
Virtual Screening in Search for a Chemical Probe for Angiotensin-Converting Enzyme 2 (ACE2)

 and
and
Abstract
:1. Introduction
2. Results and Discussion
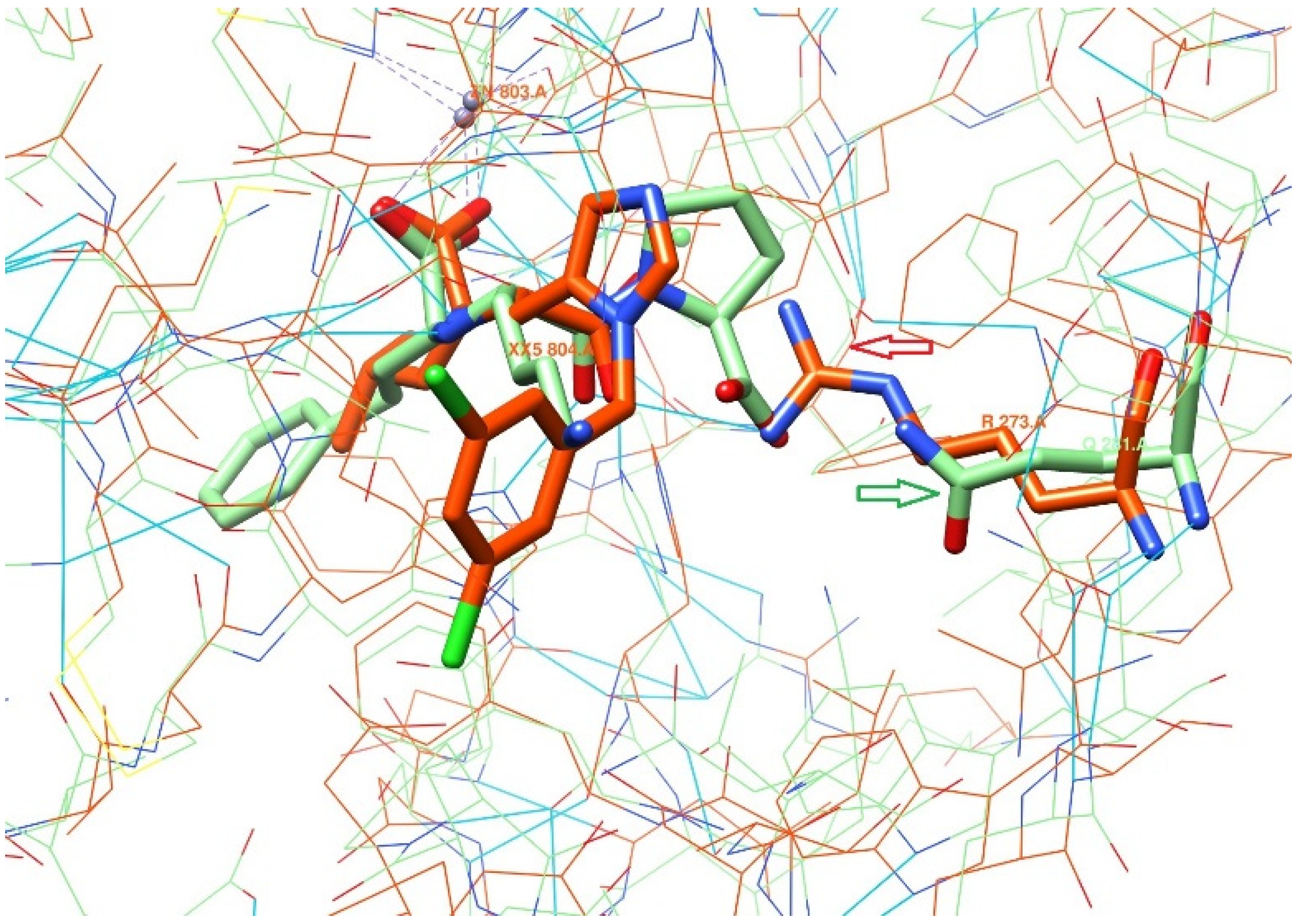
2.1. Comparison of the Binding Sites
2.2. Docking Model Preparation
- (1)
- First model:
- -
- ARG273—Interaction restraints: compulsory hydrogen bond;
- -
- The area near TYR510 is the positional/distance restraint of a spherical shape with a radius of 2 Å;
- -
- Interaction of ionic nature with Zn2+ metal.
- (2)
- Second model:
- -
- ARG273—two positional/distance restraints—two spheres with a radius of 1 Å aiming to mark the positions of the oxygen atoms in the counter-ion carboxylic group;
- -
- The area near TYR510 is the positional/distance restraint of a spherical shape with a radius of 2 Å;
- -
- Ionic interaction with Zn2 + metal.“Grids” for ACE protein have the following restraints:
- -
- Interaction with Zn2+ metal;
- -
- LYS511—compulsory hydrogen bond.
2.3. Molecular Docking, Verification of Docking Models
2.4. The Screening Workflow for the “Enamine Stock Compound Collection” (Enamine SCC, 3.2 Million Compounds)
- -
- Filtering by the SMARTS substructure, looking for molecules with at least one carboxylic group;
- -
- -
- Performing molecular docking at a minimal level of theory with tracking of the conditional assessment of the interaction for critical amino acid residues;
- -
- Molecular docking at medium accuracy and then ranking results according to the interaction energy of the ligands with the key residues of the target;
- -
- Conducting a visual inspection of the poses and selecting a set of 100 molecules with the best positioning in the binding pocket.
- -
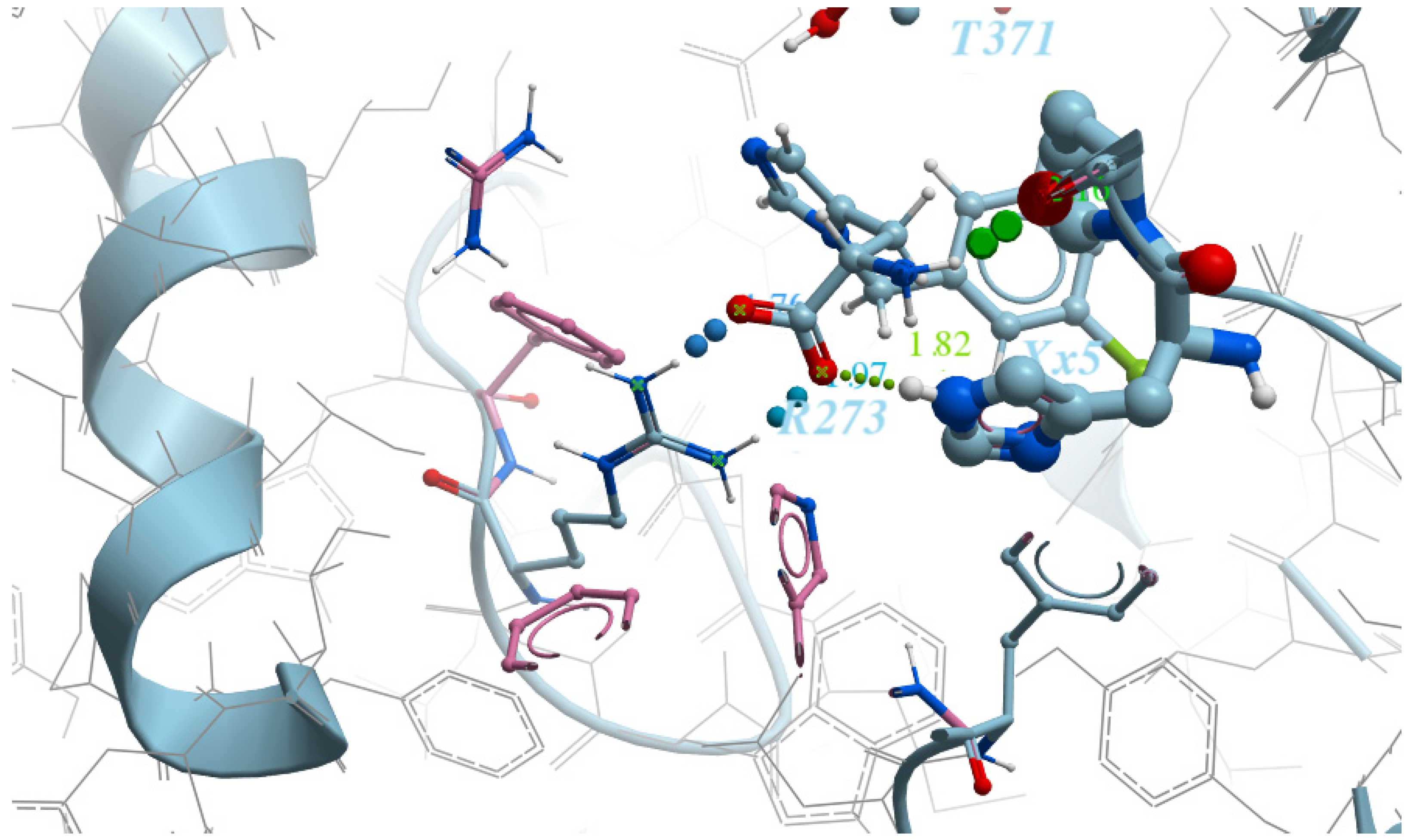
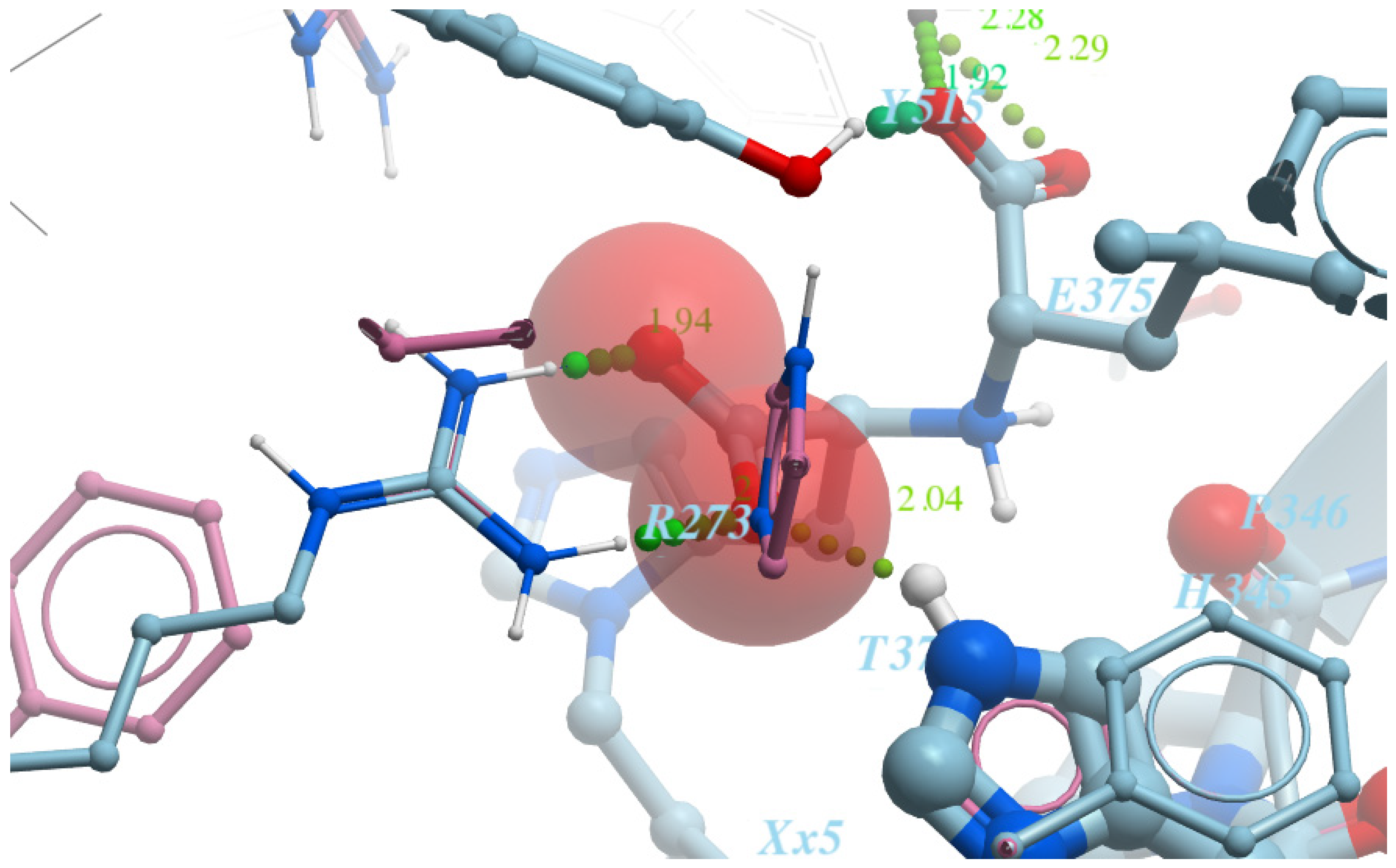
- Pose accuracy and relative distance to ARG273;
- -
- Pose accuracy and relative distance to Zn2+;
- -
- -
- The presence of a hydrogen bond with PRO346.
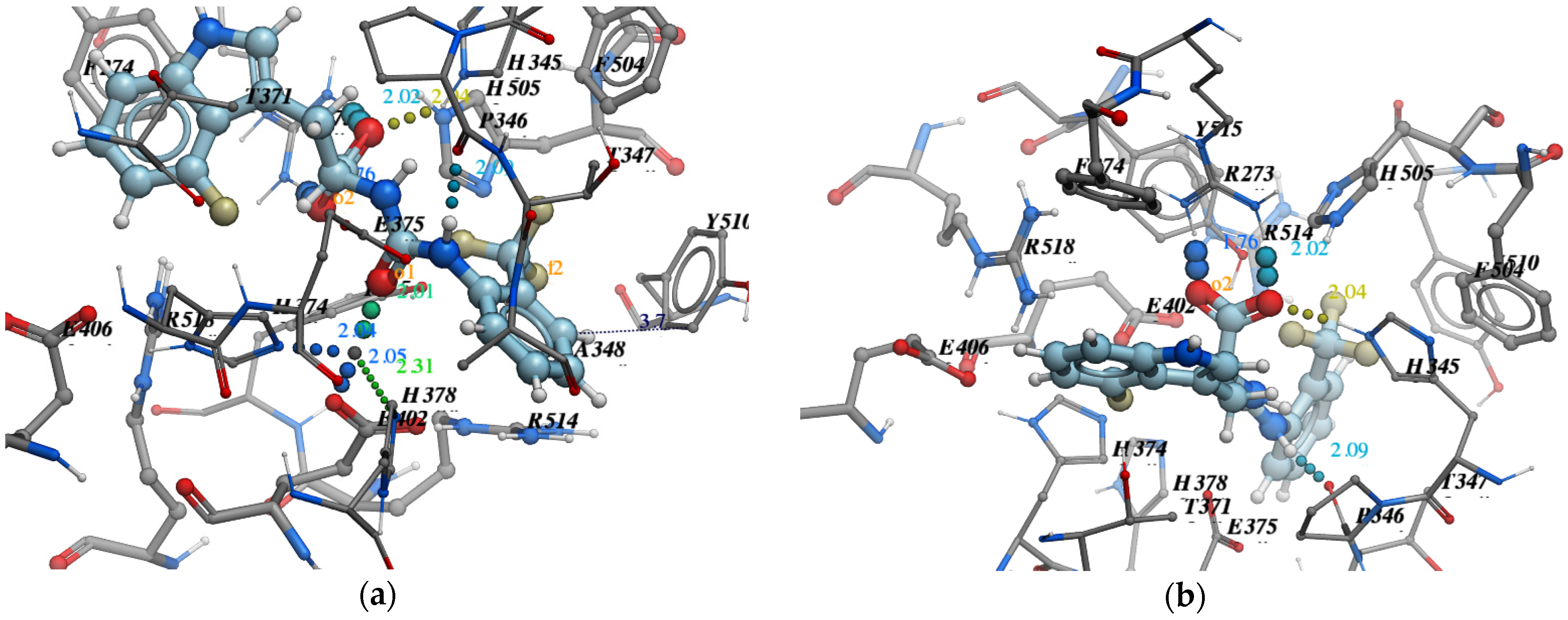
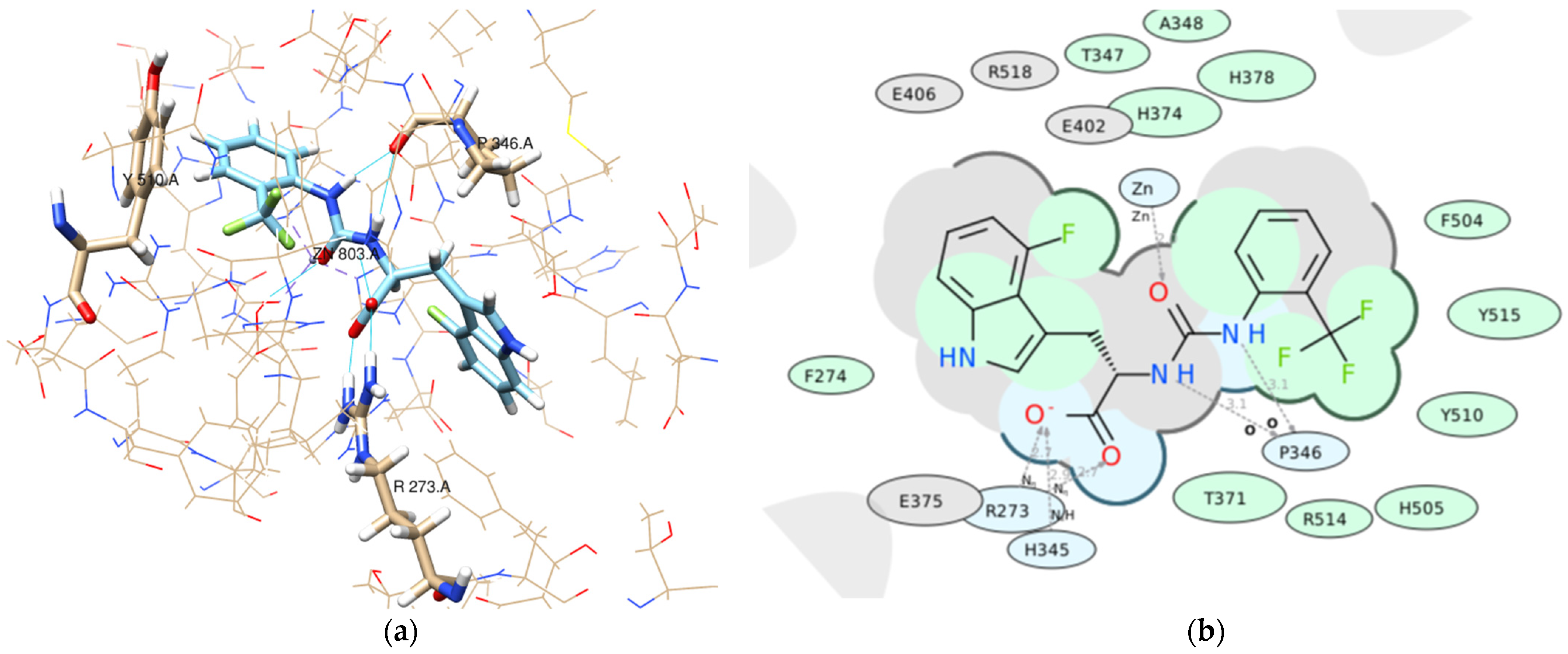
- -
- Pose accuracy and relative distance to ARG273—the hydroxyl group binds the guanidine group ARG273 like a claw, which results in a perfect H-bond;
- -
- Pose accuracy and relative distance to Zn2+—the oxygen atom binds the zinc ion, resulting in metal coordination between the partial negative charge on the oxygen atom and positively charged Zn2+;
- -
- The position of the ligand’s hydrophobic moiety in close proximity to TYR510; the hydrophobic pocket near tyrosine (TYR510) is filled with an ortho-trifluorophenyl substitute;
- -
- The presence of a hydrogen bond with PRO346—the hydrogen bond between proline and nitrogen is evident.
2.5. Screening the Enamine REAL Database Set (Enamine RDB, 1.4 Billion Compounds)
- -
- After the first SMARTS filtering (for the existence of carboxylic groups), 29 million compounds were found. Considering the size of the set, we used the chemical diversity approach (Tanimoto similarity between the molecules represented via Morgan fingerprints [32]). This narrowed down the set to 30,000 substances. After performing the docking of the 30K-set, 50 compounds were selected based on their docking score and visual inspection results. For these compounds, we performed a shape-based similarity search by means of the USRCAT (Ultrafast Shape Recognition with Credo Atom Types) method [33] and then selected those with a Tanimoto similarity of 0.4 or above. Our screening set at this step became larger, with 121K compounds.
- -
- The next step was molecular docking at the low-effort level 1 using a verified model (Section 2). The results were filtered by a docking score < −25, leaving 28,000 ligands.
- -
- Molecular docking at the standard-effort level 5 using a verified model was the next step in our screening. For a more productive virtual view, it was also first filtered by the value of closest distance in the contact analyzer (ARG273), (TYR510), (PRO346). Then the library was sorted by docking score and “cherry-picking” using the same value criteria as for working with the stock base.
- -
- After the calculation of ChemFilters (PAINS, LILLY, IN, USA) and the screening of compounds, we were left with 105 ligands.
- -
- To verify the possible non-selectivity of ligands in relation to ACE, we carried out molecular coupling of the obtained TOP105 compounds with the ACE target. Only one compound showed a score of more than 25, but the pose in it was unnatural. Therefore, we did not discard these compounds.
- -
- 105 compounds were investigated via molecular docking at a high level of effort (level 10) with high accuracy. The model used was the same as the one used previously in this work. Through an expert evaluation, paying attention to the score values, the 20 top compounds were found for specific interactions with important residues.
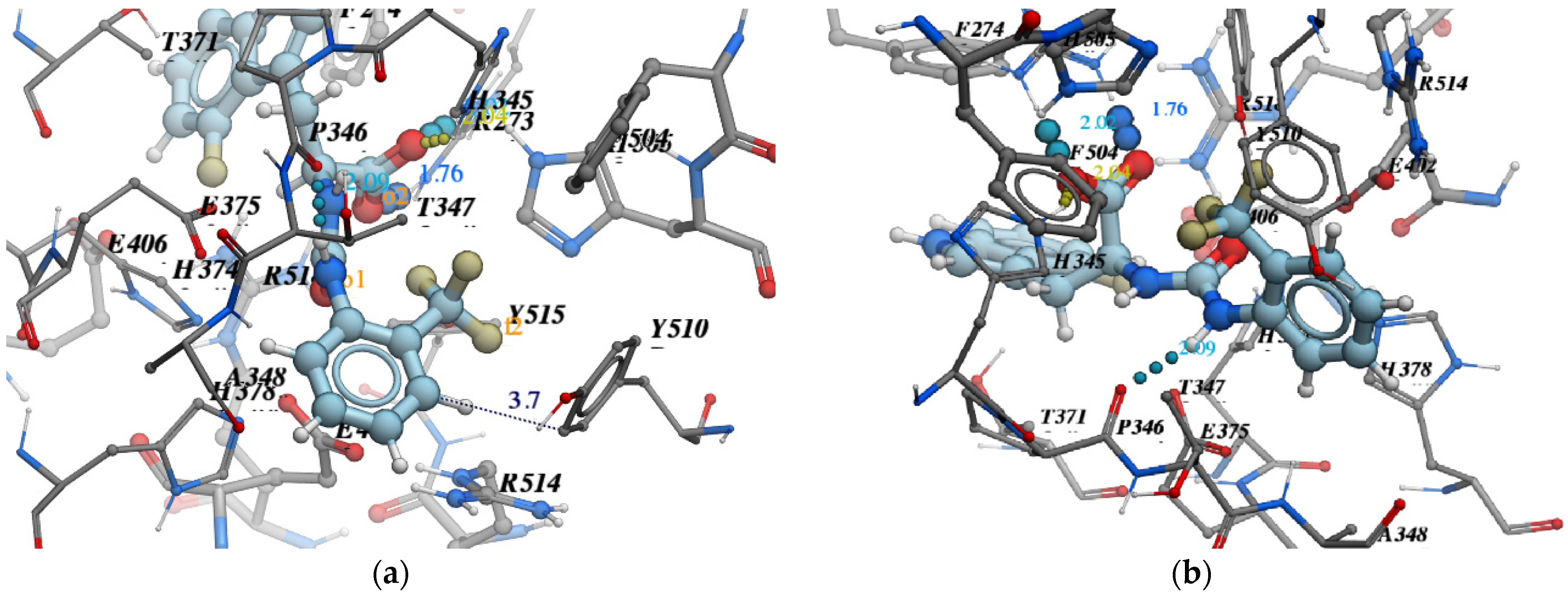
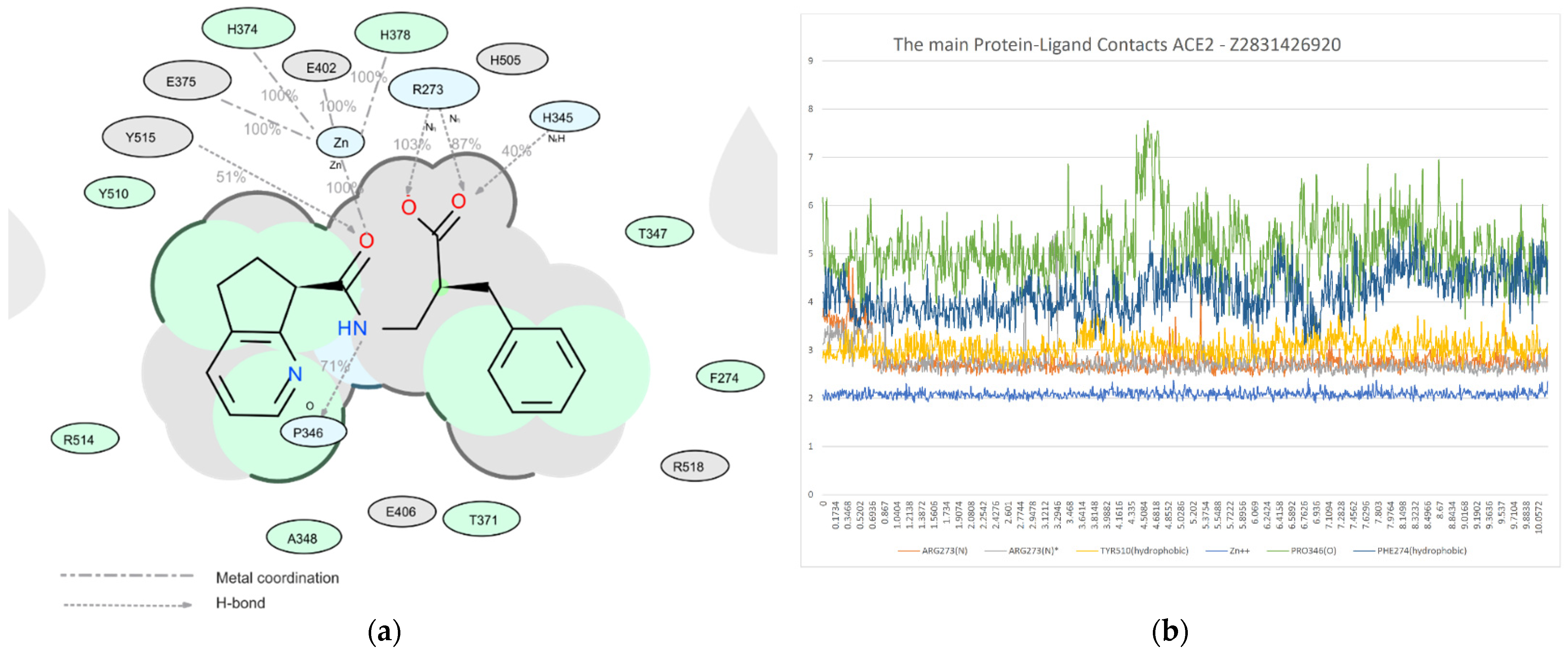
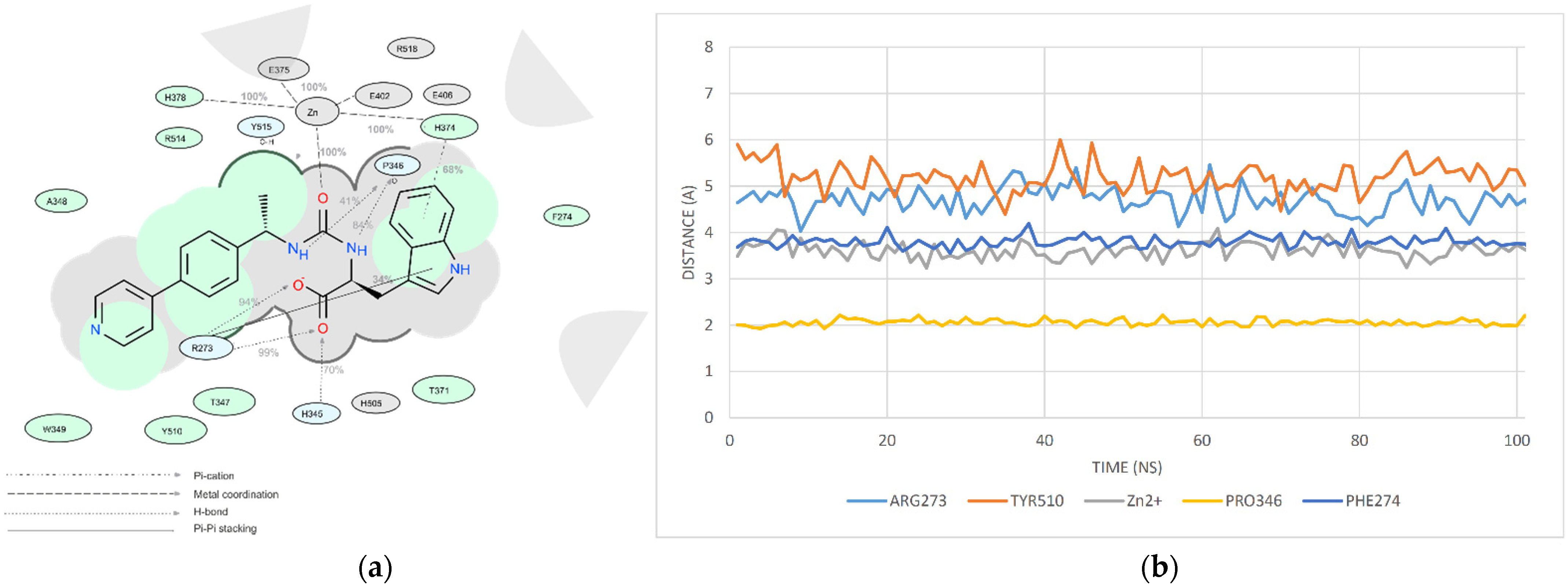
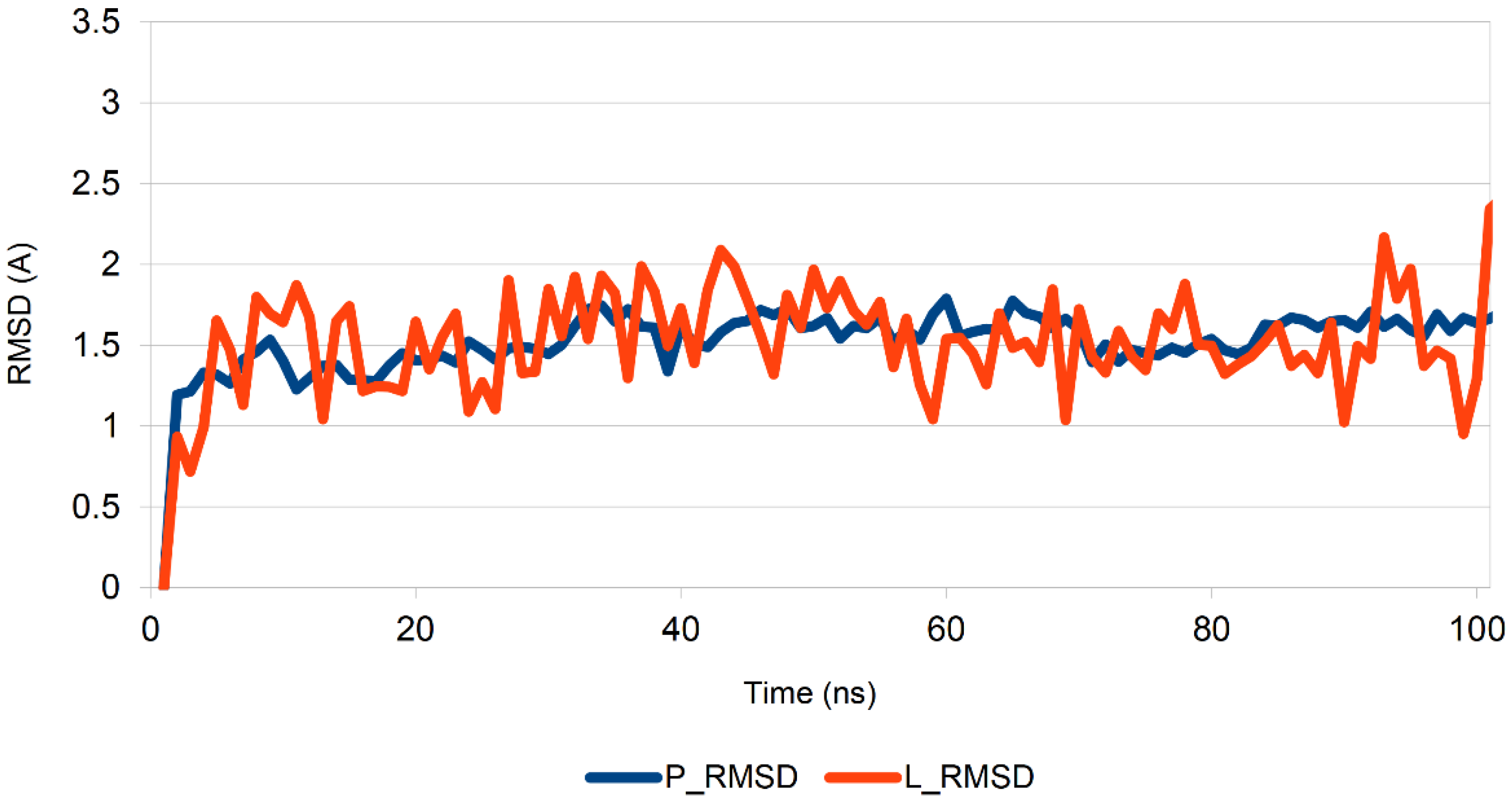
2.6. Molecular Dynamics Study
- (1)
- The RMSD of the ligand position in the complex during the MD run;
- (2)
- The estimation of the number of key contacts between the ligand and protein at each point during the dynamic period; and
- (3)
- Lifetime evolution of specific ligand–protein interactions along the MD trajectory.
3. Materials and Methods
3.1. Molecular System Setup for Docking Simulation
3.1.1. Protein Preparation
3.1.2. Ligand Preparation
3.1.3. Molecular Docking
3.2. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jiang, F.; Deng, L.; Zhang, L.; Cai, Y.; Cheung, C.W.; Xia, Z. Review of the Clinical Characteristics of Coronavirus Disease 2019 (COVID-19). J. Gen. Intern. Med. 2020, 35, 1545–1549. [Google Scholar] [CrossRef] [Green Version]
- COVID-19 Map-Johns Hopkins Coronavirus Resource Center (jhu.edu). Available online: https://coronavirus.jhu.edu/map.html (accessed on 10 September 2021).
- Byrnes, J.J.; Gross, S.; Ellard, C.; Connolly, K.; Donahue, S.; Picarella, D. Effects of the ACE2 inhibitor GL1001 on acute dextran sodium sulfate-induced colitis in mice. Inflamm. Res. 2009, 58, 819–827. [Google Scholar] [CrossRef]
- Gurley, S.B.; Coffman, T.M. Angiotensin-converting enzyme 2 gene targeting studies in mice: Mixed messages. Exp. Physiol. 2008, 93, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Volochnyuk, D.; Ryabukhin, S.V.; Moroz, Y.S.; Savych, O.; Chuprina, A.; Horvath, D.; Zabolotna, Y.; Varnek, A.; Judd, D.B. Evolution of commercially available compounds for HTS. Drug Discov. Today 2019, 24, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Zabolotna, Y.; Lin, A.; Horvath, D.; Marcou, G.; Volochnyuk, D.M.; Varnek, A. Chemography: Searching for Hidden Treasures. J. Chem. Inf. Model. 2021, 61, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Grygorenko, O.O.; Radchenko, D.S.; Dziuba, I.; Chuprina, A.; Gubina, K.E.; Moroz, Y.S. Generating Multibillion Chemical Space of Readily Accessible Screening Compounds. iScience 2020, 23, 101681. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L.; Kotz, J.D.; Li, M.; Aubé, J.; Austin, C.P.; Reed, J.C.; Rosen, H.; White, E.L.; Sklar, L.A.; Lindsley, C.W.; et al. Advancing Biological Understanding and Therapeutics Discovery with Small-Molecule Probes. Cell 2015, 161, 1252–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Warner, F.J.; Guy, J.L.; Lambert, D.W.; Hooper, N.M.; Turner, A.J. Angiotensin Converting Enzyme-2 (ACE2) and its Possible Roles in Hypertension, Diabetes and Cardiac Function. Lett. Pept. Sci. 2003, 10, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.L.; Jackson, R.M.; Acharya, K.R.; Sturrock, E.D.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme-2 (ACE2): Comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry 2003, 42, 13185–13192. [Google Scholar] [CrossRef] [PubMed]
- Dales, N.A.; Gould, A.E.; Brown, J.A.; Calderwood, E.F.; Guan, B.; Minor, C.A.; Gavin, J.M.; Hales, P.; Kaushik, V.K.; Stewart, M.; et al. Substrate-Based Design of the First Class of Angiotensin-Converting Enzyme-Related Carboxypeptidase (ACE2) Inhibitors. J. Am. Chem. Soc. 2002, 124, 11852–11853. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, P. Gliding to success. Nat. Rev. Drug Discov. 2004, 3, 299. [Google Scholar] [CrossRef]
- Ehrt, C.; Brinkjost, T.; Koch, O. A benchmark driven guide to binding site comparison: An exhaustive evaluation using tailor-made data sets (ProSPECCTs). PLoS Comput. Biol. 2018, 14, e1006483. [Google Scholar] [CrossRef]
- Konc, J.; Cesnik, T.; Konc, J.T.; Penca, M.; Janežič, D. ProBiS-database: Precalculated binding site similarities and local pairwise alignments of PDB structures. J. Chem. Inf. Model. 2012, 52, 604–612. [Google Scholar] [CrossRef]
- Leinweber, M.; Fober, T.; Strickert, M.; Baumgärtner, L.; Klebe, G.; Freisleben, B.; Hüllermeier, E. CavSimBase: A Database for Large Scale Comparison of Protein Binding Sites. IEEE Trans. Knowl. Data Eng. 2016, 28, 1423–1434. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Guerrero, G.D.; Perez-S, H.E.; Cecilia, J.M.; Garcia, J.M. Parallelization of Virtual Screening in Drug Discovery on Massively Parallel Architectures. In Proceedings of the 2012 20th Euromicro International Conference on Parallel, Distributed and Network-Based Processing, Garching, Germany, 15 February 2012; pp. 588–595. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.-C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanrikulu, Y.; Krüger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput. Aided Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminform. 2012, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Margreitter, C.; Oostenbrink, C. MDplot: Visualise Molecular Dynamics. R J. 2017, 9, 164–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshani, H.; Dehghanian, E.; Rahati, A. Enhanced GROMACS: Toward a better numerical simulation framework. J. Mol. Model. 2019, 25, 355. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Harvey, M.J.; De Fabritiis, G. High-throughput molecular dynamics: The powerful new tool for drug discovery. Drug Discov. Today 2012, 17, 1059–1062. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of Effort | CPU | Number of Ligands | Time, min | Min per Ligand |
|---|---|---|---|---|
| 1 | 1 | 250 | 31 | 0.124 |
| 5 | 1 | 250 | 110 | 0.44 |
| 10 | 1 | 250 | 220 | 0.88 |
| Description of the Step | Number of Structures Left | CPU Time (min) |
|---|---|---|
| INPUT | 1.41 bln | - |
| MedChem filter (PAINS, etc.) | 1.2 bln | 1800 |
| Ligand docking at low effort (1.) | 100,000 | 148,800,000 |
| Ligand docking at standard effort (5.) | 10,000 | 44,000 |
| Ligand docking at standard effort (5.) with ACE | 9000 | 4400 |
| Ligand docking at high effort (10.) | 200 | 7920 |
| Sort pose by docking score and visual inspection | 20 | - |
| Total CPU time (min) | - | 148,858,120 |
| Description of the Step | Number of Structures Left | CPU Time (min) |
|---|---|---|
| INPUT | 1.41 bln | - |
| Diversification 1/10 | 141 mln | 1800 |
| MedChem filter (PAINS, etc.) | 120 mln | 1800 |
| Ligand docking at low effort (1.) | - | 14,880,000 |
| Sort poses by distance to ARG273, TYR510, PRO346 | 50,000 | - |
| Ligand docking at standard effort (5.) | - | 22,000 |
| Sort poses by distance to ARG273, TYR510, PRO346 | 5000 | - |
| Ligand docking at standard effort (5.) with ACE | 4500 | 2200 |
| Ligand docking at high effort (10.) | 100 | 3960 |
| Sort poses by docking score and visual inspection | 20 | - |
| Total CPU time (min) | - | 14,909,960 |
| Description of the Step | Number of Structures Left | CPU Time (min) |
|---|---|---|
| INPUT | 1.41 bln | - |
| Filter structures contained carboxyl-group | 141 mln | 600 |
| Diversification | 120 mln | 600 |
| Ligand docking at low effort (1.) | - | 3720 |
| Sort poses by distance to ARG273, TYR510, PRO346 | 50,000 | - |
| Visual inspection of poses | - | - |
| Similarity searching by Morgan FP | 5000 | 2250 |
| Ligand docking at standard effort (5.) | 4500 | 53,240 |
| Sort poses by distance to ARG273, TYR510, PRO346 | 100 | |
| Sort poses by docking score and visual inspection | 20 | - |
| MedChem filter (PAINS, etc.) | - | |
| Ligand docking at standard effort (5.) with ACE | 46.2 | |
| Ligand docking at the level of effort (10.) | 92.4 | |
| Total CPU time (hours) | - | 60,548.6 |
| Comparison of the Estimations of CPU Time Required for Roadmaps | Difference (Number of Times) |
|---|---|
| RM1 vs. RM2 | 10 |
| RM1 vs. RM3 | 2458 |
| RM2 to RM3 | 246 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kravets, I.O.; Dudenko, D.V.; Pashenko, A.E.; Borisova, T.A.; Tolstanova, G.M.; Ryabukhin, S.V.; Volochnyuk, D.M. Virtual Screening in Search for a Chemical Probe for Angiotensin-Converting Enzyme 2 (ACE2). Molecules 2021, 26, 7584. https://doi.org/10.3390/molecules26247584
Kravets IO, Dudenko DV, Pashenko AE, Borisova TA, Tolstanova GM, Ryabukhin SV, Volochnyuk DM. Virtual Screening in Search for a Chemical Probe for Angiotensin-Converting Enzyme 2 (ACE2). Molecules. 2021; 26(24):7584. https://doi.org/10.3390/molecules26247584
Chicago/Turabian StyleKravets, Iryna O., Dmytro V. Dudenko, Alexander E. Pashenko, Tatiana A. Borisova, Ganna M. Tolstanova, Sergey V. Ryabukhin, and Dmitriy M. Volochnyuk. 2021. "Virtual Screening in Search for a Chemical Probe for Angiotensin-Converting Enzyme 2 (ACE2)" Molecules 26, no. 24: 7584. https://doi.org/10.3390/molecules26247584