15h
N-(4-Chlorophenyl)-3-methoxybenzimidoyl Chloride 15i
N-(4-Chlorophenyl)-3-methylbenzimidoyl Chloride 15j
N-(4-Chlorophenyl)-4-methylbenzimidoyl Chloride 15k
N-(4-Chlorophenyl)benzimidoyl Chloride 15l
N-(2.4-Dimethylphenyl)-4-fluorobenzimidoyl Chloride 15m
N-(2.4-Dimethylphenyl)-3-methoxybenzimidoyl Chloride 15n
3.1.4. General Procedure for the Synthesis of Ethyl 1,5-Diaryl-1H-imidazoyl Carboxylates 17a–l
Ethyl 1-(4-Bromophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carboxylate 17a
Ethyl 1-(4-Bromophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carboxylate 17b
Ethyl 1-(4-Bromophenyl)-5-(3-methoxyphenyl)-1H-imidazole-4-carboxylate 17c
Ethyl 1-(4-Bromophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carboxylate 17d
Ethyl 1-(4-Bromophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carboxylate 17e
Ethyl 1-(4-Bromophenyl)-5-phenyl-1H-imidazole-4-carboxylate 17f
Ethyl 1-(4-Chlorophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carboxylate 17g
Ethyl 1-(4-Chlorophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carboxylate 17h
Ethyl 1-(4-Chlorophenyl)-5-(3-methoxyphenyl)-1H-imidazole-4-carboxylate 17i
Ethyl 1-(4-Chlorophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carboxylate 17j
Ethyl 1-(4-Chlorophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carboxylate 17k
Ethyl 1-(4-Chlorophenyl)-5-phenyl-1H-imidazole-4-carboxylate 17l
3.1.5. General Synthetic Procedure for the Preparation of 1,5-Diaryl-1H-imidazole-4-carboxylic Acids 10a–l
1-(4-Bromophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carboxylic Acid 10a
1-(4-Bromophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carboxylic Acid 10b
1-(4-Bromophenyl)-5-(3-methoxyphenyl)-1H-imidazole-4-carboxylic Acid 10c
1-(4-Bromophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carboxylic Acid 10d
1-(4-Bromophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carboxylic Acid 10e
1-(4-Bromophenyl)-5-phenyl-1H-imidazole-4-carboxylic Acid 10f
1-(4-Chlorophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carboxylic Acid 10g
1-(4-Chlorophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carboxylic Acid 10h
1-(4-Chlorophenyl)-5-(3-methoxyphenyl)-1H-imidazole-4-carboxylic Acid 10i
1-(4-Chlorophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carboxylic Acid 10j
1-(4-Chlorophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carboxylic Acid 10k
1-(4-Chlorophenyl)-5-phenyl-1H-imidazole-4-carboxylic Acid 10l
3.1.6. General Procedure for the Synthesis of 1,5-diaryl-1H-imidazole-4-carbohydrazide Derivatives 11a–b, 11d–e, 11g–l
1-(4-Bromophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carbohydrazide 11a
1-(4-Bromophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carbohydrazide 11b
1-(4-Bromophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carbohydrazide 11d
1-(4-Bromophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carbohydrazide 11e
1-(4-Chlorophenyl)-5-(4-fluorophenyl)-1H-imidazole-4-carbohydrazide 11g
1-(4-Chlorophenyl)-5-(3-fluorophenyl)-1H-imidazole-4-carbohydrazide 11h
1-(4-Chlorophenyl)-5-(3-methoxyphenyl)-1H-imidazole-4-carbohydrazide 11i
1-(4-Chlorophenyl)-5-(3-methylphenyl)-1H-imidazole-4-carbohydrazide 11j
1-(4-Chlorophenyl)-5-(4-methylphenyl)-1H-imidazole-4-carbohydrazide 11k
1-(4-Chlorophenyl)-5-phenyl-1H-imidazole-4-carbohydrazide 11l
3.2. Modeling Protocols
3.3. Biological Evaluation Protocols
3.3.1. HIV-1 IN-LEDGF/p75 AlphaScreenTM Assay
3.3.2. Strand Transfer Assay
3.3.3. Cellular Toxicity Assay
3.3.4. Antiviral Assay
3.3.5. Multimerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Adamson, C.S.; Freed, E.O. Anti-HIV-1 Therapeutics: From FDA-approved Drugs to Hypothetical Future Targets. Mol. Interv. 2009, 9, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Mega, E.R. Alarming surge in drug-resistant HIV uncovered. Nat. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 2017, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D. Human Immunodeficiency Virus Drug Resistance: 2018 Recommendations of the International Antiviral Society–USA Panel. Clin. Infect. Dis. 2019, 68, 177–187. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Ondoa, P.; Peter, T.; Mugyenyi, P.N.; Stevens, W.S.; de Wit, T.F.R.; Hamers, R.L. Affordable HIV drug-resistance testing for monitoring of antiretroviral therapy in sub-Saharan Africa. Lancet Infect. Dis. 2016, 16, e267–e275. [Google Scholar] [CrossRef]
- Clavel, F.; Hance, A.J. HIV Drug Resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar] [CrossRef]
- Wainberg, M.A.; Zaharatos, G.J.; Brenner, B. Development of Antiretroviral Drug Resistance. N. Engl. J. Med. 2011, 365, 637–646. [Google Scholar] [CrossRef]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Cherepanov, P.; Ambrosio, A.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef] [Green Version]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 Is Essential for Nuclear and Chromosomal Targeting of HIV-1 Integrase in Human Cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.; Craigie, R. HIV Integrase Structure and Function. Adv. Appl. Microbiol. 1999, 52, 319–333. [Google Scholar] [CrossRef]
- Chen, J.C.-H.; Krucinski, J.; Miercke, L.J.W.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. USA 2000, 97, 8233–8238. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Krishnan, L.; Cherepanov, P.; Engelman, A. Structural biology of retroviral DNA integration. Virology 2011, 411, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Genet. 2012, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef]
- Sato, M.; Kawakami, H.; Motomura, T.; Aramaki, H.; Matsuda, T.; Yamashita, M.; Ito, Y.; Matsuzaki, Y.; Yamataka, K.; Ikeda, S.; et al. Quinolone Carboxylic Acids as a Novel Monoketo Acid Class of Human Immunodeficiency Virus Type 1 Integrase Inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef]
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob. Agents Chemother. 2016, 60, 7086–7097. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.-M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Shaw, S.; Demeulemeester, J.; Desimmie, B.A.; Marchand, A.; Butler, S.; Smets, W.; Chaltin, P.; Westby, M.; Debyser, Z.; et al. Small-Molecule Inhibitors of the LEDGF/p75 Binding Site of Integrase Block HIV Replication and Modulate Integrase Multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. [Google Scholar] [CrossRef] [Green Version]
- Desimmie, B.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef]
- Amadori, C.; Van Der Velden, Y.U.; Bonnard, D.; Orlov, I.; Van Bel, N.; Le Rouzic, E.; Miralles, L.; Brias, J.; Chevreuil, F.; Spehner, D.; et al. The HIV-1 integrase-LEDGF allosteric inhibitor MUT-A: Resistance profile, impairment of virus maturation and infectivity but without influence on RNA packaging or virus immunoreactivity. Retrovirology 2017, 14, 50. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLOS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef] [Green Version]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical Profile of BI 224436, a Novel HIV-1 Non-Catalytic-Site Integrase Inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef] [Green Version]
- Serrao, E.; Xu, Z.-L.; Debnath, B.; Christ, F.; Debyser, Z.; Long, Y.-Q.; Neamati, N. Discovery of a novel 5-carbonyl-1H-imidazole-4-carboxamide class of inhibitors of the HIV-1 integrase–LEDGF/p75 interaction. Bioorg. Med. Chem. 2013, 21, 5963–5972. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.W.; Debnath, B.; Christ, F.; Otake, H.; Debyser, Z.; Neamati, N. Discovery of novel inhibitors of LEDGF/p75-IN protein–protein interactions. Bioorg. Med. Chem. 2013, 21, 957–963. [Google Scholar] [CrossRef]
- Rashamuse, T.J.; Harrison, A.T.; Mosebi, S.; van Vuuren, S.; Coyanis, E.M.; Bode, M.L. Design, synthesis and biological evaluation of imidazole and oxazole fragments as HIV-1 integrase-LEDGF/p75 disruptors and inhibitors of microbial pathogens. Bioorg. Med. Chem. 2020, 28, 115210. [Google Scholar] [CrossRef]
- Tintori, C.; Demeulemeester, J.; Franchi, L.; Massa, S.; Debyser, Z.; Christ, F.; Botta, M. Discovery of small molecule HIV-1 integrase dimerization inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3109–3114. [Google Scholar] [CrossRef] [PubMed]
- Dolšak, A.; Švajger, U.; Lešnik, S.; Konc, J.; Gobec, S.; Sova, M. Selective Toll-like receptor 7 agonists with novel chromeno[3,4-d]imidazol-4(1H)-one and 2-(trifluoromethyl)quinoline/ quinazoline-4-amine scaffolds. Eur. J. Med. Chem. 2019, 179, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Zou, Y.; Khorev, O.; Jin, Y.; Zhou, H.; Zhang, Y.; Hu, D.; Ma, L.; Wang, X.; Shen, J. Synthesis of 3-Aminocoumarin and Derivatives via Copper Catalysis. Synfacts 2011, 2011, 0717. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Zhdanko, A.G.; Orru, R.V.A.; Nenajdenko, V.G. Isocyanoacetate Derivatives: Synthesis, Reactivity, and Application. Chem. Rev. 2010, 110, 5235–5331. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.L.; Gravestock, D.; Rousseau, A.L. Synthesis, Reactions and Uses of Isocyanides in Organic Synthesis. An Update. Org. Prep. Proced. Int. 2016, 48, 89–221. [Google Scholar] [CrossRef]
- Suzuki, M.; Moriya, T.; Matsumoto, K.; Miyoshi, M. A New Convenient Synthesis of 5-Amino-1,3-thiazole-4-carboxylic Acids. Synthesis 1982, 1982, 874–875. [Google Scholar] [CrossRef]
- Baxendale, I.; Ley, S.; Smith, C.; Tamborini, L.; Voica, A.-F. A Bifurcated Pathway to Thiazoles and Imidazoles Using a Modular Flow Microreactor. J. Comb. Chem. 2008, 10, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.T.; Bartlett, P.A. Regioselective Synthesis of 5-Amino-4-imidazolecarboxylates via Isonitrile Cycloaddition. Synthesis 1978, 1978, 741–742. [Google Scholar] [CrossRef]
- Nunami, K.-I.; Yamada, M.; Fukui, T.; Matsumoto, K. A Novel Synthesis of Methyl 1,5-Disubstituted Imidazole-4-carboxylates Using 3-Bromo-2-isocyanoacrylates (BICA). J. Org. Chem. 1994, 59, 7635–7642. [Google Scholar] [CrossRef]
- Aguilar, A.; Zhou, H.; Chen, J.; Liu, L.; Bai, L.; McEachern, D.; Yang, C.-Y.; Meagher, J.; Stuckey, J.; Wang, S. A Potent and Highly Efficacious Bcl-2/Bcl-xL Inhibitor. J. Med. Chem. 2013, 56, 3048–3067. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Bai, H.; Wang, Y.; Xu, X.; Xie, H.; Liu, J. Base-mediated regioselective [3+2] annulation of ketenimines and isocyanides: Efficient synthesis of 1,4,5-trisubstituted imidazoles. Chem. Commun. 2019, 55, 3821–3824. [Google Scholar] [CrossRef]
- Hu, S.; Yang, H.; Chen, Z.; Wu, X.-F. Base-mediated [3+2] annulation of trifluoroacetimidoyl chlorides and isocyanides: An improved approach for regioselective synthesis of 5-trifluoromethyl-imidazoles. Tetrahedron 2020, 76, 131168. [Google Scholar] [CrossRef]
- Lv, L.; Chen, Y.; Shatskiy, A.; Liu, J.; Liu, X.; Kärkäs, M.D.; Wang, X. Silver-Catalyzed [3+1+1] Annulation of Nitrones with Isocyanoacetates as an Approach to 1,4,5-Trisubstituted Imidazoles. Eur. J. Org. Chem. 2021, 2021, 964–968. [Google Scholar] [CrossRef]
- Schlosser, J.; Johannes, E.; Zindler, M.; Lemmerhirt, J.; Sommer, B.; Schütt, M.; Peifer, C. Novel synthesis of benzofuran- and indol-2-yl-methanamine derivatives. Tetrahedron Lett. 2015, 56, 89–94. [Google Scholar] [CrossRef]
- Deng, H.; Fang, Y. Synthesis and Agonistic Activity at the GPR35 of 5,6-Dihydroxyindole-2-carboxylic Acid Analogues. ACS Med. Chem. Lett. 2012, 3, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Guo, H.; Chen, D.; Yang, Q.; Fan, Y.; Yin, Y.; Wang, W.; Chen, D.; Wang, S.; Liu, W. Mutational biosynthesis to generate novel analogs of nosiheptide featuring a fluorinated indolic acid moiety. Org. Biomol. Chem. 2020, 18, 4051–4055. [Google Scholar] [CrossRef]
- Ren, Z.-L.; Guan, Z.-R.; Kong, H.-H.; Ding, M.-W. Multifunctional odorless isocyano(triphenylphosphoranylidene)-acetates: Synthesis and direct one-pot four-component Ugi/Wittig cyclization to multisubstituted oxazoles. Org. Chem. Front. 2017, 4, 2044–2048. [Google Scholar] [CrossRef]
- Bongards, C.; Gärtner, W. The Role of the Chromophore in the Biological Photoreceptor Phytochrome: An Approach Using Chemically Synthesized Tetrapyrroles. Accounts Chem. Res. 2010, 43, 485–495. [Google Scholar] [CrossRef]
- Latham, A.N.; Ferrence, G.M.; Lash, T.D. Metalation and Methyl Group Migration in 21-, 22-, and 23-Methylcarbaporphyrins: Synthesis and Characterization of Palladium(II), Rhodium(I), and Rhodium(III) Derivatives. Organometallics 2019, 38, 575–585. [Google Scholar] [CrossRef]
- Garrido, M.; Corredor, M.; Orzáez, M.; Alfonso, I.; Messeguer, A. Regioselective Synthesis of a Family of β-Lactams Bearing a Triazole Moiety as Potential Apoptosis Inhibitors. ChemistryOpen 2016, 5, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Genin, M.J.; Allwine, D.A.; Anderson, D.J.; Barbachyn, M.R.; Emmert, D.E.; Garmon, S.A.; Graber, D.R.; Grega, K.C.; Hester, J.B.; Hutchinson, D.K.; et al. Substituent Effects on the Antibacterial Activity of Nitrogen−Carbon-Linked (Azolylphenyl)oxazolidinones with Expanded Activity Against the Fastidious Gram-Negative OrganismsHaemophilusinfluenzaeandMoraxellacatarrhalis. J. Med. Chem. 2000, 43, 953–970. [Google Scholar] [CrossRef]
- Lai, Y.-Y.; Huang, L.-J.; Lee, K.-H.; Xiao, Z.; Bastow, K.F.; Yamori, T.; Kuo, S.-C. Synthesis and biological relationships of 3′,6-substituted 2-phenyl-4-quinolone-3-carboxylic acid derivatives as antimitotic agents. Bioorg. Med. Chem. 2005, 13, 265–275. [Google Scholar] [CrossRef]
- Caiuby, C.A.D.; de Jesus, M.P.; Burtoloso, A.C.B. α-Imino Iridium Carbenes from Imidoyl Sulfoxonium Ylides: Application in the One-Step Synthesis of Indoles. J. Org. Chem. 2020, 85, 7433–7445. [Google Scholar] [CrossRef]
- Liu, G.; Wu, X.; Shi, X. Synthesis of Polysubstituted-1,2,4-Triazoles. J. Heterocycl. Chem. 2013, 50, 39. [Google Scholar] [CrossRef]
- You, X.; Xie, X.; Sun, R.; Chen, H.; Li, S.; Liu, Y. Titanium-mediated cross-coupling reactions of 1,3-butadiynes with α-iminonitriles to 3-aminopyrroles: Observation of an imino aza-Nazarov cyclization. Org. Chem. Front. 2014, 1, 940–946. [Google Scholar] [CrossRef]
- Liu, X.; Wu, P.; Li, J.; Cui, C. Synthesis of 1,2-Borazaronaphthalenes from Imines by Base-Promoted Borylation of C–H bond. J. Org. Chem. 2015, 80, 3737–3744. [Google Scholar] [CrossRef]
- Zhou, H.; Aguilar, A.; Chen, J.; Bai, L.; Liu, L.; Meagher, J.L.; Yang, C.-Y.; McEachern, D.; Cong, X.; Stuckey, J.A.; et al. Structure-Based Design of Potent Bcl-2/Bcl-xL Inhibitors with Strong in Vivo Antitumor Activity. J. Med. Chem. 2012, 55, 6149–6161. [Google Scholar] [CrossRef] [Green Version]
- Peat, T.S.; Rhodes, D.I.; Vandegraaff, N.; Le, G.; Smith, J.A.; Clark, L.J.; Jones, E.D.; Coates, J.A.V.; Thienthong, N.; Newman, J.; et al. Small Molecule Inhibitors of the LEDGF Site of Human Immunodeficiency Virus Integrase Identified by Fragment Screening and Structure Based Design. PLoS ONE 2012, 7, e40147. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.; Mosebi, S.; Fish, M.Q.; Papathanasopoulos, M.A.; Hewer, R. Screening of the NIH Clinical Collection for inhibitors of HIV-1 integrase activity. S. Afr. J. Sci. 2018, 114, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Harada, S.; Koyanagi, Y.; Yamamoto, N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 1985, 229, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Harbut, M.B.; Vilchèze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay; Humana Press: Totowa, NJ, USA, 2011; Volume 731, pp. 237–245. [Google Scholar] [CrossRef]
- Li, M.; Mizuuchi, M.; Burke, T.R.; Craigie, R. Retroviral DNA integration: Reaction pathway and critical intermediates. EMBO J. 2006, 25, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef] [Green Version]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Compound | R1 | R2 | Yield % 1 |
| 17a | 4-F | 4-Br | 64 |
| 17b | 3-F | 4-Br | 58 |
| 17c | 3-OMe | 4-Br | 18 |
| 17d | 3-Me | 4-Br | 30 |
| 17e | 4-Me | 4-Br | 35 |
| 17f | H | 4-Br | 22 |
| 17g | 4-F | 4-Cl | 72 |
| 17h | 3-F | 4-Cl | 67 |
| 17i | 3-OMe | 4-Cl | 32 |
| 17j | 3-Me | 4-Cl | 41 |
| 17k | 4-Me | 4-Cl | 38 |
| 17l | H | 4-Cl | 42 |
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Yield % |
| 10a | 4-F | 4-Br | OH | 69 |
| 10b | 3-F | 4-Br | OH | 72 |
| 10c | 3-OMe | 4-Br | OH | 78 |
| 10d | 3-Me | 4-Br | OH | 75 |
| 10e | 4-Me | 4-Br | OH | 70 |
| 10f | H | 4-Br | OH | 63 |
| 10g | 4-F | 4-Cl | OH | 69 |
| 10h | 3-F | 4-Cl | OH | 70 |
| 10i | 3-OMe | 4-Cl | OH | 80 |
| 10j | 3-Me | 4-Cl | OH | 83 |
| 10k | 4-Me | 4-Cl | OH | 79 |
| 10l | H | 4-Cl | OH | 77 |
| 11a | 4-F | 4-Br | NHNH2 | 76 |
| 11b | 3-F | 4-Br | NHNH2 | 73 |
| 11d | 3-Me | 4-Br | NHNH2 | 64 |
| 11e | 4-Me | 4-Br | NHNH2 | 68 |
| 11g | 4-F | 4-Cl | NHNH2 | 66 |
| 11h | 3-F | 4-Cl | NHNH2 | 73 |
| 11i | 3-OMe | 4-Cl | NHNH2 | 68 |
| 11j | 3-Me | 4-Cl | NHNH2 | 62 |
| 11k | 4-Me | 4-Cl | NHNH2 | 63 |
| 11l | H | 4-Cl | NHNH2 | 58 |
| Compound | Biochemical Assays | |
|---|---|---|
| HIV-1 IN-LEDGF (%) Inhibition at 100 µM (AlphaScreenTM) 1* | Strand Transfer Activity (%) Inhibition at 100 µM (ELISA) 1* | |
| 10a | 83 ± 3.36 | 44 ± 1.13 |
| 10b | 76 ± 2.83 | 24 ± 3.39 |
| 10c | 51 ± 4.16 | 0 |
| 10d | 84 ± 0.29 | 9 ± 9.66 |
| 10e | 82 ± 0.36 | 2 ± 9.00 |
| 10f | 17 ± 2.55 | 16 ± 4.89 |
| 10g | 47 ± 3.44 | 0 |
| 10h | 54 ± 10.05 | 0 |
| 10i | 74 ± 9.29 | 0 |
| 10j | 78 ± 8.99 | 12 ± 4.26 |
| 10k | 85 ± 3.79 | 1.4 ± 6.07 |
| 10l | 35 ± 2.26 | 39 ± 1.80 |
| 11a | 51 ± 9.16 | 0 |
| 11b | 50 ± 2.01 | 0 |
| 11d | 67 ± 9.00 | 1 ± 5.89 |
| 11e | 89 ± 1.38 | 0 |
| 11g | 0 | 0 |
| 11h | 59 ± 3.22 | 0 |
| 11i | 50 ± 9.61 | 0 |
| 11j | 33 ± 6.89 | 9± |
| 11k | 78 ± 8.96 | 43 ± 6.20 |
| 11l | 0 | 0 |
| MUT 101 | 90 ± 0.52 | - |
| Raltegravir | - | 95 ± 0.98 |
| Compound | Antiviral Activity at 100 µM (%) HIV-1 Subtype C 1* | Cytotoxicity CC50 (μM) |
|---|---|---|
| 10a | 15 | 113.9 |
| 10b | 9 | 67.1 |
| 10c | 32 | 81.5 |
| 10d | 11 | 100.1 |
| 10e | 20 | 108.1 |
| 10f | 0 | 39.0 |
| 10g | 0 | 199.7 |
| 10h | 4 | 168.8 |
| 10i | 0 | >200 |
| 10j | 0 | 135.7 |
| 10k | 5 | 103.2 |
| 10l | 0 | >200 |
| 11a | 36 | >200 |
| 11b | 35 | 158.2 |
| 11d | 33 | 58.4 |
| 11e | 24 | 78.6 |
| 11g | 31 | >200 |
| 11h | 40 | 50.4 |
| 11i | 10 | 91.2 |
| 11j | 18 | 37.9 |
| 11k | 23 | 47.6 |
| 11l | 26 | 172.7 |
| Raltegravir | 83 at 10 µM | - |
| Auranofin | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashamuse, T.J.; Fish, M.Q.; Coyanis, E.M.; Bode, M.L. Studies towards the Design and Synthesis of Novel 1,5-Diaryl-1H-imidazole-4-carboxylic Acids and 1,5-Diaryl-1H-imidazole-4-carbohydrazides as Host LEDGF/p75 and HIV-1 Integrase Interaction Inhibitors. Molecules 2021, 26, 6203. https://doi.org/10.3390/molecules26206203
Rashamuse TJ, Fish MQ, Coyanis EM, Bode ML. Studies towards the Design and Synthesis of Novel 1,5-Diaryl-1H-imidazole-4-carboxylic Acids and 1,5-Diaryl-1H-imidazole-4-carbohydrazides as Host LEDGF/p75 and HIV-1 Integrase Interaction Inhibitors. Molecules. 2021; 26(20):6203. https://doi.org/10.3390/molecules26206203
Chicago/Turabian StyleRashamuse, Thompho J., Muhammad Q. Fish, E. Mabel Coyanis, and Moira L. Bode. 2021. "Studies towards the Design and Synthesis of Novel 1,5-Diaryl-1H-imidazole-4-carboxylic Acids and 1,5-Diaryl-1H-imidazole-4-carbohydrazides as Host LEDGF/p75 and HIV-1 Integrase Interaction Inhibitors" Molecules 26, no. 20: 6203. https://doi.org/10.3390/molecules26206203