
Amino Acylguanidines as Bioinspired Catalysts for the Asymmetric Aldol Reaction

Department of Biological Chemistry, Institute of Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, E08034 Barcelona, Spain
Molecules 2021, 26(4), 826; https://doi.org/10.3390/molecules26040826
Submission received: 23 December 2020
/
Revised: 1 February 2021
/
Accepted: 3 February 2021
/
Published: 5 February 2021
(This article belongs to the Special Issue Organocatalysis II: Bioinspired and Supramolecular Catalysts)
Abstract
:The binding and stabilizing effect of arginine residues in certain aldolases served as inspiring source for the development of a family of amino acylguanidine organocatalysts. Screening and optimization led to identify the threonine derivative as the most suitable catalyst for the asymmetric aldol addition of hydroxyacetone, affording the syn diastereomer in high ee. In contrast, the proline derivative yielded the anti diasteromer. MMFF models suggest the presence of an extensive hydrogen bonding network between the acylguanidinium group and the reaction intermediates.

1. Introduction
Aldolases can proceed through two general mechanisms: In class I aldolases, a lysine residue is in charge of the enamine formation with the substrate, which is then able to react with an aldehyde. In contrast, in class II aldolases, substrate enolization takes place through binding to a divalent cation that acts as Lewis acid. Moreover, numerous electrostatic and hydrogen bonding interactions with other residues contribute to the reaction outcome in both reaction mechanisms. In particular, an arginine residue is an additional important parameter in both aldolase classes, participating in the stabilization and orientation of the intermediates through its guanidine group [1,2,3]. This feature made me think that amino guanidines, and in particular, amino acid derived acylguanidines, could become convenient organocatalysts for the asymmetric aldol reaction, exploiting the enamine formation ability of class I aldolases and the intermediates stabilization ability provided by the arginine residue in some class I and II aldolases (Figure 1A,B).
Chiral guanidines have been used successfully for some time as asymmetric organocatalysts [4,5]. Indeed, during our ongoing program on supramolecular and bioinspired organocatalysts [6,7,8], we already developed an N-alkyl acylguanidine able to operate in water under emulsion conditions. Remarkable rate acceleration and stereoselectivity were obtained for the aldol reaction of cyclohexanone and aromatic aldehydes [9].
In this communication, a novel family of bioinspired organocatalysts is presented [10] (Figure 1C) based on the hydrogen bonding features of the acylguanidinium group and its application in the asymmetric aldol reaction of hydroxyacetone, a typical substrate for class I aldolases and thoroughly studied in bioinspired catalysis and organocatalysis. Selected examples are given in references [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29].
2. Results and Discussion
Having in mind that the new catalysts were intended to work under homogeneous conditions, a series of N-Boc-amino acids were derivatized to acylguanidines 3a–e by coupling with N-Boc guanidine and subsequent deprotection with trifluoroacetic acid (Scheme 1). Yields in the range 66–86% were obtained for the coupling reaction after purification by flash chromatography, whereas quantitative yields were found in the deprotection step. Details are given in the Supporting Information.
It is worth mentioning that final catalysts 3a–e were obtained as trifluoroacetate salts. In particular, 2.2–2.4 equivalents of TFA were present in respect to the acylguanidine, as determined by 1H/19F NMR using 2,2,2-trifluoroethanol as internal standard. This result indicates that 3a–e exist as dications, and that both the amine and the guanidine group are protonated. Indeed, 1H-NMR spectra of 3a–e in CD3CN show two wide singlets downfield (typically between 7.5–9.5 ppm) integrating 2 H each that are assigned to the protonated N–C(NH2)=NH2+ guanidinium group. On the other hand, the amide N-H and the protonated amino group are difficult to detect as they appear together as very broad signals of low intensity. This behavior indicates fast exchange at the NMR time scale, and therefore higher acidity than the guanidium group.
This set of organocatalysts was subsequently tested in the asymmetric aldol reaction of hydroxyacetone and p-nitrobenzaldehyde. Initial screening conditions involved the use of THF as solvent and 10 mol% catalyst loading at 0 °C (Table 1). The Phe derivative 3a yielded the highest conversion towards product (53% conv.) with moderate enantioselectivities for the two aldol diastereomers (entry 1). The Val and Ala derivatives 3b and 3c gave lower conversion but higher enantioselectivity for the aldol reaction: up to 72% ee was obtained for the syn aldol with 3b and 68% ee with 3c (entries 3 and 5). In turn, the Thr organocatalyst 3d provided the lowest conversion, just 17%, but the highest ee (82% ee) for the syn diastereomer (entry 7). It must be noted that the syn diastereomer predominates in the reactions catalyzed by these four organocatalysts, although in a moderate 2 to 1 ratio, approximately. Finally, the Pro acylguanidine 3e catalyst provided a 31% conversion and 62% ee for the anti aldol product, which predominated in this case at a 2.5 to 1 ratio over the syn diastereomer (Entry 9) (Table 1).
The reaction was repeated by letting them warm up slowly from 0 °C to rt. Yields effectively increased this time in all cases (Entries 2, 4, 6, 8, and 10, Table 1), but stereoselectivity did not significantly improve except for two cases: for catalyst 3d, dr increased from 1/2.5 to 1/3.8 (anti/syn) although ee stayed the same (Entry 8). For catalyst 3e, dr increased a little bit, as well as the ee (Entry 10, Table 1). Similar reaction conditions but adding NaHCO3 to obtain the free amino groups of catalysts 3a–e at rt only produced deleterious results (See Supplementary).
Given the important solvent effects commonly observed in organocatalysis, solvent was optimized next. The Thr derivative 3d, which gave the highest stereoselectivity before, was used. We expected to increase ee to >90% ee along a significant rate acceleration due to the solvent (Table 2).
Unfortunately, with these results in hand, it was clear that the expected concomitant rate acceleration and stereoselectivity increase was not achieved. However, excellent ee and dr for the syn diastereomer was found with DMF (Entry 2, Table 2), only approached by results obtained in ethanol.
In the next step, the concentration was optimized. We have showed recently how dilution was an important parameter to reach actually higher reaction rates and enantioselectivity in organocatayzed Michael additions [30,31]. In the current case, results are summarized in Table 3. Reactions were carried out at rt for 24 h, using this time a 20 mol% catalyst loading to further favor reaction acceleration.
In terms of stereoselectivity, best results were obtained under diluted conditions (Entry 1, Table 3), which allowed the aldol reaction to proceed with an anti/syn dr of 1/4.4 and 91% ee. Upon concentration, stereoselectivity slowly eroded even though conversion increased a bit (Compare entries 1 to 3). Finally, under neat conditions, wherein hydroxyacetone in excess acted as the only solvent, conversion did not improve and stereoselectivity was especially poorer (Entry 4). This drawback seems to be due to the absence of DMF. As I have shown in Table 2, the solvent plays a fundamental role in stereoselectivity.
Therefore, to achieve practical conditions with DMF, catalyst loading of 3d was increased to 20 mol% at the same time that the reaction was carried out entirely at rt while increasing reaction time to 2 days. In this way, high conversion and stereoselectivity were eventually observed (Table 4, Entry 1). It must be noted that results at 0 °C implied only a slight increase in ee, whereas a sharp fall in conversion was observed (Entry 2).
Other aldehydes with electron withdrawing groups were tested as well: the 2,4-dichloro derivative showed excellent conversion (99%), diastereoselectivity (anti/syn 1/12) and enantiomeric excess (94% ee), whereas o-fluorobenzaldehyde gave lower yield (64% conversion) but still excellent enanioselectivity (97% ee for the syn diastereomer, See Entries 3 and 4, Table 4). The less electron withdrawing m-chlorobenzaldehyde, however, yielded lower conversion after three days of reaction, although still with good levels of stereoselectivity (58% conversion and 89% ee, entry 5, Table 4). In turn, benzaldehyde, which is even less reactive, gave a sluggish reaction with only 18% conversion after 4 days and poorer ee (86% ee) (Entry 6). This trend of reactivity and enantioselectivity was already noted by us for hydrophobic acylguanidines operating in aqueous medium [9].
Since the Pro derivative 3e yielded the anti aldol diastereomer, it was tested again under the new conditions (20 mol%, DMF at rt for 2 days). Excellent yield but moderate stereoselectivity were observed, in any case lower than that of the threonine derivative 3d (Entry 7, Table 4).
To assess the functionality of the acylguanidine group, the benchmark asymmetric aldol reaction was carried out using the parent free amino acids as catalysts under identical reaction conditions. L-proline yielded lower conversion and regioselectivity than 3e (76% conversion taking into account 18% of the iso regioisomer vs. 95% conv.) but higher enantiomeric excess (92% vs. 77% ee, entries 7 and 8). In contrast, reaction with L-threonine was sluggish, with just 11% conversion after 2 days compared to 94% conversion for 3d. Moreover, enantioselectivity was also much poorer for l-Thr, affording 69% ee as compared to 91% ee for 3d (Entries 1 and 9, Table 4). Therefore, the acylguanidinium group provides a convenient enhancement of the catalytic characteristics of threonine, although this is not so clear for proline, at least in terms of ee.
Finally, a few words on the absolute configuration of the aldol products: the absolute configuration of the major syn diastereomer obtained with catalyst 3d was determined by comparison of its HPLC trace with that of the product arising from asymmetric dihydroxylation of (E)-4-(4-nitrophenyl)but-3-en-2-one using Sharpless’ AD mix α [25,32]. It corresponds to (3R,4S), as depicted throughout this paper. It must be noted that the anti diastereomer has opposite configuration depending on whether the catalyst is 3a–e or 3d. The anti configuration shown throughout this paper corresponds to the major enantiomer of the anti diastereomer obtained with catalysts 3a–e. Therefore, the major compound obtained with proline derivative 3d corresponds to the (3S,4S) isomer (See Figure 2 bottom). The HPLC traces and the assignment of configuration are shown in the Supplementary Materials section.
Indeed, the absolute configuration of the predominant products match those obtained through established models for these reactions [23,25]. In Figure 2, the molecular mechanics models for reactions catalyzed by 3d and 3e (not transition states) are shown. Interestingly, a stabilizing hydrogen bonding network amongst the acylguanidinium moiety, the enamine N and the aldehyde carbonyl O seems to be an important feature of these catalysts [33]. However, the exact stabilizing effect is not known, since it would require high level computational studies out of the scope of this communication.
3. Conclusions
In conclusion, a novel family of amino acid derived acylguanidines for the asymmetric aldol reaction of hydroxyacetone have been synthesized, characterized, and optimized. These compounds were isolated as diprotonated species (ammonium-guanididium) and the threonine derivative 3d afforded high levels of stereoselectivity for the syn aldol diasteromer, surpassing the characteristics of free threonine. In contrast, the proline derivative 3e afforded the anti diastereomer, but with less practical results. The operation of these catalysts was inspired by the mode of action of some natural aldolases wherein arginine residues participate in the stabilization of intermediates through hydrogen bonding. Actually, simple molecular mechanics models do suggest the presence of a widespread hydrogen bonding network amongst the acylguanidinium moiety and the reaction intermediates that would be responsible for the observed reactivity and stereoselectivity. Further applications of this class of organocatalysts are under investigation and will be reported at due course.
Supplementary Materials
The following are available online, experimental procedures and characterization data, along with 1H and 13C NMR spectra of catalysts 3a–e. Chiral HPLC traces.
Funding
Financial support from the Spanish Ministry of Science and Innovation/Spanish Research Agency (Project RTI2018-096182-B-I00) and Generalitat de Catalunya/AGAUR (Project 2017 SGR 208) is gratefully acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References
- Huang, G.-T.; Yu, J.-S.K. Catalytic roles of histidine and arginine in pyruvate class II aldolase: A perspective from QM/MM metadynamics. ACS Catal. 2017, 7, 8130–8133. [Google Scholar] [CrossRef]
- Bolt, A.; Berry, A.; Nelson, A. Directed evolution of aldolases for exploitation in synthetic organic chemistry. Arch. Biochem. Biophys. 2008, 474, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Coincon, M.; Wang, W.; Sygusch, J.; Seah, S.Y.K. Crystal Structure of Reaction Intermediates in Pyruvate Class II Aldolase. J. Biol. Chem. 2012, 287, 36208–36221. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Feng, X.; Liu, X. Chiral guanidines and their derivatives in asymmetric synthesis. Chem. Soc. Rev. 2018, 47, 8525–8540. [Google Scholar] [CrossRef]
- Selig, P. Guanidines as Reagents and Catalysts, I. (Top. Heterocycl. Chem. 50); Selig, P., Ed.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Serra-Pont, A.; Alfonso, I.; Jimeno, C.; Solà, J. Dynamic assembly of a zinc-templated bifunctional organocatalyst in the presence of water for the asymmetric aldol reaction. Chem. Commun. 2015, 51, 17386–17389. [Google Scholar] [CrossRef] [Green Version]
- Serra-Pont, A.; Alfonso, I.; Solà, J.; Jimeno, C. An efficient dynamic asymmetric catalytic system within a zinc-templated network. Chem. Commun. 2019, 55, 7970–7973. [Google Scholar] [CrossRef] [Green Version]
- Serra-Pont, A.; Alfonso, I.; Solà, J.; Jimeno, C. A copper-templated, bifunctional organocatalyst: A strongly cooperative dynamic system for the aldol reaction. Org. Biomol. Chem. 2017, 15, 6584–6591. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso, A.M.; Catot, A.; Alfonso, I.; Jimeno, C. Intramolecular hydrogen bonding guides a cationic amphiphilic organocatalyst to highly stereoselective aldol reactions in water. RSC Adv. 2015, 5, 62331–62335. [Google Scholar] [CrossRef]
- Itoh, S.; Sonoike, S.; Kitamura, M.; Aoki, S. Design and synthesis of chiral Zn2+ complexes mimicking natural aldolases for catalytic C-C bond forming reactions in aqueous solution. Int. J. Mol. Sci. 2014, 15, 2087–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimchuk, V.V.; Kucherenko, A.S.; Fakhrutdinov, A.N.; Medvedev, M.G.; Nelyubina, Y.V.; Zlotin, S.G. Towards Sustainable Amino Acid Derived Organocatalysts for Asymmetric syn-Aldol Reactions. Eur. J. Org. Chem. 2017, 17, 2540–2544. [Google Scholar] [CrossRef]
- Gerasimchuk, V.V.; Romanov, R.R.; Woo, G.H.T.; Dmitriev, I.A.; Kucherenko, A.S.; Zlotin, S.G. Novel L-threonine-based ionic liquid supported organocatalyst for asymmetric syn-aldol reactions: Activity and recyclability design. Arkivoc 2017, 3, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Henseler, A.H.; Ayats, C.; Pericàs, M.A. An enantioselective recyclable polystyrene-supported threonine-derived organocatalyst for aldol reactions. Adv. Synth. Catal. 2014, 356, 1795–1802. [Google Scholar] [CrossRef]
- Li, S.; Wu, C.; Fu, X.; Miao, Q. Cysteine-Based Organocatalysts for the Highly Efficient Direct Stoichiometric anti- and syn-Aldol Reactions. Ind. Eng. Chem. Res. 2011, 50, 13711–13716. [Google Scholar] [CrossRef]
- Wu, C.; Fu, X.; Li, S. A highly efficient, large-scale, asymmetric direct aldol reaction employing simple threonine derivatives as recoverable organocatalysts in the presence of water. Eur. J. Org. Chem. 2011, 1291–1299. [Google Scholar] [CrossRef]
- Wu, C.; Fu, X.; Li, S. Simple and inexpensive threonine-based organocatalysts for the highly diastereo- and enantioselective direct large-scale syn-aldol and anti-Mannich reactions of α-hydroxyacetone. Tetrahedron Asymmetry 2011, 22, 1063–1073. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Z.; Ye, Z.; Qian, S.; Zhao, G. Highly efficient organocatalyzed direct asymmetric aldol reactions of hydroxyacetone and aldehydes. Adv. Synth. Catal. 2009, 351, 158–162. [Google Scholar] [CrossRef]
- Chen, X.-H.; Luo, S.-W.; Tang, Z.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. Organocatalyzed highly enantioselective direct aldol reactions of aldehydes with hydroxyacetone and fluoroacetone in aqueous media: The use of water to control regioselectivity. Chem. Eur. J. 2007, 13, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Xu, H.; Zhang, L.; Li, J.; Cheng, J.-P. Highly enantioselective direct syn- and anti-Aldol reactions of dihydroxyacetones catalyzed by chiral primary amine catalysts. Org. Lett. 2008, 10, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chimni, S.S. Organocatalyzed direct asymmetric aldol reaction of isatins in water: Low catalyst loading in command. Tetrahedron 2013, 69, 5197–5204. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, S.; Kumar, V.; Singh Chimni, S. Asymmetric syn-selective direct aldol reaction of protected hydroxyacetone catalyzed by primary amino acid derived bifunctional organocatalyst in the presence of water. Org. Biomol. Chem. 2011, 9, 2731–2742. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, J.; Pasternak, M.; Gut, B.; Gryzlo, B.; Mlynarski, J. Direct Asymmetric Aldol Reactions Inspired by Two Types of Natural Aldolases: Water-Compatible Organocatalysts and ZnII Complexes. J. Org. Chem. 2012, 77, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Popik, O.; Pasternak-Suder, M.; Lesniak, K.; Jawiczuk, M.; Gorecki, M.; Frelek, J.; Mlynarski, J. Amine-Catalyzed Direct Aldol Reactions of Hydroxy- and Dihydroxyacetone: Biomimetic Synthesis of Carbohydrates. J. Org. Chem. 2014, 79, 5728–5739. [Google Scholar] [CrossRef]
- Hoffmann, T.; Zhong, G.; List, B.; Shabat, D.; Anderson, J.; Gramatikova, S.; Lerner, R.A.; Barbas, C.F., III. Aldolase Antibodies of Remarkable Scope. J. Am. Chem. Soc. 1998, 120, 2768–2779. [Google Scholar] [CrossRef]
- Ramasastry, S.S.V.; Zhang, H.; Tanaka, F.; Barbas, C.F., III. Direct Catalytic Asymmetric Synthesis of anti-1,2-Amino Alcohols and syn-1,2-Diols through Organocatalytic anti-Mannich and syn-Aldol Reactions. J. Am. Chem. Soc. 2007, 129, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Guillena, G.; Hita, M.d.C.; Nájera, C. Organocatalyzed direct aldol condensation using L-proline and BINAM-prolinamides: Regioselective, diastereoselective and enantioselective controlled synthesis of 1,2-diols. Tetrahedron Asymmetry 2006, 17, 1027–1031. [Google Scholar] [CrossRef]
- Sarkar, D.; Harman, K.; Ghosh, S.; Headley, A.D. Chiral amine organocatalysts for the syn-aldol reaction involving substituted benzaldehydes and hydroxyacetone. Tetrahedron Asymmetry 2011, 22, 1051–1054. [Google Scholar] [CrossRef]
- Czarnecki, P.; Plutecka, A.; Gawronski, J.; Kacprzak, K. Simple and practical direct asymmetric aldol reaction of hydroxyacetone catalyzed by 9-amino Cinchona alkaloid tartrates. Green Chem. 2011, 13, 1280–1287. [Google Scholar] [CrossRef]
- Demuynck, A.L.W.; Vanderleyden, J.; Sels, B.F. Direct asymmetric syn-aldol reactions of linear aliphatic ketones with primary amino acid-derived diamines. Adv. Synth. Catal. 2010, 352, 2421–2426. [Google Scholar] [CrossRef]
- Günler, Z.I.; Alfonso, I.; Jimeno, C.; Pericàs, M.A. Concentration Effect in the Asymmetric Michael Addition of Acetone to β-Nitrostyrenes Catalyzed by Primary Amine Thioureas. Synthesis 2017, 49, 319–325. [Google Scholar]
- Günler, Z.I.; Companyó, X.; Alfonso, I.; Burés, J.; Jimeno, C.; Pericàs, M.A. Deciphering the roles of multiple additives in organocatalyzed Michael additions. Chem. Commun. 2016, 52, 6821–6824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M.; Xu, D.; et al. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Al-Taie, Z.S.; Anetts, S.R.; Christensen, J.; Coles, S.J.; Horton, P.N.; Evans, D.M.; Jones, L.F.; de Kleijne, F.F.J.; Ledbetter, S.M.; Mehdar, Y.T.H.; et al. Proline derived guanidine catalysts forge extensive H-bonded architectures: A solution and solid state study. RSC Adv. 2020, 10, 22397–22416. [Google Scholar] [CrossRef]
Figure 1.
Mechanism of class I fructose bis(phosphate) aldolase (A) and class II pyruvate aldolase (B). Bioinspired amino acid derived acylguanidinium organocatalysts (C).
Figure 1.
Mechanism of class I fructose bis(phosphate) aldolase (A) and class II pyruvate aldolase (B). Bioinspired amino acid derived acylguanidinium organocatalysts (C).

Scheme 1.
Synthesis of amino acid acylguanidines 3a–e.

Figure 2.
MMFF models for the asymmetric addition of hydroxyacetone to p-nitrobenzandehyde catalyzed by 3d (top) and 3e (bottom). Hydrogen bonding showed as red dotted lines.
Figure 2.
MMFF models for the asymmetric addition of hydroxyacetone to p-nitrobenzandehyde catalyzed by 3d (top) and 3e (bottom). Hydrogen bonding showed as red dotted lines.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Screening of acylguanidines 3a–e in the asymmetric aldol reaction.
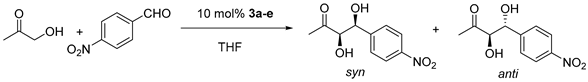
| Entry. | Catalyst | Conversion/% c | d.r. anti/syn c | ee anti/% d | ee syn/% d |
|---|---|---|---|---|---|
| 1 | 3a | 53 a | 1/1.9 a | 50 a | 55 a |
| 2 | 70 b | 1/2 b | 37 b | 40 b | |
| 3 | 3b | 27 a | 1/1.4 a | 59 a | 72 a |
| 4 | 43 b | 1/2 b | 41 b | 68 b | |
| 5 | 3c | 25 a | 1/2.3 a | 43 a | 60 a |
| 6 | 44 b | 1/2.3 b | 45 b | 48 b | |
| 7 | 3d | 17 a | 1/2.5 a | 52 a | 82 a |
| 8 | 33 b | 1/3.8 b | 51 b | 81 b | |
| 9 | 3e | 31 a | 2.5/1 a | 62 a | 35 a |
| 10 | 46 b | 2.7/1 b | 69 b | 41 b |
a Performed at 0 °C for 24 h. b Performed from 0 °C to rt, for 24 h. c Determined by 1H NMR. d Determined by HPLC on a chiral stationary phase (ID column).
Table 2.
Effect of solvent in the asymmetric aldol reaction catalyzed by 3d.

| Entry | Solvent | Conversion/% c | d.r. anti/syn c | ee syn/% d |
|---|---|---|---|---|
| 1 | THF | 33 | 1/3.8 | 81 |
| 2 | DMF | 19 | 1/5.0 | 92 |
| 3 | CH2Cl2 a | 3 | 1/1.4 | 57 |
| 4 | CH3CN | 41 | 1/2.3 | 80 |
| 5 | AcOEt b | 46 | 1/2.0 | 67 |
| 6 | iPrOH b | 59 | 1/2.0 | 79 |
| 7 | EtOH | 23 | 1/2.8 | 89 |
| 8 | water | N. R. | - | - |
a 15 mol% catalyst loading. b 20 mol% catalyst loading. c Determined by 1H NMR. d Determined by HPLC on a chiral stationary phase (ID column). N. R. No reaction.
Table 3.
Effect of concentration in the aldol reaction catalyzed by 3d.
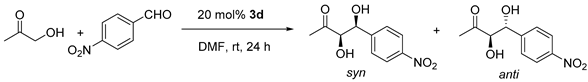
| Entry | Conc./M a | Conv./% c | dr anti/syn c | ee syn/% d |
|---|---|---|---|---|
| 1 | 0.18 | 75 | 1/4.4 | 91 |
| 2 | 0.31 | 75 | 1/4.1 | 90 |
| 3 | 0.46 | 81 | 1/3.7 | 88 |
| 4 | 1.43 b | 71 | 1/1.6 | 57 |
ap-Nitrobenzaldehyde concentration. b Neat (No DMF). c Determined by 1H NMR. d Determined by HPLC on a chiral stationary phase (ID column).
Table 4.
Optimized conditions for the asymmetric aldol reaction.
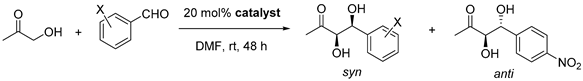
| Entry | Catalyst | Aldehyde | Conversion/% d | d.r. anti/syn d | ee Major/% e |
|---|---|---|---|---|---|
| 1 | 3d |  | 94 | 1/4.5 | 91 |
| 2 | 26 a | 1/4.4 a | 93 a | ||
| 3 | 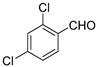 | 99 | 1/12 | 94 | |
| 4 | 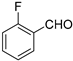 | 64 | 1/4.2 | 97 | |
| 5 b |  | 58 b | 1/4.5 b | 89 b | |
| 6 c |  | 18 c | 1/3 c | 86 c | |
| 7 | 3e | 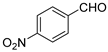 | 95 | 3.7/1 | 77 |
| 8 | L-Pro | 76 f | 3.1/1 | 92 | |
| 9 | L-Thr | 11 | 1/3.7 | 69 |
a Performed at 0 °C. b 3 days reaction. c 4 days reaction. d Determined by 1H NMR. e Determined by HPLC on a chiral stationary phase. f Comprises 18% of the iso regioisomer.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jimeno, C. Amino Acylguanidines as Bioinspired Catalysts for the Asymmetric Aldol Reaction. Molecules 2021, 26, 826. https://doi.org/10.3390/molecules26040826
AMA Style
Jimeno C. Amino Acylguanidines as Bioinspired Catalysts for the Asymmetric Aldol Reaction. Molecules. 2021; 26(4):826. https://doi.org/10.3390/molecules26040826
Chicago/Turabian StyleJimeno, Ciril. 2021. "Amino Acylguanidines as Bioinspired Catalysts for the Asymmetric Aldol Reaction" Molecules 26, no. 4: 826. https://doi.org/10.3390/molecules26040826