
Expanding the Reactive Sulfur Metabolome: Intracellular and Efflux Measurements of Small Oxoacids of Sulfur (SOS) and H2S in Human Primary Vascular Cell Culture

 , , , and
, , , and
Abstract
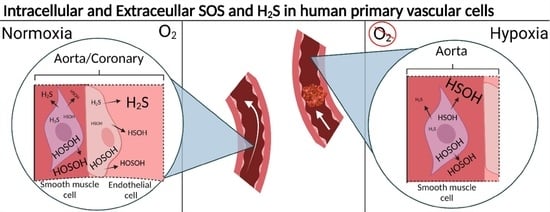
:
1. Introduction
2. Results
2.1. Measurements of Intracellular Concentrations
2.2. Measurements of Extracellular Concentrations
2.3. Hypoxia Induced Changes of Endogenous and Effluxed Analytes in HAOSMCs
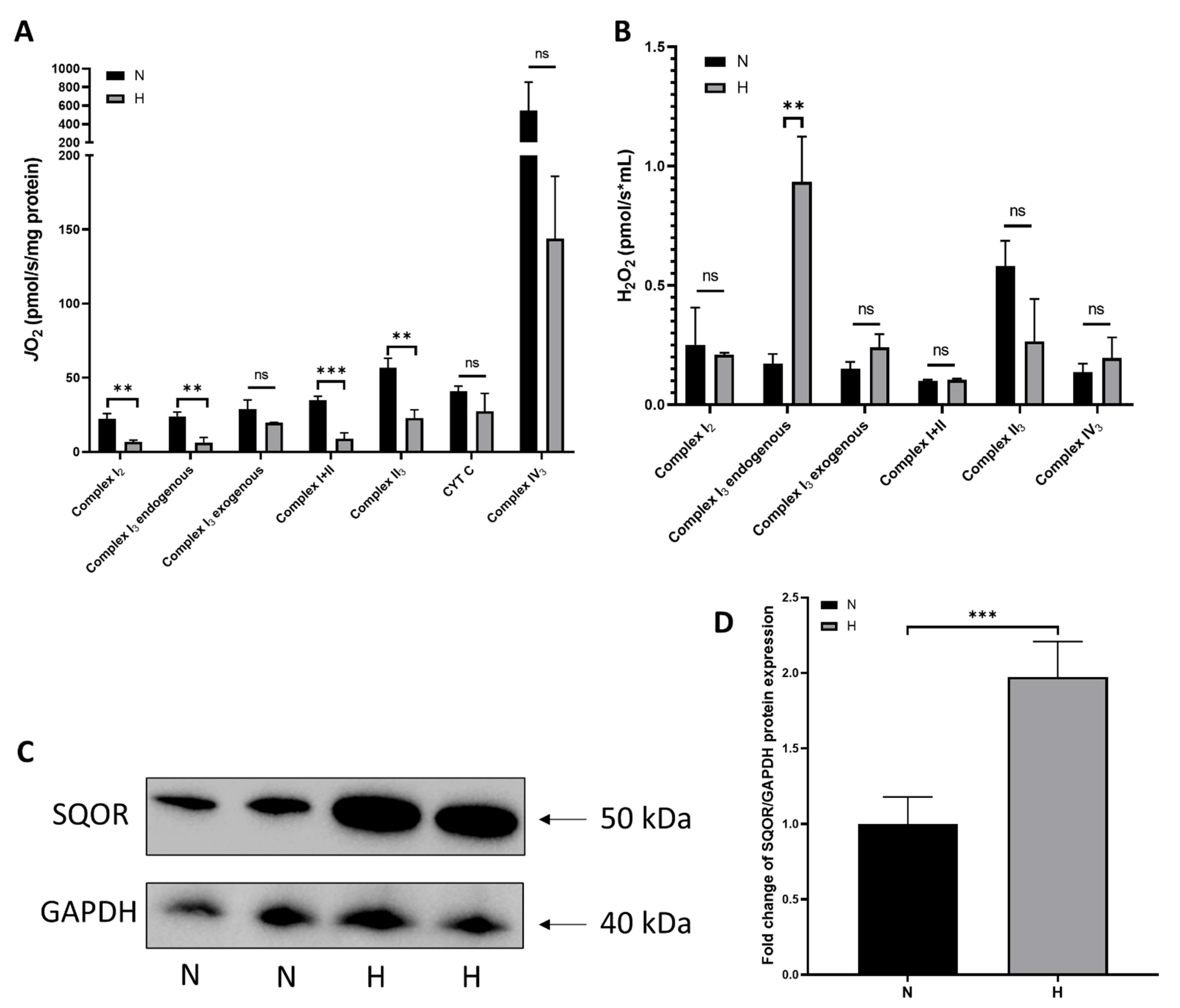
2.4. Hypoxia-Induced Changes in Mitochondrial Function and ROS production
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Electrophilic and Nucleophilic Trapping Procedures
4.4. Efflux Measurements
4.5. Cell Culture in Hypoxic Chamber
4.6. Sample Preparation
4.7. Mass Spectrometry
4.8. Mitochondrial Respiration and ROS Productions
4.9. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yang, G.; Wang, R. H2S and blood vessels: An overview. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide, Handbook of Experimental Pharmacology; Moore, P.K., Whiteman, M., Eds.; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.-S.; Yong, Q.C.; Pan, T.-T.; Feng, Z.-N.; Ali, M.Y.; Zhou, S.; Moore, P.K. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J. Pharmacol. Exp. Ther. 2006, 316, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Kevil, C.G. Nitric oxide and hydrogen sulfide regulation of ischemic vascular remodeling. Microcirculation 2016, 23, 134–145. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Yang, W.; Jia, X.; Yang, G.; Duridanova, D.; Cao, K.; Wang, R. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab. Investig. 2009, 89, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Zou, C.-G. Redox regulation and reaction mechanism of human cystathionine-β-synthase: A PLP dependent hemesensor protein. Arch. Biochem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.-Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.-S. A review of hydrogen sulfide synthesis, Metabolism, and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Prabhudesai, S.; Koceja, C.; Dey, A.; Eisa-Beyg, S.; Leigh, N.R.; Bhattacharya, R.; Mukherjee, P.; Ramchandran, R. Cystathionine β-synthase is necessary for axis development in vivo. Front. Cell Dev. Biol. 2018, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Li, Z.; Sharp, T.E., 3rd; Polhemus, D.J.; Carnal, J.; Moles, K.H.; Tao, Y.-X.; Elrod, J.; Pfeilschifter, J.; Beck, K.F.; et al. Endothelial cell cystathionine γ-lyase expression level modulates exercise capacity, vascular function, and myocardial ischemia reperfusion injury. J. Am. Heart Assoc. 2020, 9, e017544. [Google Scholar] [CrossRef]
- Lindsei, K.; Sarna, Y.L.S.; Karmin, O. The CBS/CSE system: A potential therapeutic target in NAFLD? Can. J. Physiol. Pharmacol. 2015, 93, 1–11. [Google Scholar]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.-L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Ito, T.; Kitamura, H.; Nishino, T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: Confocal laser fluorescence and immunoelectron-microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 1998, 110, 243–250. [Google Scholar] [CrossRef]
- Billaut-Laden, I.; Allorge, D.; Crunelle-Thibaut, A.; Rat, E.; Cauffiez, C.; Chevalier, D.; Houdret, N.; Guidice, J.-M.L.; Broly, F. Evidence for a functional genetic polymorphism of the human thiosulfate sulfurtransferase (Rhodanese), a cyanide and H2S detoxification enzyme. Toxicology 2006, 225, 1–11. [Google Scholar] [CrossRef]
- Testai, L.; D’Antongiovanni, V.; Piano, I.; Martelli, A.; Citi, V.; Duranti, E.; Virdis, A.; Blandizzi, C.; Gargini, C.; Breschi, M.; et al. Different patterns of H2S/NO activity and cross-talk in the control of the coronary vascular bed under normotensive or hypertensive conditions. Nitric Oxide 2015, 47, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Szabo, C.; Ichinose, F.; Ahmed, A.; Whiteman, M.; Papapetropoulos, A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharm. Sci. 2015, 36, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox. Signal. 2011, 15, 373–378. [Google Scholar] [CrossRef]
- Peter, E.A.; Shen, X.; Shah, S.H.; Pardue, S.; Glawe, J.D.; Zhang, W.W.; Reddy, P.; Akkus, N.I.; Varma, J.; Kevil, C.G. Plasma free H2S levels are elevated in patients with cardiovascular disease. J. Am. Heart Assoc. 2013, 2, e000387. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.L.; Wu, H.-C.; Li, Z.-L.; Geng, B.; Tang, C.-S. Changes of the new gaseous transmitter H2S in patients with coronary heart disease. Di 1 Jun Yi Da Xue Xue Bao = Acad. J. First Med Coll. PLA 2005, 25, 951. [Google Scholar]
- Siebert, N.; Cantre, D.; Eipel, C.; Vollmar, B. H2S contributes to the hepatic arterial buffer response and mediates vasorelaxation of the hepatic artery via activation of K(ATP) channels. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1266–G1273. [Google Scholar] [CrossRef] [Green Version]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Marletta, M.A. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr. Opin. Chem. Biol. 2012, 16, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Tatiana, V.; Mishanina, M.L.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-TRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.; Nasuno, R.; Yoshikawa, Y.; Jung, M.; Ida, T.; Matsunaga, T.; Morita, M.; Takagi, H.; Motohashi, H.; Akaike, T. Mitochondrial cysteinyl-TRNA synthetase is expressed via alternative transcriptional initiation regulated by energy metabolism in yeast cells. J. Biol. Chem. 2019, 294, 13781–13788. [Google Scholar] [CrossRef]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox. Signal. 2015, 22, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef] [Green Version]
- McCook, O.; Radermacher, P.; Volani, C.; Asfar, P.; Ignatius, A.; Kemmler, J.; Möller, P.; Szabó, C.; Whiteman, M.; Wood, M.E.; et al. H2S during circulatory shock: Some unresolved questions. Nitric Oxide 2014, 15, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lancaster, J.R., Jr. Chemical foundations of hydrogen sulfide biology. Nitric Oxide 2014, 30, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilboa-Garber, N. Direct spectrophotometric determination of inorganic sulfide in biological materials and in other complex mixtures. Anal. Biochem. 1971, 43, 129–133. [Google Scholar] [CrossRef]
- Siegel, L.M. A direct microdetermination for sulfide. Anal. Biochem. 1965, 11, 126–132. [Google Scholar] [CrossRef]
- Yu, F.; Han, X.; Chen, L. Fluorescent probes for hydrogen sulfide detection and bioimaging. Chem. Commun. 2014, 50, 12234–12249. [Google Scholar] [CrossRef]
- Henthorn, H.A.; Pluth, M.D. Mechanistic insights into the H2S-mediated reduction of aryl azides commonly used in H2S detection. J. Am. Chem. Soc. 2015, 137, 15330–15336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Kolluru, G.K.; Yuan, S.; Kevil, C. Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzym. 2015, 554, 31–45. [Google Scholar]
- Nagy, P.; Doka, E.; Ida, T.; Akaike, T. Measuring reactive sulfur species and thiol oxidation states: Challenges and cautions in relation to alkylation-based protocols. Antioxid. Redox Signal. 2020, 33, 1174–1189. [Google Scholar] [CrossRef]
- Hamid, H.A.; Tanaka, A.; Ida, T.; Nishimura, A.; Matsunaga, T.; Fujii, S.; Morita, M.; Sawa, T.; Fukuto, J.M.; Nagy, P.; et al. Polysulfide stabilization by tyrosine and hydroxyphenyl-containing derivatives that is important for a reactive sulfur metabolomics analysis. Redox Biol. 2019, 21, 2213–2317. [Google Scholar] [CrossRef]
- Takata, T.; Jung, M.; Matsunaga, T.; Ida, T.; Morita, M.; Motohashi, H.; Shen, X.; Kevil, C.G.; Fukuto, J.M.; Akaike, T. Methods in sulfide and persulfide research. Nitric Oxide 2021, 116, 47–64. [Google Scholar] [CrossRef]
- Bradford, G.; Hill, C.R.; Oh, J.-Y.; Johnson, M.S.; Landar, A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic. Biol. Med. 2009, 47, 675–683. [Google Scholar]
- Shi, Y.; Carroll, K.S. Parallel evaluation of nucleophilic and electrophilic chemical probes for sulfenic acid: Reactivity, selectivity and biocompatibility. Redox Biol. 2021, 46, 102072. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Milena, H.; Ida, T.; Mijuskovic, A.; Dustin, C.M.; Bogdándi, V.; Fukuto, J.M.; Dick, T.P.; Nagy, P.; Li, J. Cysteine perthiosulfenic acid (Cys-SSOH): A novel intermediate in thiol-based redox signaling? Redox Biol. 2018, 14, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.; Thomas-Lopez, D.; Jung, Y.; et al. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metab. 2019, 30, 1152–1170. [Google Scholar] [CrossRef] [PubMed]
- Bogdándi, V.; Ida, T.; Sutton, T.R.; Bianco, C.; Ditrói, T.; Koster, G.; Henthorn, H.A.; Minnion, M.; Toscano, J.P.; van der Vliet, A.; et al. Speciation of reactive sulfur species and their reactions with alkylating agents: Do we have any clue about what is present inside the cell? Br. J. Pharmacol. 2019, 176, 646–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, S.; Rehder, C.R.B. Cysteine sulfenic Acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 2010, 49, 7748–7755. [Google Scholar]
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1*. J. Biol. Chem. 2009, 284, 6476–6485. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.R.; Farmer, P.J. Chemical trapping and characterization of small oxoacids of sulfur (SOS) generated in aqueous oxidations of H2S. Redox Biol. 2018, 14, 485–491. [Google Scholar] [CrossRef]
- Kumar, M.R.; Farmer, P.J. Characterization of polysulfides, polysulfanes, and other unique species in the reaction between GSNO and H2S. Molecules 2019, 24, 3090. [Google Scholar] [CrossRef] [Green Version]
- Kallis, G.-B.; Holmgren, A. Differential Reactivity of the Functional Sulfhydryl Groups of Cysteine32 and Cysteine-35 Present in the Reduced Form of Thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [Google Scholar] [CrossRef]
- Koike, S.; Nishimoto, S.; Ogasawara, Y. Cysteine persulfides and polysulfides produced by exchange reactions with H2S protect SH-SY5Y cells from methylglyoxal-induced toxicity through Nrf2 activation. Redox Biol. 2017, 12, 530–539. [Google Scholar] [CrossRef]
- Islam, T.A.; Miller-Martini, D.D.; Horowitz, P.M. Mutation of cysteine 254 facilitates the conformational changes accompanying the interconversion of persulfide-substituted and persulfide-free rhodanese. J. Biol. Chem. 1994, 269, 7908–7913. [Google Scholar] [CrossRef]
- Francoleon, N.E.; Carrington, S.J.; Fukuto, J.M. The reaction of H2S with oxidized thiols: Generation of persulfides and implications to H2S biology. Arch. Biochem. Biophys. 2011, 516, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Scrivner, O.; Kumar, M.R.; Sorokolet, K.; Wong, A.; Kebaara, B.; Farmer, P.J. Characterization of endogenous and extruded H2S and small oxoacids of sulfur (SOS) in cell cultures. ACS Chem. Biol. 2021, 16, 1413–1424. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, M.; Krämer, A.C.; Miotto, G.; Zaccarin, M.; Zhang, H.; Ursini, F. Protein cysteine oxidation in redox signaling: Caveats on sulfenic acid detection and quantification. Arch. Biochem. Biophys. 2017, 617, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Tao, B.-B.; Liu, S.-Y.; Zhang, C.-C.; Fu, W.; Cai, W.-J.; Wang, Y.; Shen, Q.; Wang, M.-J.; Chen, Y.; Zhang, L.-J.; et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045–Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox Signal. 2013, 19, 448–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an endothelial NAD+-H2S signaling network is a reversible cause of vascular aging. Cell 2018, 173, 74–89. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pan, L.; Zhuo, Y.; Gong, Q.; Rose, P.; Zhu, Y. Hypoxia-inducible factor-1a is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol. Pharm. Bull. 2010, 33, 1550–1554. [Google Scholar] [CrossRef] [Green Version]
- Greaney, J.L.; Kutz, J.L.; Shank, S.W.; Jandu, S.; Santhanam, L.; Alexander, L.M. Impaired hydrogen sulfide–mediated vasodilation contributes to microvascular endothelial dysfunction in hypertensive adults. Hypertension 2017, 69, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Materazzi, S.; Zagli, G.; Nassini, R.; Bartolini, I.; Romagnoli, S.; Chelazzi, C.; Benemei, S.; Coratti, A.; De Gaudio, A.R.; Patacchini, R. Vasodilator activity of hydrogen sulfide (H2S) in human mesenteric arteries. Microvasc Res. 2017, 109, 38–44. [Google Scholar] [CrossRef]
- Patel, S.; Fedinec, A.L.; Liu, J.; Weiss, M.A.; Pourcyrous, M.; Harsono, M.; Parfenova, H.; Leffler, C.W. H2S mediates the vasodilator effect of endothelin-1 in the cerebral circulation. Vasc. Biol. Microcirc. 2018, 315, H1759–H1764. [Google Scholar] [CrossRef] [Green Version]
- la Fuente, J.M.; Fernandez, A.; Pepe-Cardoso, A.J.; Martínez-Salamanca, J.I.; Louro, N.; Angulo, J. L-cysteine/hydrogen sulfide pathway induces cGMP-dependent relaxation of corpus cavernosum and penile arteries from patients with erectile dysfunction and improves arterial vasodilation induced by PDE5 inhibition. Eur. J. Pharmacol. 2019, 863, 172675. [Google Scholar] [CrossRef]
- Rushing, A.M.; Donnarumma, E.; Polhemus, D.J.; Au, K.R.; Victoria, S.E.; Schumacher, J.D.; Li, Z.; Jenkins, J.S.; Lefer, D.J.; Goodchild, T.T. Effects of a novel hydrogen sulfide prodrug in a porcine model of acute limb ischemia. J. Vasc. Surg. 2019, 69, 1924–1935. [Google Scholar] [CrossRef]
- Wang, R. Shared signaling pathways among gasotransmitters. Proc. Natl. Acad. Sci. USA 2012, 109, 8801–8802. [Google Scholar] [CrossRef] [Green Version]
- Baragatti, B.; Ciofini, E.; Sodini, D.; Luin, S.; Scebba, F.; Coceani, F. Hydrogen sulfide in the mouse ductus arteriosus: A naturally occurring relaxant with potential EDHF function. Vasc. Biol. Microcirc. 2013, 304, H927–H934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovgaard, N.; Gouliaev, A.; Aalling, M.; Simonsen, U. The role of endogenous H2S in cardiovascular physiology. Curr. Pharm. Biotechnol. 2011, 12, 1385–1393. [Google Scholar] [CrossRef]
- Wang, R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009, 76, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, G.; Cai, D.; Pan, B.; Lin, Y.; Li, X.; Ling, S.L.; Liao, Z.X.; Wang, H. H2S inhibition of chemical hypoxia-induced proliferation of HPASMCs is mediated by the upregulation of COX-2/PGI2. Int. J. Mol. Med. 2014, 33, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.; Wang, Y.; Liu, J.; Yu, X.; Sun, Y.; Yang, F.; Dong, S.; Wu, J.; Zhao, Y.; Xu, C.; et al. Exogenous H2S modulates mitochondrial fusion–fission to inhibit vascular smooth muscle cell proliferation in a hyperglycemic state. Cell Biosci. 2016, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Li, H.; Tang, G.; Wu, L.; Zhao, K.; Cao, Q.; Xu, C.; Wang, R. Increased neointimal formation in cystathionine gamma-lyase deficient mice: Role ofhydrogen sulfide inα5β1-integrin and matrix metalloproteinase-2 expression insmooth muscle cells. J. Mol. Cell. Cardiol. 2012, 52, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1–21. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wu, L.; Liang, W.; Wang, R. Direct stimulation of KATP channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 2005, 68, 1757–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucci, M.; Papapetropoulos, A.; Vellecco, V.; Zhou, Z.; Pyriochou, A.; Roussos, C.; Roviezzo, F.; Brancaleone, V.; Cirino, G. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arter. Thromb. Vasc. Biol. 2010, 30, 1998–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, M.; Li, L.; Kostetski, I.; Chu, S.H.; Siau, J.L.; Bhatia, M.; Moore, P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.; Calvert, J.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nubel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Yong, R.; Searcy, D.G. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 129, 129–137. [Google Scholar] [CrossRef]
- Jensen, B.S.; Pardue, S.; Duffy, B.; Kevil, C.G.; Staples, J.F.; Fago, A. Suppression of mitochondrial respiration by hydrogen sulfide in hibernating 13-lined ground squirrels. Free Radic. Biol. Med. 2021, 169, 181–186. [Google Scholar] [CrossRef]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide catabolism ameliorates hypoxic brain injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef]
- Malagrino, F.; Zuhra, K.; Mascolo, L.; Mastronicola, D.; Vicente, J.B.; Forte, E.; Giuffre, A. Hydrogen sulfide oxidation: Adaptive changes in mitochondria of SW480 colorectal cancer cells upon exposure to hypoxia. Oxid. Med. Cell. Longev. 2019, 2019, 8102936. [Google Scholar] [CrossRef]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Wang, Z.; Zhang, M.; Huang, C.; Song, Y.; Xu, F.; Zhang, J.; Li, J.; He, M.; Li, Y.; et al. SQR mediates therapeutic effects of H2S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci Adv. 2020, 6, eaaz5752. [Google Scholar] [CrossRef]
- Nicholson, C.K.; Calvert, J.W. Hydrogen Sulfide and Ischemia - Reperfusion Injury. Pharmacol Res. 2010, 62, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Bir, S.C.; Kolluru, G.K.; McCarthy, P.; Shen, X.; Pardue, S.; Pattillo, C.B.; Kevil, C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor–dependent angiogenesis. J. Am. Heart Assoc. 2012, 1, e004093. [Google Scholar] [CrossRef] [Green Version]
- Islam, K.N.; Polhemus, D.J.; Donnarumma, E.; Brewster, L.P.; Lefer, D.J. Hydrogen sulfide levels and nuclear factor-erythroid 2-related factor 2 (NRF2) activity are attenuated in the setting of critical limb ischemia (CLI). J. Am. Heart Assoc. 2015, 4, e001986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, M.M.; Hui, C.K.; Whiteman, M.; Wood, M.E.; Adcock, I.; Kirkham, P.; Michaeloudes, C.; Chung, K.F. Hydrogen sulfide inhibits proliferation and release of IL-8 from human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K. Acute vs. chronic vs. cyclic hypoxia: Their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialò, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The role of protein persulfidation in brain aging and neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef] [PubMed]
- Bithi, N.; Link, C.; Henderson, Y.O.; Kim, S.; Yang, J.; Li, L.; Wang, R.; Willard, B.; Hine, C. Dietary restriction transforms the mammalian protein persulfidome in a tissue-specific and cystathionine γ-lyase-dependent manner. Nat. Commun. 2021, 12, 1745. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, G. H2S signaling and extracellular matrix remodeling in cardiovascular diseases: A tale of tense relationship. Nitric Oxide 2021, 116, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Ismaeel, A.; Miserlis, D.; Papoutsi, E.; Haynatzki, G.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Casale, G.P.; Pipinos, I.I.; Koutakis, P. Endothelial cell-derived pro-fibrotic factors increase TGF-β1 expression by smooth muscle cells in response to cycles of hypoxia-hyperoxia. Biochim. Biophys. Acta Mol. Basis. Dis. 2021, 1868, 166278. [Google Scholar] [CrossRef] [PubMed]
- Krumschnabel, G.; Fontana-Ayoub, M.; Sumbalova, Z.; Heidler, J.; Gauper, K.; Fasching, M.; Gnaiger, E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 2015, 1264, 245–261. [Google Scholar] [PubMed]
- Mohanty, J.G.; Jaffe, J.S.; Schulman, E.S.; Raible, D.G. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods 1997, 202, 133–141. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | H2S (pM) | HSOH (pM) | HOSOH (pM) | Total [H2S + SOS] (pM) |
|---|---|---|---|---|
| HAOSMC | 2940 ± 78.8 | 1240 ± 73.4 | 5270 ± 87.3 | 9450 ± 63.1 |
| HCASMC | 1480 ± 136 | 1030 ± 12.3 | 1846.8 ± 102 | 4360 ± 170 |
| HAOEC | 75.8 ± 7.37 | 73.4 ± 0.901 | 87.3 ± 5.29 | 237 ± 9.12 |
| HCAEC | 110 ± 5.62 | 79.3 ± 0.960 | 130 ± 9.29 | 319 ± 10.9 |
| Cell Type | H2S/HSOH/HOSOH Ratios | Sulfomic Index | Total [H2S + SOS] (pM) |
|---|---|---|---|
| HAOSMC | 1:0.42:1.79 | 1.27 | 9450 ± 63.1 |
| HCASMC | 1:0.70:1.25 | 1.08 | 4360 ± 170 |
| HAOEC | 1:0.93:1.11 | 1.04 | 237 ± 9.12 |
| HCAEC | 1:0.72:1.18 | 1.06 | 319 ± 10.9 |
| HEK293 * | 1:0.28:0.34 | 0.70 | 2610 ± 700 |
| HeLa * | 1:0.11:0.11 | 0.27 | 82.9 ± 4.55 |
| A375 * | 1:0.03:0.06 | 0.14 | 2920 ± 466 |
| Cell Type | H2S (pM/h) | HSOH (pM/h) | HOSOH (pM/h) | Total [H2S + SOS] (pM/h) |
|---|---|---|---|---|
| HAOSMC | 6810 ± 900 | 3500 ± 1200 | 3300 ± 230 | 13,600 ± 1520 |
| HCASMC | 4430 ± 139 | 2150 ± 548 | 8870 ± 2120 | 15,500 ± 2190 |
| HAOEC | 1150 ± 742 | 1540 ± 1090 | 622 ± 404 | 3310 ± 1380 |
| HCAEC | 398 ± 209 | 88.6 ± 3.99 | 346 ± 45.6 | 833 ± 214 |
| Cell Type | H2S/HSOH/HOSOH Ratios | Sulfomic Index | Total [H2S + SOS] (pM) |
|---|---|---|---|
| HAOSMC | 1:0.51:0.48 | 0.74 | 13,600 ± 1520 |
| HCASMC | 1:0.49:2.00 | 1.29 | 15,500 ± 2190 |
| HAOEC | 1:1.34:0.54 | 0.84 | 3310 ± 1380 |
| HCAEC | 1:0.22:0.87 | 0.94 | 833 ± 214 |
| HEK293 * | 1:0.05:0.37 | 0.55 | 2610 ± 700 |
| HeLa * | 1:0.04:0.30 | 0.47 | 82.9 ± 4.55 |
| A375 * | 1:0.017:0.057 | 0.12 | 5930 ± 899 |
| Growth Condition | Total S-Species [H2S+SOS] | |
|---|---|---|
| Intracellular (pM) | Extracellular (pM)/h | |
| Normoxia | 9450 ± 63.1 | 13,540 ± 301 |
| Hypoxia | 5161 ± 247 | 14,770 ± 371 |
| Growth Condition | H2S/HSOH/HOSOH Ratios | Sulfomic Index | ||
|---|---|---|---|---|
| Intracellular | Efflux | Intracellular | Efflux | |
| Normoxia | 1:0.42:1.79 | 1:0.51:0.48 | 1.27 | 0.74 |
| Hypoxia | 1:1.20:1.68 | 1:2.38:0.79 | 1.18 | 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scrivner, O.; Ismaeel, A.; Kumar, M.R.; Sorokolet, K.; Koutakis, P.; Farmer, P.J. Expanding the Reactive Sulfur Metabolome: Intracellular and Efflux Measurements of Small Oxoacids of Sulfur (SOS) and H2S in Human Primary Vascular Cell Culture. Molecules 2021, 26, 7160. https://doi.org/10.3390/molecules26237160
Scrivner O, Ismaeel A, Kumar MR, Sorokolet K, Koutakis P, Farmer PJ. Expanding the Reactive Sulfur Metabolome: Intracellular and Efflux Measurements of Small Oxoacids of Sulfur (SOS) and H2S in Human Primary Vascular Cell Culture. Molecules. 2021; 26(23):7160. https://doi.org/10.3390/molecules26237160
Chicago/Turabian StyleScrivner, Ottis, Ahmed Ismaeel, Murugaeson R. Kumar, Kristina Sorokolet, Panagiotis Koutakis, and Patrick J. Farmer. 2021. "Expanding the Reactive Sulfur Metabolome: Intracellular and Efflux Measurements of Small Oxoacids of Sulfur (SOS) and H2S in Human Primary Vascular Cell Culture" Molecules 26, no. 23: 7160. https://doi.org/10.3390/molecules26237160