
Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications
by
 ,
,
Ricardo Cerón-Camacho
1 ,
,
Manuel A. Roque-Ramires
2,
Alexander D. Ryabov
3 and
Ronan Le Lagadec
2,* 1
CONACyT-Instituto Mexicano del Petróleo, Eje Central Lázaro Cárdenas 152, Col. San Bartolo Atepehuacán, México D.F. 07730, Mexico
2
Instituto de Química, UNAM, Circuito Exterior s/n, Ciudad Universitaria, Coyoacán, Ciudad de México 04510, Mexico
3
Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(6), 1563; https://doi.org/10.3390/molecules26061563
Submission received: 18 February 2021
/
Revised: 8 March 2021
/
Accepted: 9 March 2021
/
Published: 12 March 2021
(This article belongs to the Special Issue Influencers in Organometallic Chemistry—A Themed Issue Honoring Dr. Michel Pfeffer)
Abstract
:The synthesis of cyclometalated osmium complexes is usually more complicated than of other transition metals such as Ni, Pd, Pt, Rh, where cyclometalation reactions readily occur via direct activation of C–H bonds. It differs also from their ruthenium analogs. Cyclometalation for osmium usually occurs under more severe conditions, in polar solvents, using specific precursors, stronger acids, or bases. Such requirements expand reaction mechanisms to electrophilic activation, transmetalation, and oxidative addition, often involving C–H bond activations. Osmacycles exhibit specific applications in homogeneous catalysis, photophysics, bioelectrocatalysis and are studied as anticancer agents. This review describes major synthetic pathways to osmacycles and related compounds and discusses their practical applications.
1. Introduction
Osmium is the heaviest and rarest stable element in the earth’s crust, with a concentration of approximately 0.05 part per billion. Perhaps the latter accounts for why the coordination and organometallic chemistry of osmium are less advanced than that of other transition metals. Common precursors, such as OsCl3 or M2[OsCl6], are significantly more expensive than their respective ruthenium precursors. Osmium is often associated with highly toxic OsO4, widely used as an oxidizing agent in organic chemistry and as a staining agent in transmission and scanning electron microscopies [1,2,3]. Fortunately, osmium chemistry is not limited to its oxides, and numerous coordination, organometallic, and cluster compounds have been prepared, though their chemistry is not as deeply explored as that of ruthenium [4]. The osmium chemistry was not extensively reviewed in the literature. The first review was written by Griffith in 1987 [5]. Some osmium compounds were also compiled in Gmelin Handbuch der Anorganischen Chemie in the 1980s [6]. In the 2011 edition of the Encyclopedia of Inorganic Chemistry, two articles were devoted to coordination and organometallic chemistry of osmium [7,8]. To our knowledge, the chemistry of cyclometalated osmium species was not specifically reviewed, in contrast with the extended coverage of the chemistry of other metalacycles in the last several decades [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23].
This review attempts to fill the existing gap in covering the cyclometalation chemistry of osmium compounds as they are presented in Figure 1 with M = Os. The metal is usually σ-bonded to a carbon atom and coordinated to E, where E is generally N, P, O, S, As, Se. Undoubtedly, the impact of Michel Pfeffer’s work on the development of cyclometalation reactions was fundamental, and many of his publications, first on palladium then on ruthenium metalacycles, set the path for countless publications [24,25,26,27].
2. Historical Background
Ruthenium and osmium belong to group 8 metals, and they frequently form similar complexes. The oxidation states vary from 0 to VIII, though more common are oxidation states between II to IV [4]. Osmium forms stronger metal-metal and metal-carbon bonds, and hence osmium complexes are generally less active catalysts than analogous ruthenium species. Many osmium complexes are made via different or more difficult routes than their ruthenium analogs. Publications on osmacycles started to grow after 2000, prior to which they were nearly nonexistent. Their number is considerably lower than the number of reports on ruthenacycles (Figure 2).
The first osmacycle prepared by Bruce in 1973 was made from [Os3(CO)12] and benzo[h]quinoline. A mixture of mononuclear isomers 1 was obtained in low yields after 230 h at reflux in light petroleum [28]. In 1981, Jameson described cyclometalated formazan derivatives of osmium 2 [29]. Desrosiers prepared a hydride osmium–phosphine complex 3 by intramolecular C−H activation in 1986 [30]. Cyclometalated piano-stool derivative of benzoic acid 4 was reported by Kisenyl in 1987 [31]. Later, mononuclear and dinuclear pincer osmium complexes 5, 6, dinuclear Ru-Os cyclometalated derivative 7 [32,33], cationic mononuclear osma(II)cycle with PPh3 8 [34], and osmacycle 9 from polyquinoline [35] were obtained. Structures of compound 5–11 are shown in Table 1.
3. Main Precursors
3.1. [OsX2(PR3)3]
Complexes [OsX2(PR3)3], a family of 16 electron osmium(II) precursors, are widely used for the synthesis of osmacycles. Among them, [OsBr2(PPh3)3] is particularly valuable.
3.1.1. mono-Osmacycles
Chakravorty synthesized phenolate osmium complex 11 from 2,6-diformyl-4-methylphenol and [OsBr2(PPh3)3] [36]. The authors proposed the oxidative addition of the aldehyde, followed by decarbonylation, to form an unstable OsIV intermediate. Subsequent reductive elimination of HX yields the cyclometalated complex [36]. Related [OsBr(C~O)(PPh3)2(CO)] complexes 12 were similarly prepared, and the substitution of bromide generated cationic species 13 (Figure 3) [37].
The mechanism of cyclometalation of 2-vinylpyridine by [MCl2(PPh3)3] (M = Ru, Os) was explored theoretically for the ruthenium case [38]. The lower reactivity of osmium allowed to isolate several intermediates proposed. The reaction of [OsCl2(PPh3)3] with an excess of 2-vinylpyridine in the presence of Cs2CO3 and NaBF4 results in cyclometalation of the ligand with liberation of PPh3 to produce 14. The cationic complex 15 is formed via substitution of the chloride by a second equivalent of 2-vinylpyridine in presence of NaBF4. In the absence of NaBF4, the neutral cyclometalated complex 14 generates isomers 16 and 17 (Figure 4) [38].
3.1.2. Pincer and bis-Cyclometalated Complexes
[OsBr2(PPh3)3] reacts with 2-(2′,6′-dimethylphenylazo)-4-methylphenol in refluxing 2-methoxiethanol in the presence of NEt3 to produce osmium(III) complex 24. The authors consider a coordination of the ligand to osmium(II) in a tridentate C~N~O fashion, followed by the oxidation to osmium(III) in 18 (Figure 5). Thereafter, the loss of HX affords a carbene species 19. In the presence of water, the carbene intermediate could transform into the OsH-C=O fragment (22) after elimination of H2 in intermediate 21. A posterior reductive elimination of a half equivalent of hydrogen from 22, followed by the migration of a CO fragment (23), would produce the final cyclometalated complex [Os(C~N~O)(CO)] (24) [39].
Wen and co-workers prepared cyclometalated [Os(P~C~P)Cl(PPh3)] complexes 25 and 26 (Figure 6) [40]. They react with H2 to afford dihydrido species. The corresponding carbonyl derivatives can also be prepared in the presence of CO [41].
The diphosphine {1,3,5-(CH3)3-2,6-(iPr2-PCH2)2C6H} was also reacted with [OsCl2(PPh3)3]. The metal promotes the cleavage of one C−CH3 bond with liberation of methane (Figure 7). In the presence of H2, dihydride heptacoordinated complex 27 is formed, while [OsH(Cl)(PPh3)3] generates a pentacoordinated complex 28 [41].
The N,N’-bis(phosphinomethyl)dihydropermidine-type ligands H2C(NCH2PR2)2C10H6 (R = Cy, Ph) react with [OsCl2(PPh3)3] yielding the P~C~P pincer complexes 29 and 30. The latter may undergo the second C−H activation to form the corresponding carbenes. Figure 8 shows the proposed mechanism for the double C−H activation [42].
Other examples involving C−H activation are shown in Figure 9. Majumder et al. synthesized osmacycles 36 and 37 from 2-(arylazo)phenols and [OsBr2(PPh3)3] [43], and Das found an unusual cyclometalation of N-arylbenzohydroxamic acids leading to 38 [44].
Bis-pincer complex 39 was obtained from [OsCl2(PPh3)3] and diphosphine 2,6-(CH2PPh2)2C6H3 at a ligand-to-complex ratio of 3:1 (Route 1 in Figure 10). On the other hand, the ratio of 1:1 provides mono-pincer complex 25. An exchange of chloride by triflate affords 40 and a subsequent reaction with the second equivalent of the ligand leads to bis-pincer complex 39 (Route 2 in Figure 10) [45].
3.1.3. Others Osmacycles
Gong studied the interaction between HC≡CCH(OH)CH=CH2 and [OsCl2(PPh3)3] in THF leading to η2-allyl alcohol osmacycle complexes, which could further be converted to osmabenzene, cyclic osmium η2-allene, osmafuran and α,β-unsaturated ketone complexes in a one-pot reaction [46] (Figure 11). It is worth mentioning that the “donor center” in such osmacycles is a C=C double bond, narrowly fitting into the definition of a metalacycle. Nevertheless, the authors classify the complexes as osmacycles. The cyclometalation takes place through a nucleophilic attack of PPh3 at the coordinated alkyne [47]. This method results in efficient preparations of conjugated osmacycles in high yields and allows the synthesis of complexes containing a phosphonium moiety incorporated in the metalacycle. Osmabenzenes were prepared in a similar way [47]. The η2-alkyne-coordinated alcohol complex 41 can be transformed under acidic conditions to a η2-coordinated α,β-unsaturated ketone 42. Additionally, 41 reacted with triphenylphosphine to give 43 in presence of Bu4NX (X = Cl, Br) with elimination of OH-. The efficient cyclometalation to give 45 with η2-coordinated allyl alcohol was also performed. Upon thermal treatment, 45 converts to a mixture of four conjugated osmacycles (42, 46–48) [48]. Osmafuran 49 was prepared similarly [48] (Figure 11).
3.2. [OsX2(CO)(PR3)3]
Related precursors [OsX2(CO)(PR3)2] were also explored. The oxidative addition of the ortho C−H bond of the phenyl group of imine Ph2C=NH to [Os(Cl)(Ph)(PiPr3)2(CO)] followed by the reductive elimination of benzene and the coordination of the iminic nitrogen generates osmacycle 50 (Figure 12). The process could be viewed as an unusual σ-bond metathesis [49].
Clark et al. studied by X-ray crystallography four cyclometalated osmium complexes (51–55) obtained via the transmetalation reaction between hydride precursors [OsH(Cl)(CO)(PPh3)3] and organomercurial derivatives of 2-phenylpyridine. A series of related complexes (56–58) in which the phenylpyridine ligand was modified by electrophilic substitutions was also reported (Figure 13) [50].
Bennett et al. also showed that the reaction between [Hg(o-C6H4PPh2)2] and [MH(Cl)(CO)(PPh3)3] (M = Ru, Os) form the corresponding four-membered metalacycles 59, elemental mercury and free triphenylphosphine (Figure 14) [51]. It was suggested that the reaction occurs through the oxidative addition of Hg−Ar to a coordinatively unsaturated intermediate generated by the dissociation of PPh3 and the posterior reductive elimination of ArH and Hg [52].
3.3. Osmium Hydride Complexes
Esteruelas and coworkers have extensively been working on C−H bond activations mediated by transition metals. Their results represent an important step in the development of new synthetic routes through C−H bond activations by osmium. Notably, osmium hydrides complexes favor high oxidation states (4 and 6) and display a wider range of stoichiometries and structures than ruthenium derivatives. Osmium hydride complexes can also exhibit lability of the hydride ligands under thermal conditions [53]. Particularly, the hexahydride osmium(VI) precursor, [OsH6(PiPr3)2], was widely used to prepare a variety of osmacycles. For example, this hexahydride osmium complex is capable of generating a tetrahydride [OsIVH4(PiPr3)2] species and one hydrogen molecule by thermal activation [53]. The tetrahydride complex can then coordinate the oxygen atom of the carbonyl group of an aromatic ketone and subsequently activates C−H or C−F bonds for the corresponding cyclometalation step [54].
3.3.1. [OsH6(PR3)2]
Studies by NMR revealed that [OsH6(PiPr3)2] reacts with cycloalkyl or phenyl methyl ketone to form cyclometalated compounds 60, 61 and 62 (Table 2) [49,50,54,55]. The reactivity of 61 and related complexes will be discussed in Section 4.2. The reaction of [OsH6(PiPr3)2] with 2-vinylpiridine affords 63, and a secondary C−H bond activation generates complex 64 [56]. The C(sp3)−H activation occurs when [OsH6(PiPr3)2] reacts with 8-methylquinoline. Activation of the methyl group and elimination of two H2 molecules gives 65. Experiments with methyl deuterated 8-methylquinoline and DFT calculations confirmed the mechanism [57].
The same hexahydride osmium precursor in the presence of 0.5 eq of 2,6-bis{1-[(4-methylphenyl)imino]ethyl}pyridine is involved in a double C−H activation of the pyridine ring producing a dimetalatricyclic system 66 (Table 2). The product was characterized by an X-ray study and the presence of three fused rings was confirmed [58]. Other bimetallic osmacycles were prepared from [OsH6(PiPr3)2] by activation of the ortho C−H bond of N,N’-di- and tetraphenyl bipyridines. Starting from 2-phenylpyridine, compound 67 was first obtained (Table 2). Using 2,2′-diphenyl-4,4′-bipyridine in 1:3 and 3:1 ratios with respect to osmium allowed to isolate mononuclear 68 and dinuclear 69 osmacycles, respectively (Table 2). The corresponding phenyl and anthracenyl derivatives of bipyridine also form dinuclear osmacycles (70, 71) [59]. Complex [OsH6(PiPr3)2] and 2-azetidinones substituted at 4-position by an N-heterocycle and in 3-position by a phenoxy group afford corresponding cyclometalated complexes (72, 73). This reaction is associated with the metal-mediated degradation of the ligand which involves two C−H bond activations and a parallel C−N and C−C rupture within the four-membered rings [60,61] (Table 2). Double cyclometalation takes place when [OsH6(PiPr3)2] reacts with 2,6-diphenylpyridine to give tridentate C~N~C pincer compound 74. In the presence of HBF4 the latter can suffer the addition of one proton at the metalated carbon to form the cationic mono-cyclometalated complex, which eliminates 2,6-diphenylpyridine in the presence of acetonitrile to generate the corresponding coordination derivative. When the ligand is 2-phenoxy-6-phenylpyridine, a double C−H activation also occurs. Five- and six-membered rings 75 are produced (Table 2) [62]. Other reactions with [OsH6(PiPr3)2] involve the cyclometalation of azole ligands by selective C−H bond activation which can discern the coordination of N over “N−H side” in the ligand (76, 77). Such behavior is also observed with 2-phenylthiazole and 2-phenylbenzothiazole, where the cyclometalation occurs at the “C−N side” [63]. The reaction of the hexahydride precursor with benzonitrile in toluene promotes the C−H activation and the reduction of the triple C≡N bond forming the corresponding imine. The resulting complex 78 is produced in a 75% yield. The substitution of PiPr3 by PPh3 in such complexes was also studied [64]. Complex [OsH6(PiPr3)2] can also activate directly the 8-position in 2-, 3-, 6- and 7-methylquinolines to obtain corresponding [OsH3{κ2-C8,N-(quinilinyl)}(PiPr3)2] and [OsH3{κ2-C8,N-(quinilinyl-n-Me)}(PiPr3)2] (79, n = 2, 3, 6, 7) complexes. It is interesting to note that a four-membered cyclometalated fragment is formed in all cases (Table 2) [65]. Complex [OsH6(PiPr3)2] reacts with 2-vinylpyridine to form an osmacycle 80 where C(sp2)−H activation of the vinyl is observed (Table 2) [56].
A curious reductive cyclometalation occurs when the dihydride 16e OsIV complex [OsH2Cl2(PiPr3)2] reacts with an excess of allylamine. Cyclometalated OsII species 81 is formed alongside with the [HPiPr3]Cl salt (Figure 15). The final product is coordinated by two molecules of allyl amine. One of them is cyclometalated through the insertion of the alkene fragment into the Os−H bond. The mechanism proposed by the authors suggests a C−H activation to justify the [HPiPr3]Cl formation. This cyclometalated complex 81 is the starting material to form a variety of other complexes by either ligand metathesis or CO and allene insertions [66].
3.3.2. bis- and tris-Osmacycles
Several osmacycles that contain two osmium–carbon σ-bonds (bis-cyclometalated), as well as some rare examples with three osmium–carbon σ-bonds (tris-cyclometalated) were prepared from [OsH6(PiPr3)2]. The first examples were classified by Esteruelas et al. as “multiple C−H bond activations”. Complex [OsH6(PiPr3)2] reacts with phenyl-substituted pyrimidines or triazines to form the corresponding [Os(C~N~C)] pincer derivatives. The reaction with phenyl pyrimidines leads to mono-cyclometalated complexes along with the pincer species. The phenyl triazine ligand forms pincer complex 85 but can also generate a dinuclear bis-cyclometalated osmium complex 86 (Figure 17) [68].
More recently, Esteruelas et al. described tris-cyclometalated osmium complex 87 produced from 3 eq of N,N’-diphenylbenzimidazolium chloride [Ph2BImH]Cl and [OsH6(PiPr3)2] in the presence of a base. In acidic medium (HBF4), one cyclometalated phenyl group of 87 is protonated and a reductive elimination of hydride with another phenyl group forms a cationic octaedral complex 88 stabilized by two agostic interactions between osmium and C−H bonds (Figure 18) [69]. The replacement of chloride by BF4- in the N,N’-diphenylbenzimidazolium salt promoted the tridentate coordination to form 89. Monocyclometalated complex 90 was made using of one equivalent of [Ph2BImH]Cl. However, complex 87 could be obtained through the subsequent reaction with another equivalent of the ligand [69].
3.4. [Os(η6-arene)Cl2]2
The [Os(η6-arene)Cl2]2 dimers usually display similar reactivity as their widely used ruthenium analogues. The bridging chloride bonds are readily broken in polar solvents or in the presence of coordinating ligands. Monomers formed were used to prepare a variety of osmacycles [70].
3.4.1. mono-Osmacycles
In 2003, piano-stool neutral 92 and cationic 93 osmacycles were prepared from the dimeric [Os(η6-C6H6)(μ-Cl)Cl]2 precursor (Figure 19) [71]. We are particularly proud that the first experiments were carried out in the lab of Michel Pfeffer in Strasbourg. Needless to say that background for this work was previously created by Michel through the thorough investigation of the chemistry of matching ruthenium complexes [72,73,74,75]. Coordinated benzene and acetonitrile in 93 are easily substituted by 2,2′-bipyridines or 1,10-phenantroline to form octahedral cationic complexes (94, 95, 96) [71]. Complex 92 can also be synthetized through a transmetalation reaction with organomercurial derivatives and the [Os(η6-C6H6)(μ-Cl)Cl]2 dimer [71]. Similarly, series of complexes bearing cyclometalated 2-phenylpyridine and N,N-dimethylbenzylamines with different electron-donating groups such as 4-OMe (97b) and 3,5-(OMe)2 (97c) were prepared with the aim to modulate the electron density at the osmium center [76].
Transmetalation between [Os(η6-C6H6)(μ-Cl)Cl]2 and silver(I) compounds is exemplified by the reaction of the 1-phenyl-3-methyl-1H-benzimidazolium [AgPhMeBIm]I complex. Intermediate 98 forms first followed by the metalation of the phenyl group in the presence of Al2O3. Piano-stool mono-cyclometalated complex 99 is the final product (Figure 20) [77]. It takes two days for triazolium silver salts to react with [Os(η6-C6H6)(μ-Cl)Cl]2 to give structurally similar complex 100 after treatment with KPF6 [78]. This methodology was also successfully employed to prepare cyclometalated complexes of iridium and ruthenium [78].
3.4.2. bis- and tris-Osmacycles
A few bis- and tris-cyclometalated osmium complexes have been described [79,80]. Such compounds differ from pincer complexes, as they contain two or three cyclometalated ligands, respectively. Complexes [Os(phpy)2(bpy)]PF6 (102) and [Os(phpy)3] (101) were prepared from organomercurial Hg(phpy)2 and mono-cyclometalated complex 92. Their electrochemical properties were compared with those of the corresponding coordination complex [Os(bpy)3](PF6)2 (103) and mono-cyclometalated complex [Os(phpy)(bpy)2]PF6 (94), (Figure 19). Each Os−C bond decreases the OsII/OsIII reduction potential by ca. 500 mV ranging from 888 mV for coordination compound 103 to −958 mV (vs. Ag/AgCl) for tris-cyclometalated complex 101 [79,80] (Figure 21).
3.5. Other Precursors
Complexes [cation]2[OsX6] and [OsX3(tterpy)] (tterpy = 4′-(4-tolyl)-2,2′:6′,2″-terpyridine) were also used as osmacycle precursors. Complex [Et4N]2[OsCl6] reacts with 1,3-(CH2PtBu2)2C6H4 in the presence of NEt3 affording 16e pincer complexes [OsCl(P~C~P)(CO)] (108) and [OsCl(H2)(P~C~P)] (109) [81]. It is suggested that OsIV is first reduced to OsII by methanol to produce CO. When the osmium precursor and the ligand are heated in 2-propanol, the solvent is not decarbonylated and dihydride complex 109 is obtained (Figure 23) [81].
Complex [Et4N]2[OsCl6] reacts also with 1,5-bis(di-terbutylphosphino)pentane in the presence of NEt3 and H2 in 2-pentanol to give P~C~P alkyl complex 110. A posterior thermal treatment initiates second C−H bond activation via α-elimination to give carbene complex 111 (Figure 24) [82].
4. Representative Reactions of Osmacycles
4.1. Reactivity of Chelate and Pincer Complexes
Ligand substitution in 59a (see also Figure 14) yields neutral mono-osmacycle 124 [51]. On the other hand, ruthenium analogue 59b displayed a different behavior since a parallel insertion of CO into the Ru−C bond in addition to ligand substitution accounts for the formation of 126 (Figure 27). In order to force the CO insertion into the Os−C bond, complex 59a was treated with AgSbF6. However, the cationic species 125, product of the substitution of the chloride ligand, was obtained [51].
Reactions of 2-phenylpyridine osma(II)cycle 51 are summarized in Figure 28 [50]. The cyclometalated ligand is activated by OsII towards electrophilic substitution allowing the introduction of diverse functional groups. Nitration occurs at both the 4- and 6-position of the phenyl ring, whereas bromination takes place at the 4-position only. Complex 51 reacts also with CO2/H+ or Bu3SnCl as shown in Figure 28 [50].
Gusev et al. studied the reactivity of P~C~P pincer compounds (Figure 29) [81]. Complex 108 reacts with NaBH4 and the mixture of two hydrides 128 and 129 is obtained. A chloride for iodide exchange takes place in 109 in the presence of MeI. The reaction of 109 with NaBH4 gives trihydride 131. Complex 132 with two H2 molecules is also obtained in the presence of H2 at −80 °C; at higher temperatures one of the coordinated H2 undergoes oxidative addition to yield complex 133 [81].
Reactions of P~C~P pincer complex 25 with a series of alkynes afford carbene and carbyne osmacycles (Figure 30) [40]. Complex 25 is also convertible to monohydride 141 and trihydride 142 in the presence of NaH (Figure 30). In an excess of phenylacetylene, 142 gives 143 with three incorporated phenylacetylene ligands. Remarkably, the coordination of all three is different including acetylide- and vinylidene-binding motifs. Vinylidene complex 135 reacts with Tl(OAc) to form 144. Chloride is being replaced by acetate which acts as a bidentate ligand [85].
Related P~C~P pincer complex 26 reacts with H2 giving rise to dihydride species 145. Carbonyl derivatives 146 and 147 can also be prepared in the presence of CO (Figure 31) [41].
Dihydride [OsH2(PiPr3)2(C~C′~N)] (73) reacts with HBF4 to yield [OsH2(PiPr3)2(C~C’’~N)]BF4 (148) where the carbon in α position to the central carbon of the pincer ligand was protonated by the acidic medium [60]. Similarly, C~N~C’ pincer complexes 74 and 75 accept one proton at a metalated carbon to form cationic mono-cyclometalated complexes, which further react with nitriles as shown in Figure 32 [62].
4.2. Reactions of Hydride Complexes
Hydrides in osmacycles promote reactions of adjacent ligands. For example, complex 80 reacts with HBF4 to give 63 with regenerated 2-vinylpyridine, the Os-trihydride unit being transformed into the Os-hydride-dihydrogen functionality. Subsequent treatment of 63 at 50 °C in the presence of Ph2CO induces the reduction of the double bond and formation of 64. The elimination of benzophenone in acetonitrile at room temperature gives 153 [56]. In the case of trihydride complex 61 (Figure 33), addition of HBF4, however, affords dihydrogen derivative 154, which is converted to cationic complexes (155, 156) in the presence of NaCl or CsF, the cyclometalated fragment being unaffected [86].
4.3. Ligand Substitution in Osmacycles
Piano-stool osmacyle 93 is a versatile precursor for a series of octahedral 2,2′-bipyridine, 1,10-phenanthroline and MeCN complexes (Figure 34). The substitutions are due to the lability of the coordinated benzene in polar solvents such as MeCN and MeOH. Cyclometalated N,N-dimethylbenzylamine derivatives 97 behave similarly affording complexes 159–162 [71,76,79,80].
5. Applications of Osmacycles
Osmacycles find applications in many areas including homogeneous catalysts; they are used as chemical sensors, luminescent materials, and anticancer agents. Relevant examples are summarized below.
5.1. Catalysis
Although ruthenium and palladium cyclometalated complexes have been widely investigated in homogeneous catalysis [19,20,21,88,89,90], osmium analogues are not as widely used. Perhaps they have not yet demonstrated their potential. However, there are a few examples where osmacycles show good catalytic activity and look like promising candidates for new applications. In particular, binding of H2 to 25 affords 175 with η2-bound H2 (Figure 37) which might be a key intermediate in catalytic hydrogenation [91].
Hexamerization of phenylacetylene at pincer complex 25 is a promising reaction which gives a mixture of isomers of diosmium complex 176 with a μ-1,2-bis(η5-cyclopentadienyl)-1,2-diphenylethane bridging ligand (Figure 38) [92].
Baratta et al. contributed significantly to the homogeneous catalysis by cyclometalated Ru and Os complexes. Benzo[h]quinoline C~N~N osmium pincers were tested as hydrogenation catalysts. Three osmium complexes [OsX(C~N~N)(dppb)] (177) (X = Cl, H, OR; dppb = 1,4-bis(diphenylphosphino)butane) were studied in catalytic hydrogen transfer from isopropanol to asymmetric ketones. Using a catalyst charge of 0.005% mol, conversions above 94% in less than 30 min with TOFs of 105–106 h−1 were achieved. The ruthenium analogues of 177 were slightly more active for the same process, with conversions above 97% within 10 min, presenting TOFs values in the order of 106 h−1 [93]. Chiral ligands in 178 were tested for the asymmetric hydrogenation of prochiral ketones and conversions up to 92% with 74–94% enantiomeric excess (ee) were reported [94]. Structural modifications of the benzo[h]quinoline ligands gave two new series of chiral osmium complexes 179. The complexes with a Josiphos ligand were used in the hydrogenation of acetophenone with conversions up to 97% with TOFs around 104 h−1 and ee up to 86%. In the same work, the ruthenium analogue of 179a was also studied. Conversions up to 95% with TOFs around 104 h−1 and ee between 90 and 99% were obtained. However, in the case of ruthenium the reaction was performed at 40 °C instead of 70 °C for the osmium catalyst [95]. Different C~N~N pincer ligands were also used, keeping the Josiphos ligand system in complexes 180. These were used for catalytic ketone reduction. Conversions were above 93% with TOFs 104–105 h−1 and ee between 90 and 99%. The ruthenium analogues were also obtained, and the catalytic activity for both metals was comparable [96]. The results are summarized in Figure 39.
Osmium C~N~N pincer complexes 177 (X = Cl) and 178a were employed in dehydrogenation of alcohols to form the corresponding ketones and H2. The dehydrogenation of α-tetralol using a catalyst charge of 0.4% mol was run with 36–44% conversions in 24 h. Their ruthenium analogues were also tested and were found to be more active, with conversions between 90–93% at the same reaction time [97]. The osmium C~N~N pincer complexes 181 and 182 were used for the racemization of alcohols with good results. In this case, a similar catalytic activity was observed for both ruthenium and osmium analogues (Figure 40) [98].
Fluorinated pincer complex [OsH(CF3P~C~P)(cod)] (183) (Figure 41) obtained from [Os(cod)(η3-2-methylallyl)2] was tested in alkane dehydrogenation. The catalyst showed a longer lifetime than its ruthenium analogue. The dehydrogenation of cyclooctane in the presence of tert-butylethene as a hydrogen donor was carried out. A significant production of cyclooctene was detected within a few minutes at 200 °C [99].
Esteruelas’ group reported on the metalation of imidazolium salts using [OsH6(PiPr3)2]. Stable compounds 185 and 187, like possible intermediates in catalytic hydrogenation, were isolated (with yields from 83 to 89%) through the osmium reduction in the presence of HBF4. The authors also explored the substitution of the η2-bonded molecular hydrogen by a coordinating molecule like acetonitrile [100].
The metalation of 1-(2-methoxy-2-oxoethyl)-3-methylimidazolium chloride afforded cyclometalated acyl complex 188 which coordinates dioxygen, dihydrogen, and carbon monoxide (Figure 42). Complex 188 was used as a catalyst for alcoholysis and hydrolysis of pinacolborane. Using a 2% mol catalyst charge for the alcoholysis reaction, TOFs between 62 and 3644 h−1 were obtained at 50% conversion. For the hydrolysis carried out with the same amount of catalyst, TOFs were 473–1648 h−1 at 50% [101]. The 2-(aminomethyl)pyridine-based complexes 190 were used in the imine-to-amine hydrogenation (Figure 43). Depending on the reaction conditions, conversions between 26 and 99% were observed. In this work, the ruthenium equivalents of 190 were also obtained. In general, under the same reaction conditions, the osmium complexes were found to be less active than the corresponding ruthenium derivatives [102].
Esteruelas et al. described the heterobinuclear iridium–osmium compound [(PiPr3)2(H)2Ir{μ-(κ2-Npy,Nimine-BMePI-κ2-Nimine,C4iso)}OsH3(PiPr3)2] (191) incorporating 1,3-bis(6′-methylpyridyl-2′-imino)isoindoline (HBMePI) as a bridging ligand. This complex was tested in the base-free dehydrogenation of secondary alcohols using 7% mol of catalyst. Conversions up to 84% were obtained. Interestingly, heterobimetallic complex 191 proved to be more active than the corresponding monometallic components taken independently (Figure 44) [103].
The resolution of a helical chiral octahedral osmium complex afforded optically pure Λ (192) and Δ (193) enantiomers (Figure 45). The Δ enantiomer was tested as a catalyst in two processes, viz. C(sp3)–H aminations of sulfonylazide and azidoformate. For the former, a product yield of 96% was observed using 2% mol of catalyst, with an enantiomeric ratio of 92:8 (Figure 45). Under similar conditions, a yield of 86% was registered in the case of azidoformate with the enantiomeric ratio of 89:11. This is the first example of an osmium complex with a central (helicoidal) chirality successfully used in asymmetric catalysis [104]. It is important to mention that Meggers’ group also developed highly effective enantioselective catalytic processes with metal-centered chiral octahedral ruthenium and rhodium complexes [105,106,107].
Octahedral cyclometalated 2-phenylpyridine RuII and OsII complexes were investigated as catalysts for the atom transfer radical polymerization (ATRP). Polymerization of styrene was catalyzed by ruthenium complexes though the osmium counterparts were not able to mediate the polymerization and just traces of the polymer were observed [108].
5.2. Chemical Sensors and Biosensors
Oxidoreductases are enzymes that catalyze oxidative and reductive reactions. They are used in various amperometric biosensors, including glucometers [109]. Active sites of the enzymes do not usually exchange electrons with an electrode and therefore low-molecular-weight compounds are commonly used to move the electrons. Known as electron shuttles or mediators, they are often transition-metal complexes. In particular, cyclometalated ruthenium derivatives were successfully coupled with a number of oxidoreductases such as glucose oxidase, glucose and alcohol dehydrogenases, or peroxidases [110,111,112]. The MII/III reduction potential is an essential feature for optimal performance of biosensors [113], and since the potentials are tunable as it is illustrated in Inset to Figure 21 [80], the corresponding osmium-based mediators were exploited. In particular, a series of octahedral osmium(II) complexes of the general formula [Os(N~C)(N~N)(MeCN)2]PF6 (159, 161) and [Os(N~C)(N~N)2]PF6 (94, 95, 162, 163) were investigated, where C~N is cyclometalated 2-phenylpyridine or N,N-dimethylbenzylamine and the N~N ligands are 2,2′-bipyridines or 1,10-phenanthrolines. The complexes were evaluated as electron shuttles with glucose oxidase (GO) for potential applications in amperometric enzymatic glucose sensors. All complexes showed extremely fast electron transfer with the enzyme, with the second-order rate constants k2 for bimolecular oxidation of reduced glucose oxidase in the range of (0.67 − 2.90) × 106 M−1 s−1 (Figure 46 and Table 3) [76].
Discussed above mono- [Os(phpy)(bpy)2]PF6 (94), bis- [Os(phpy)2(bpy)]PF6 (102) and tris-cyclometalated [Os(phpy)3] (101) derivatives turned out to be attractive objects for electrochemical studies. Using related coordination complex [Os(bpy)3](PF6)2 (103), it was shown that the OsII/OsIII reduction potential significantly decreases as the number of Os−C bonds increases. Rates of electron transfer between the osmium center and the active sites of oxidoreductases including horseradish peroxidase (HRP) were investigated (Figure 47). Cyclic voltammetry was used to estimate the rate constants. In the case of HRP, the results showed that the rate constants k2 are higher than k3 for all compounds, i.e., electrons move faster from OsII to compound I than to compound II (see Figure 47). The value of both rate constants increases in the series [Os(bpy)3](PF6)2 < [Os(phpy)(bpy)2)]PF6 (94) < [Os(phpy)2(bpy)]PF6 (102) and then decline for the tris-cyclometalated complex (101). It should be mentioned that all complexes are very reactive. The Monte Carlo docking simulations helped to evaluate how the complexes approach the active site of HRP [80].
Bis-cyclometalated complex [Os(phpy)2(bpy)]PF6 (102) was tested as a mediator in a prototype of an amperometric biosensor to quantify hydrogen peroxide (Figure 48) An iron(III)-TAML (TAML = tetraamido macrocyclic ligand) catalyst used as an alternative to peroxidase enzymes was immobilized on an electrode surface with the osmium mediator. The activity and sensitivity of such a device were similar to those of a biosensor based on HRP enzyme [114].
5.3. Electronic Properties and Photophysics
Due to a unique combination of spectroscopic, photophysical, photochemical and electrochemical properties, octahedral RuII and OsII polypyridyl complexes have been extensively studied in various fields such as photochemical conversion of solar energy, photocatalysis and in molecular electronic devices. Their properties can be modulated by modifying the ligand structures and by introducing ancillary ligands [115,116]. The major goal is to enhance excited state lifetimes of the compounds at room temperature by increasing the energy gap between the radiative 3MLCT and quenching 3MC states by destabilizing the 3MC state using cyclometalated ligands [117,118]. However, the enhanced spin-orbit coupling of the third-row elements when compared to the second-row elements usually makes optoelectronic properties of osmium complexes notably different from those of ruthenium derivatives [119]. Nevertheless, as we discuss in this section, switching from ruthenium to osmium has proven to be successful in generating promising materials.
Sierra et al. synthesized dinuclear cyclometalated trihydride osmium(IV) complexes (69–71, 194, 195) (Figure 49) which were studied by cyclic voltammetry. Two quasi-reversible processes corresponding to the OsIV/OsV and OsV/OsVI redox features were observed. The OsIV complexes showed emission around 360 nm, with quantum yields being between 0.005 and 0.020. The excitation spectra suggested that a ligand-centered transition is at the origin of the emission. Moreover, spectrochemical studies showed that the emission spectra remained very similar upon OsIV to OsV oxidation at 0.05 V vs. Fc+/Fc, since only a slight increase in intensity was observed, with quantum yields from 0.013 to 0.026. Nevertheless, when a potential of 1.10 V was applied to carry out the oxidation of OsV to OsVI, a bathochromic shift, from around 360 to around 400 nm, was observed along with an increase in the luminescence intensity with quantum yields around 0.030–0.110. DFT calculations suggest that the photophysical properties vary due to the conversion of trihydride species to monohydride-dihydrogen derivatives during sequential oxidation processes [59].
Kapturkiewicz et al. prepared the series of cationic osmium complexes 196 of the general formula [OsCl(N~C)(PPh3)2(CO)] (N~C is a cyclometalated substituted 2-phenylpyrididine) and measured their UV-visible absorption and emission spectra (Figure 50). The complexes showed emission at 455–532 nm attributed to 3MLCT transitions. Importantly, only the solids emitted light at room temperature; the compounds in solution were emissive only at a low temperature (77 K). The emission lifetimes are relatively long, between 10 and 20 μs [120].
Bis-(P~C~P) pincer osmium complex 39 (Figure 10) has a strong absorption at 250 nm attributed to the ligand π→π* transitions. The emission at 546 nm in a dilute solution of 2-MeTHF at 77 K with a quantum yield of 0.6 and a lifetime of 8.0 μs suggests phosphorescence. The solid complex is also luminescent at room temperature at 556 nm, with a quantum yield of 0.03 and a lifetime of 0.3 μs. The electronic structure of the complex and related transitions were explored by DFT [45].
Carbene pincer ruthenium and iridium compounds display remarkable luminescent properties [121]. Related osmium C~C’~C complexes 197 and 198 were obtained by reacting [OsCl4(N~N)] precursors (N~N = 2,2′-bypiridine, 1,10-phenanthroline, 4,4′-diphenyl-2,2′-bypiridine) with bis-imidazolium or bis-benzimidazolium hexafluorophosphates in refluxing ethylene glycol as a source of the CO ligand (Figure 51). The UV-visible absorption data showed intense and high-energy absorption at λ lower than 330 nm and a less intense band at λ > 330 nm. The complexes are also emissive upon photoexcitation with emission maxima in the red region (674−731 nm). The quantum yields reported were between 10−4 and 10−2 and emission lifetimes in the order of 10−1 μs. The photophysical parameters are sensitive to the modification of N~N and C~C’~C parts, suggesting that the emissive-excited state should involve both fragments and the emission occurs due to the energy dissipation of d(Os)→π*[N~N] MLCT transitions [122].
The photophysical studies of the C~C’~C mono- and bis-pincer compounds (199–204) in Figure 51 revealed an intense absorption between 341 and 363 nm along with a less intense band between 396 and 405 nm. TD-DFT calculations allowed to attribute the high energy bands to interligand charge-transfer transitions and the low energy bands to metal-to-ligand charge-transfer processes. Particularly, the bis-pincer complexes 202–204 exhibited emission in the blue-green region (475–578 nm), both in the solid state at room temperature and in toluene solution (at room temperature and 77 K) with lifetimes in the range of 10–29 μs. The best quantum yield is observed for complex 202 (R = CF3) with a value of 0.62. This last complex 202 (R = CF3), with emission in the blue region, was chosen for the fabrication of a model OLED device, obtaining promising results [123].
The osmium complexes 205 and 206 with a tetradentate C~C~C~C ligand (Cphenyl~Ccarbene~Ccarbene~Cphenyl) in Figure 51 can be viewed as bis-cyclometalated compounds. The complexes with two coordinated DMSO ligands provide options for their replacement by bidentate diphosphines or dienes. Complexes bearing 1,2-bis(diphenylphosphino)benzene emit light upon photoexcitation in the solid state at room temperature and in solution of 2-MeTHF at 77 K. The bands between 630 and 549 nm were attributed to the π-π* HOMO (Os and NHC fragment) to LUMO (diphenylphosphino) and metal-to-ligand (osmium to phosphino ligand) charge-transfer transitions. In addition, the absorption spectra contained an intense band at 330 nm and a less intense band at 404 nm attributed to metal-to-ligand charge-transfer transitions [124].
Bis-tridentate cyclometalated complexes [Os(N~N~N)(N~C~N)]PF6 (207, 208) have been tested as molecular wires, and compounds 208, in which the N~C~N ligand with a triarylamine substituent is separated from the metal center by an oligophenyl bridge, were of special interest (Figure 52). This is due to the presence of two electrochemically active centers, the metal and the triarylamine substituent which can undergo a N•+/N0 process (neutral nitrogen to positive radical nitrogen). These molecular wires show two anodic redox features between +0.1 and +0.9 V vs. Ag/AgCl, the first one assigned to the OsII/OsIII process and the second one to the N•+/N0 process, and an electron transfer from the neutral amine to the OsIII center was observed in the one-electron oxidized form [OsIII-N] [125].
Esteruelas et al. studied the luminescent properties of C~C~N pincer complexes derived from 2-azetidones (see Table 2). At room temperature, complex (73-L22) showed yellow emission in the solid state at 571 nm (lifetime 1.1 μs) and at 562 nm in a toluene solution (lifetime 2.0 μs), as well as green emission at 77K at 540 nm (lifetime 6.6 μs) [60]. Complexes 74 and 75 in Table 2 are phosphorescent upon photoexcitation in a poly(methyl methacrylate) (PMMA) film and in 2-MeTHF at room temperature and 77 K. Both compounds emit in the yellow region. The 2,6-diphenylpyridine complex 74 is more efficient, the quantum yields being 0.28 (film) and 0.56 (solution). The introduction of oxygen between the pyridine and phenyl ring in 75 lowered the emission (quantum yields below 0.10). Curiously, the iridium analogue of 75 showed the opposite trend [126].
The absorption spectra of C~N~N and C~N~C pincer complexes 209–212 in Figure 53 are similar. They present two main bands between 279 and 317 nm and between 364 and 394 nm. TD-DFT calculations allowed to assign the high energy absorptions to ligand-centered transitions while the low energy absorptions were assigned to metal-to-ligand charge-transfer processes. The compounds were emissive upon photoexcitation in the solid state at room temperature and in 2-MeTHF solution both at room temperature and at 77 K. The use of pincer ligands allowed to obtain more rigid structures reducing both the energy dissipation and the difference between the excited state and the ground state structures. As a consequence, enhanced quantum yields in the range of 0.08–0.59 were obtained, lifetimes being 1.5−5.0 μs in the solid state and 0.9–5.8 μs in solution [127].
The electronic structure of osmium bis-pincer complexes 213 with N~C~N cyclometalated pyrene ligands (Figure 54) was explored by DFT and TD-DFT. It was found that HOMO orbitals are mainly osmium in character. The LUMO orbitals were largely associated with ligand contributions. There are two intense bands absorption spectra between 282 and 372 nm assigned to intraligand transition from the pyrene and terpyridine fragments. Two other intense bands between 410 and 447 nm were associated with intraligand charge-transfer (ILCT) transitions. The bands observed between 513 and 556 nm were due to MLCT transitions [128]. These experimental results launched a theoretical study which helped to optimize photophysical features of the compounds. It was concluded that compounds 214 in Figure 54 should display optimal properties [129].
DFT calculations performed for bimetallic complex 215 shown in Figure 54 suggested that the HOMO orbital is contributed by pyrene and metal centers. The LUMO orbital has contributions from the pyrene moiety and pyridine rings. Two intense bands observed between 457 and 507 nm were assigned to ligand-to-ligand charge-transfer transitions. An absorption band between 800 and 1000 nm was assigned to metal-to-ligand (pyrene) charge-transfer transitions. An electronic coupling between the neighboring osmium centers affects cyclic voltammograms of osmium compounds and brings about two reversible OsII/OsIII redox features overlapping at −0.15 V (vs. Fc/Fc+). A similar behavior is observed for ruthenium but not for iridium complex [130].
5.4. Anticancer and Biological Properties
Transition metal derivatives have been extensively studied in medicinal chemistry, often with emphasis on their anticancer activity. Organometallic complexes are currently receiving special attention [131,132,133,134,135,136,137]. The leadership belongs to ruthenium compounds including the clinical studies of metal-based anticancer drugs [138,139,140,141]. Omae published a comprehensive review on anticancer properties of cyclometalated complexes with a few osmium examples [132]. Pfeffer and Gaiddon dedicated a review to ruthenacycles [142]. Examples of a few osmacycles were included in the recent review by Zhang devoted to biological properties of osmium compounds [131]. Here, we show the most recent examples of osmium cyclometalated complexes used in biological assays.
Meggers and Xia compared the anticancer activity of a series of osmium derivatives, including complexes 43, 49, 164, 174, a new η2-allene compound (216, Figure 55), and other non-cyclometalated complexes. The cytotoxic activity in HeLa cancer cells was determined first by the evaluation of the half-maximum effective concentration (EC50) at which viability of the cells is reduced to 50% after 24 h incubation. Observed values for EC50 were between 1 and 30 μM. The best results were obtained with allene complex 216 which exhibited cytotoxic activity with EC50 = 1 μM. Because of this and its high stability, studies of this complex were prioritized. The compound was tested against Burkitt-like lymphoma (BJAB), showing an inhibition of the proliferation at concentrations lower than 1 μM. Another assay revealed that the compound induces apoptosis in BJAB cells by DNA fragmentation. The results showed that even a 0.3 μM concentration, 216 induced DNA fragmentation in 25% of the cells, with the highest DNA fragmentation observed in 75% of the cells at 5 μM. In addition, other experiments showed that apoptosis occurs through the intrinsic mitochondrial pathway. The compound was also tested in leukemia cells (Nalm6), Vcr-resistant NAlm6 cells and Dau-resistant Nalm6 cells (the last two are drug-resistant leukemia cells) showing a lower activity, with EC50 higher than 1 μM, but with effective activity at 5μM [143].
The successful use of ruthenacycles and osmacycles as efficient electron shuttles for oxidoreductase enzymes described in Section 5.2 prompted the investigation of the potential use of these compounds for biological applications. Gaiddon and Pfeffer evaluated the cytotoxic properties of such compounds on series of human cancer cell lines. An exhaustive work was published where a series of osmium cyclometalated compounds was tested in in vitro cell growth inhibition in A172 glioblastoma cell line. The structures of the compounds studied are summarized in Figure 56. First results with the piano-stool complexes (92, 93, 217–219) showed moderate cytotoxic activity with IC50 values of 10−100 μM. The octahedral complexes with bidentate cyclometalated ligands (94, 158, 159, 220–224) exhibited very promising results with IC50 values in the nanomolar range. Pincer complexes (225, 226) showed very poor cytotoxic activity, with IC50 values 21−375 μM, which could be attributed to the presence of three labile acetonitrile ligands. Interestingly, the pincer complexes with polypyridine ligands (227, 228) showed very good results with IC50 values between 0.3 and 4.1 μM. There was no correlation between the cytotoxicity and structure. However, the reduction potentials were measured for all complexes and the results showed that the compounds with the potentials between 0.3 and 0.6 V (vs. SCE) displayed the lowest IC50 values. The lipophilicity was also determined and the complexes with the highest activity showed log(Po/w) values around 2. These results allowed to hypothesize that the compounds could strongly modify the metabolism of the cells by interactions with oxidoreductases and this would partially explain the relationship between the activity and the reduction potentials [144]. Another important target of such studies is to discover molecules that can overcome drug resistance induced by cisplatin. Recently, the mechanisms governing the variability in the cytotoxicity of two ruthenium cyclometalated compounds and their osmium equivalents were studied (Figure 34, complexes 95 and 159). Their anticancer properties in vitro and in vivo were first evaluated and genes involved in their sensibility/resistance were identified by correlating their cytotoxicity with transcriptomic data of 60 cancer cell lines. Docking and functional studies demonstrated that inhibition of known resistance mechanisms, ABCB1 export and EGFR expression, allowed to improve the activity of cyclometalated complexes. Interestingly, switching from ruthenium to osmium favored the cytotoxicity while reducing the sensibility to the ABCB1 export mechanism [145].
Kandioller developed the coordination of a series of 4-phenyl-1,2,3-triazole-based ligands to ruthenium and osmium to prepare complexes 229 in Figure 57. Their cytotoxic activity was evaluated on three cancer cell lines: non-small-cell lung cancer (A549), colon adenocarcinoma (SW480) and human ovarian carcinoma (CHI/PA-1). The free ligands did not show cytotoxic activity within the studied concentrations range. However, the ruthenium derivatives bearing ligands a and b showed significant cytotoxicity with IC50 values around 13 μM in A549, 7 μM in SW480 and 4 μM in CHI/PA-1. Osmium counterparts displayed an even higher activity, ligands a and b being also the most active, with IC50 around 6 μM for A549, 3.6 μM for SW480, and 1 μM for CHI/PA-1 cells. The topoisomerase IIα inhibition was investigated but no compound was capable of inhibiting its function, indicating that the mechanism of action does not follow this pathway. The cell cycle distribution in SW480 in response to treatment with the complexes was determined and only the osmium derivatives exhibited elimination of the S phase fraction, information relevant in order to understand the mechanism of action [146].
Using a triazole-based ligand, Makhubela prepared a series of half-sandwich ruthenium, rhodium, iridium and osmium complexes (230), Figure 57. The complexes were tested on different cancer cell lines (leukemia MT4, cervical cancer HeLa, kidney adenocarcinoma HEK293 and lung cancer A549), and the CC50 (cytotoxic concentration with 50% of the maximum decrease in cell viability) was calculated. The ruthenium derivative (230a) exhibited a moderate activity against A549, but the osmium compound (230b) was much more active with a CC50 value of 6.4 μM, comparable to cisplatin used as reference. However, its activity was very modest against the other cancer cell lines. Interestingly, the rhodium and iridium derivatives were more effective against all cancer cell lines when compared to their ruthenium and osmium analogues [147].
Benzimidazole-based ligands were used by Ruiz to prepare the series of new osmium complexes 231, Figure 57. Their antiproliferative activity was tested in six different human cancer cell lines (including cisplatin-resistant cell lines), as well as non-tumorigenic human endothelial hybrid cells and Buffalo green monkey cells. All osmium compounds showed a higher cytotoxic activity than cisplatin in all cancer cell lines, with IC50 values lower than 10 μM. The cytotoxicity against the non-cancer cells was very similar to cisplatin. The cellular concentrations of both metals were measured in A2780 cells and the results showed that the osmium cellular uptake is ten times higher than for platinum. Other studies revealed that the compounds caused a decrease of the ROS levels and, as a result, an affectation in the G0/G1 phase in the cell cycle is observed [148].
In a very recent work, Gómez-Gallego, Sierra and Esteruelas explored the reaction between osmium precursor [OsH6(PiPr3)2] and nucleosides. Cyclometalated complexes 232–235 were obtained in good yields (Figure 58). The authors mentioned that this methodology could be applied to the functionalization of oligonucleotides with promising potential biological applications [149].
Many more biological applications of osmium-based complexes remain to be discovered and exploited. For instance, the unique photophysical properties of osmacycles (see Section 5.3) will undoubtedly open the field for further research on their use as PDT agents. On the other hand, deeper in vivo studies are needed to confirm properties such as cellular uptake ability, cellular distribution, as well as to determine essential pharmacokinetic data.
6. Conclusions
As this review demonstrates, even though osmacycles chemistry is still in a growing stage, cyclometalated osmium compounds are essential in many aspects. The broadest chemistry of osmium and the low reactivity of osmium compounds grants determination and studies of a variety of mechanisms, which in turn provide tools to design novel complexes with a wide variety of relevant properties and applications. Due to the enormous number of molecules, known or readily accessible, that can lead to cyclometalation reactions, unique applications of osmacycles will see the light in the near future. Many cyclometalated osmium derivatives have shown remarkable catalytic activity in various reactions, principally hydrogenations and dehydrogenations, often similar and in some cases superior to those reported for analogous ruthenium systems. Such results exhibit the potential of osmacycles for the design of new highly effective and robust catalytic systems for chiral and non-chiral processes. On the other hand, new osmium-containing cyclometalated materials will continue to be studied for their activity in biological systems, and they will likely be involved in the development of innovative chemical sensors, biosensors and improved drugs.
Funding
We wish to thank DGAPA–UNAM (IN-207419) and CONACyT (A1-S-15068) for funding our research projects on osmium chemistry.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to all colleagues and students who have been involved in our works on osmium chemistry. Their names are listed in the references.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviation
| 2-MeTHF | 2-methyl-tetrahydrofuran |
| A2780 | Human ovarian cancer cell line |
| A549 | Human lung cancer cell line |
| AND/OR | Boolean operator for browsing in databases |
| ATRP | Atom Transfer Radical Polymerization |
| B3LYP | Becke three-parameter exchange and Lee–Yang–Parr correlation |
| BJAB | Burkitt-like lymphoma |
| Bn | Benzyl substituent |
| bpy | 2,2′-bipyridine |
| C~C’~N | Pincer ligand with Carbon-Carbon-Nitrogen, as donor atoms |
| C~C~C | Pincer ligand with Carbon-Carbon-Carbon, as donor atoms |
| C~C~C~C | Tetradentate Ligand with Carbon-Carbon-Carbon-Carbon as donor atoms |
| C~E | Bidentate ligand with Carbon and E as donor atoms |
| C~N | Bidentate ligand with Carbon and Nitrogen as donor atoms |
| C~N~C | Pincer ligand with Carbon-Nitrogen-Carbon as donor atoms |
| C~N~N | Pincer ligand with Carbon-Nitrogen-Nitrogen as donor atoms |
| C~N~O | Pincer ligand with Carbon-Nitrogen-Oxygen as donor atoms |
| C~O | Bidentate ligand with Carbon and Oxygen as donor atoms |
| CC50 | 50% Cytotoxic concentration |
| CHI/PA-1 | Human ovarian carcinoma cells |
| cod | 1,4-cyclo-octadiene |
| DFT | Density Funtional Theory |
| DMSO | Dimethyl sulphoxide |
| DNA | Deoxyribonucleic acid |
| dpbH | 1,3-di(2-pyridyl)benzene |
| dppb | 1,4-bis(diphenylphosphino)butane |
| E | Donor atom such as: N, P, O, S, As, Se |
| EC50 | 50% Effective concentration |
| GO | Glucose oxidase |
| HBMePI | 1,3-bis(6′-methylpyridyl-2′-imino)isoindoline |
| HEK293 | Kidney adenocarnocinoma cell line |
| HeLa | Cervical cancer cell line |
| HOMO | Highest Occupied Molecular Orbital |
| HRP | Horseradish peroxidase |
| IC50 | 50% Inhibitory concentration |
| ILCT | Intraligand Charge-Transfer |
| log(Po/w) | N-Octanol-water partition coefficient |
| LUMO | Lowest Unoccupied Molecular Orbital |
| Mebib | 2-deprotonated form of 1,3-bis(N-methylbenzimidazolyl)benzene |
| Mebip | bis(N-methylbenzimidazolyl)pyridine) |
| MLCT | Metal-to-Ligand Charge Transfer |
| MT4 | Leukemia cell line |
| N~C~N | Pincer ligand with Nitrogen-Carbon-Nitrogen as donor atoms |
| N~N | Bidentate ligand with Nitrogen-Nitrogen as donor atoms |
| N~N~N | Terdentate ligand where the atom donors are Nitrogen |
| Nalm6 | Leukemia cell line |
| NHC | N-Heterocyclic Carbene |
| NHE | Normal Hydrogen Electrode |
| NMR | Nuclear Magnetic Resonance |
| OLED | Organic Light-Emitting Diode |
| Otf | Triflate |
| P~C(H)~P | Ligand with only phosphorous as donor atoms |
| P~C~P | Pincer ligand with phosphorous–carbon–phosphorous as donor atoms |
| PDT | Photodynamic Therapy |
| PhMeBIm | 1-Phenyl-3-methyl-1H-benzimidazolium |
| phen | 1,10-Phenanthroline |
| phpyH | 2-Phenylpyridine |
| PMMA | Polymethylmethacrylate |
| ROS | Reactive Oxygen Species |
| SCE | Saturated Calomel Electrode |
| SW480 | Colon adenocarcinoma cell line |
| TAML | Tetra-Amido Macrocyclic Ligand |
| TD-DFT | Time-Dependent Density Functional Theory |
| TOF | Turnover Frequency |
| tterpy | 4′-tolyl-2,2′,6′,2”-terpyridine ligand |
References
- Griffith, W.P. Osmium tetroxide and its applications. Platin. Met. Rev. 1974, 18, 94–96. [Google Scholar]
- Smith, M. Organic Synthesis, 4th ed.; Academic Press: Cambridge, MA, USA, 2016; ISBN 9780128007204. [Google Scholar]
- Bozzola, J.J.; Russell, L.D. Electron Microscopy: Principles and Techniques for Biologists, 2nd ed.; Jones and Bartlett: Boston, MA, USA, 1999. [Google Scholar]
- Lay, P.A.; Harman, W.D. Recent advances in Osmium chemistry. Adv. Inorg. Chem. 1991, 37, 219–379. [Google Scholar] [CrossRef]
- Griffith, W.P.; Wilkinson, R.D.G.; McCleverty, J.A. (Eds.) Comprehensive Coordination Chemistry: The Synthesis, Reactions, Properties & Applications of Coordination Compounds; Comprehensive Coordination Chemistry; G. Wilkins; Pergamon: Oxford, UK, 1987; Volume 4. [Google Scholar]
- Warncke, R. Gmelin Handbuch der Anorganischen Chemie; Osmium; Springer: Berlin/Heidelberg, Germany, 1980; Volume 1, No. 66. [Google Scholar]
- Collinson, S.R.; Schröder, M. Osmium: Inorganic & coordination chemistry. Encycl. Inorg. Bioinorg. Chem. 2011, 1–24. [Google Scholar] [CrossRef]
- Shapley, P.A. Osmium: Organometallic chemistry. Encycl. Inorg. Bioinorg. Chem. 2011, 3960–3978. [Google Scholar] [CrossRef]
- Bruce, M.I. Cyclometalation reactions. Angew. Chem. Int. Ed. Engl. 1977, 16, 73–86. [Google Scholar] [CrossRef]
- Omae, I. Organometallic intramolecular-coordination compounds containing a nitrogen donor ligand. Chem. Rev. 1979, 79, 287–321. [Google Scholar] [CrossRef]
- Omae, I. Cyclometalation Reactions: Five-Membered Ring Products as Universal Reagents; Springer: Sayama, Japan, 2014; ISBN 9784431546047. [Google Scholar]
- Michon, C.; Macintyre, K.; Corre, Y.; Agbossou-Niedercorn, F. Pentamethylcyclopentadienyl Iridium(III) metallacycles applied to homogeneous catalysis for fine chemical synthesis. ChemCatChem 2016, 8, 1755–1762. [Google Scholar] [CrossRef]
- Leist, M.; Kerner, C.; Ghoochany, L.T.; Farsadpour, S.; Fizia, A.; Neu, J.P.; Schön, F.; Sun, Y.; Oelkers, B.; Lang, J.; et al. Roll-over cyclometalation: A versatile tool to enhance the catalytic activity of transition metal complexes. J. Organomet. Chem. 2018, 863, 30–43. [Google Scholar] [CrossRef]
- Zi, G. Recent developments in actinide metallacycles. Chem. Commun. 2018, 54, 7412–7430. [Google Scholar] [CrossRef]
- Ryabov, A.D. The exchange of cyclometalated ligands. Molecules 2021, 26, 210. [Google Scholar] [CrossRef]
- Ryabov, A.D. Mechanisms of intramolecular activation of carbon-hydrogen bonds in transition-metal complexes. Chem. Rev. 1990, 90, 403–424. [Google Scholar] [CrossRef]
- Van Der Boom, M.E.; Milstein, D. Cyclometalated phosphine-based pincer complexes: Mechanistic insight in catalysis, coordination, and bond activation. Chem. Rev. 2003, 103, 1759–1792. [Google Scholar] [CrossRef]
- Dupont, J.; Consorti, C.S.; Spencer, J. The potential of palladacycles: More than just precatalysts. Chem. Rev. 2005, 105, 2527–2571. [Google Scholar] [CrossRef]
- Dupont, J.; Pfeffer, M. (Eds.) Palladacycles: Synthesis, Characterization and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; ISBN 9783527623211. [Google Scholar]
- Djukic, J.-P.; Sortais, J.-B.; Barloy, L.; Pfeffer, M. Cycloruthenated compounds—Synthesis and applications. Eur. J. Inorg. Chem. 2009, 2009, 817–853. [Google Scholar] [CrossRef]
- Albrecht, M. Cyclometalation using d-block transition metals: Fundamental Aspects and recent trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Nam, W. Photofunctional triplet excited states of cyclometalated Ir(III) complexes: Beyond electroluminescence. Chem. Soc. Rev. 2012, 41, 7061–7084. [Google Scholar] [CrossRef]
- Cutillas, N.; Yellol, G.S.; De Haro, C.; Vicente, C.; Rodríguez, V.; Ruiz, J. Anticancer cyclometalated complexes of platinum group metals and gold. Coord. Chem. Rev. 2013, 257, 2784–2797. [Google Scholar] [CrossRef]
- Dehand, J.; Pfeffer, M. Cyclometallated compounds. Coord. Chem. Rev. 1976, 18, 327–352. [Google Scholar] [CrossRef]
- Dehand, J.; Mutet, C.; Pfeffer, M. Reactivite d’anilines tertiaires vis-a-vis de complexes du palladium: Dealkylation de l’azote et cyclopalladation. J. Organomet. Chem. 1981, 209, 255–270. [Google Scholar] [CrossRef]
- Pfeffer, M. Reactions of cyclopalladated compounds and alkynes: New pathways for organic synthesis? Recl. Des Trav. Chim. Des. Pays-Bas 1990, 109, 567–576. [Google Scholar] [CrossRef]
- Pfeffer, M. Selected applications to organic synthesis of intramolecular C-H activation reactions by transition metals. Pure Appl. Chem. 1992, 64, 335–342. [Google Scholar] [CrossRef]
- Bruce, M.I.; Goodall, B.L.; Stone, F.G. Cyclometallation reactions. J. Organomet. Chem. 1973, 60, 343–349. [Google Scholar] [CrossRef]
- Jameson, G.B.; Muster, A.; Robinson, S.D.; Wingfield, J.N.; Ibers, J.A. Cyclometalated formazan derivatives of ruthenium and Osmium: Structure of Ru((o-C6H4)N=NC(Ph)=NNPh)(CO)(PPH3)2. Inorg. Chem. 1981, 20, 2448–2456. [Google Scholar] [CrossRef]
- Desrosiers, P.J.; Shinomoto, R.S.; Flood, T.C. Intra- and intermolecular activation of carbon-hydrogen bonds in a tetrakis(trimethylphosphine)Osmium(II) system. J. Am. Chem. Soc. 1986, 108, 1346–1347. [Google Scholar] [CrossRef]
- Kisenyi, J.M.; Sunley, G.J.; Cabeza, J.A.; Smith, A.J.; Adams, H.; Salt, N.J.; Maitlis, P.M. The cyclometallation of benzoic acid to give rhodium, iridium, and Osmium C,O-benzoates. X-Ray structure determination of the dibenzoate [(C5Me5)Rh(OOCPh)2(H2O)]. J. Chem. Soc. Dalton Trans. 1987, 2459–2466. [Google Scholar] [CrossRef]
- Beley, M.; Chodorowski, S.; Collin, J.-P.; Sauvage, J.-P.; Flamigni, L.; Barigelletti, F. Luminescent dinuclear complexes containing Ruthenium(II)- and Osmium(II)-terpyridine-type chromophores bridged by a rigid biscyclometalating ligand. Inorg. Chem. 1994, 33, 2543–2547. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.P.; Sauvage, J.P. Highly coupled mixed-valence dinuclear ruthenium and Osmium complexes with a bis-cyclometalating terpyridine analog as bridging ligand. Inorg. Chem. 1993, 32, 4539–4543. [Google Scholar] [CrossRef]
- Wanandi, P.W.; Tilley, T.D. Osmium alkyl and silyl derivatives with cyclopentadienyl(phosphine) and pentamethylcyclopentadienyl(phosphine) ligand sets. Organometallics 1997, 16, 4299–4313. [Google Scholar] [CrossRef]
- Mamo, A.; Stefio, I.; Poggi, A.; Tringali, C.; DiPietro, C.; Campagna, S. Ruthenium(II) and Osmium(II) complexes with new terdentate polyquinoline and cyclometalating ligands. Synthesis, NMR characterization, luminescence properties, and electrochemical behavior. New J. Chem. 1997, 21, 1173–1185. [Google Scholar]
- Chakravorty, A. New organometallic chemistry of ruthenium and Osmium. Proc. Indian Acad. Sci. Chem. Sci. 1999, 111, 469–477. [Google Scholar] [CrossRef]
- Panda, B.K.; Chakravorty, A. Chemistry of a family of Osmium(II) metallacycles incorporating isonitrile coordination. Indian J. Chem. 2005, 44, 1127–1132. [Google Scholar]
- Zhang, L.; Dang, L.; Wen, T.B.; Sung, H.H.-Y.; Williams, I.D.; Lin, Z.; Jia, G. Cyclometalation of 2-Vinylpyridine with MCl2(PPh3)3 and MHCl(PPh3)3 (M = Ru, Os). Organometallics 2007, 26, 2849–2860. [Google Scholar] [CrossRef]
- Acharyya, R.; Peng, S.-M.; Lee, G.-H.; Bhattacharya, S. An unprecedented oxidative migration of a methyl group from 2-(2′,6′-dimethylphenylazo)-4-methylphenol mediated by ruthenium and Osmium. Inorg. Chem. 2003, 42, 7378–7380. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.B.; Cheung, Y.K.; Yao, J.; Wong, W.-T.; Zhou, Z.Y.; Jia, G. Vinylidene and carbyne complexes derived from the reactions of OsCl(PPh3)(PCP) (PCP = 2,6-(PPh2CH2)2C6H3) with terminal acetylenes. Organometallics 2000, 18, 3803–3809. [Google Scholar] [CrossRef]
- Gauvin, R.M.; Rozenberg, H.; Shimon, L.J.W.; Milstein, D. Synthesis and structure of new Osmium-PCP complexes. Osmium-mediated C-C bond activation. Organometallics 2001, 20, 1719–1724. [Google Scholar] [CrossRef]
- McQueen, C.M.A.; Hill, A.F.; Ma, C.; Ward, J.S. Ruthenium and Osmium complexes of dihydroperimidine-based N-heterocyclic carbene pincer ligands. Dalton Trans. 2015, 44, 20376–20385. [Google Scholar] [CrossRef]
- Majumder, K.; Peng, S.-M.; Bhattacharya, S. Cyclometallation and N=N bond cleavage of 2-(arylazo)phenols by Osmium. Synthesis, structure and redox properties. J. Chem. Soc. Dalton Trans. 2001, 33, 284–288. [Google Scholar] [CrossRef]
- Das, A.; Basuli, F.; Falvello, L.R.; Bhattacharya, S. Unusual transformation of N-arylbenzohydroxamic acids mediated by Osmium. Formation of organometallic complexes of Osmium(III). Inorg. Chem. 2001, 40, 4085–4088. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Djurovich, P.I.; Haiges, R.; Thompson, M.E. Synthesis and photophysical characterization of a bis-pincer Osmium complex. Polyhedron 2014, 84, 136–143. [Google Scholar] [CrossRef]
- Gong, L.; Lin, Y.; Wen, T.B.; Zhang, H.; Zeng, B.; Xia, H. Formation of four conjugated osmacyclic species in a one-pot reaction. Organometallics 2008, 27, 2584–2589. [Google Scholar] [CrossRef]
- Xia, H.; He, G.; Zhang, H.; Wen, T.B.; Herman, H.; Sung, Y.; Williams, I.D.; Jia, G. Osmabenzenes from the reactions of HC≡CCH(OH)C≡CH with OsX2(PPh3)3 (X = Cl, Br). J. Am. Chem. Soc. 2004, 126, 6862–6863. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Gong, L.; Xu, H.; He, X.; Wen, T.B.; Xia, H. Nine-membered osmacycles derived from metathesis reactions between Alkynes and an osmafuran. Organometallics 2009, 28, 1524–1533. [Google Scholar] [CrossRef]
- Buil, M.L.; Esteruelas, M.A.; Goni, E.; Oliván, M.; Oñate, E. Displacement of phenyl and styryl ligands by benzophenone imine and 2-vinylpyridine on Ruthenium and Osmium. Organometallics 2006, 25, 3076–3083. [Google Scholar] [CrossRef]
- Clark, A.M.; Rickard, C.E.F.; Roper, W.R.; Wright, L.J. Electrophilic substitution reactions at the phenyl ring of the chelated 2-(2‘-Pyridyl)phenyl ligand bound to Ruthenium(II) or Osmium(II). Organometallics 1999, 18, 2813–2820. [Google Scholar] [CrossRef]
- Bennett, M.A.; Clark, A.M.; Contel, M.; Rickard, C.E.F.; Roper, W.R.; Wright, L.J. Cyclometallated complexes of ruthenium and Osmium containing the o-C6H4PPh2 ligand. J. Organomet. Chem. 2000, 601, 299–304. [Google Scholar] [CrossRef]
- Rickard, C.E.F.; Roper, W.R.; Taylor, G.E.; Waters, J.M.; Wright, L.J. Coordinatively unsaturated σ-aryl complexes of Ruthenium(II) and Osmium(II). J. Organomet. Chem. 1990, 389, 375–388. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; López, A.M.; Oliván, M. Polyhydrides of platinum group metals: Nonclassical interactions and σ-bond activation reactions. Chem. Rev. 2016, 116, 8770–8847. [Google Scholar] [CrossRef] [Green Version]
- Barrio, P.; Castarlenas, R.; Esteruelas, M.A.; Lledós, A.; Maseras, F.; Oñate, E.; Tomàs, J. Reactions of a hexahydride-Osmium Complex with aromatic ketones: C−H activation versus C−F activation. Organometallics 2001, 20, 442–452. [Google Scholar] [CrossRef]
- Barrio, P.; Castarlenas, R.; Esteruelas, M.A.; Oñate, E. Triple C-H activation of a cycloalkyl ketone using an Osmium-hexahydride complex. Organometallics 2001, 20, 2635–2638. [Google Scholar] [CrossRef]
- Buil, M.L.; Esteruelas, M.A.; Garcés, K.; Oliván, M.; Oñate, E. Understanding the formation of N-H tautomers from α-substituted pyridines: Tautomerization of 2-ethylpyridine promoted by Osmium. J. Am. Chem. Soc. 2007, 129, 10998–10999. [Google Scholar] [CrossRef] [PubMed]
- Baya, M.; Eguillor, B.; Esteruelas, M.A.; Lledós, A.; Olivan, M.; Oñate, E. Coordination and rupture of methyl C(sp3)-H bonds in Osmium-polyhydride complexes with δ agostic interaction. Organometallics 2007, 26, 5140–5152. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Masamunt, A.B.; Oliván, M.; Oñate, E.; Valencia, M. Aromatic diosmatricyclic nitrogen-containing compounds. J. Am. Chem. Soc. 2008, 130, 11612–11613. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Fernández, I.; Gómez-Gallego, M.; Martín-Ortíz, M.; Molina, P.; Oliván, M.; Otón, F.; Sierra, M.A.; Valencia, M. Mono- and dinuclear Osmium N,N′-di- and tetraphenylbipyridyls and extended bipyridyls. Synthesis, structure and electrochemistry. Dalton Trans. 2013, 42, 3597. [Google Scholar] [CrossRef]
- Casarrubios, L.; Esteruelas, M.A.; Larramona, C.; Muntaner, J.G.; Oñate, E.; Sierra, M.A. 2-Azetidinones as precursors of pincer ligands: Preparation, structure, and spectroscopic properties of CC′N-Osmium complexes. Inorg. Chem. 2015, 54, 10998–11006. [Google Scholar] [CrossRef] [Green Version]
- Casarrubios, L.; Esteruelas, M.A.; Larramona, C.; Lledós, A.; Muntaner, J.G.; Oñate, E.; Ortuño, M.A.; Sierra, M.A. Mechanistic insight into the facilitation of β-lactam fragmentation through metal assistance. Chem. A Eur. J. 2015, 21, 16781–16785. [Google Scholar] [CrossRef] [PubMed]
- Eguillor, B.; Esteruelas, M.A.; Lezáun, V.; Oliván, M.; Oñate, E. Elongated dihydrogen versus compressed dihydride in Osmium complexes. Chem. A Eur. J. 2017, 23, 1526–1530. [Google Scholar] [CrossRef] [Green Version]
- Eguillor, B.; Esteruelas, M.A.; Fernández, I.; Gómez-Gallego, M.; Lledós, A.; Martín-Ortiz, M.; Oliván, M.; Oñate, E.; Sierra, M.A. Azole assisted C-H bond activation promoted by an Osmium-polyhydride: Discerning between N and NH. Organometallics 2015, 34, 1898–1910. [Google Scholar] [CrossRef] [Green Version]
- Buil, M.L.; Cardo, J.J.F.; Esteruelas, M.A.; Fernández, I.; Oñate, E. An entry to stable mixed phosphine-Osmium-NHC polyhydrides. Inorg. Chem. 2016, 55, 5062–5070. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Larramona, C.; Oñate, E. Osmium-mediated direct C-H bond activation at the 8-position of quinolines. Organometallics 2016, 35, 1597–1600. [Google Scholar] [CrossRef] [Green Version]
- Baya, M.; Esteruelas, M.A.; Oñate, E. Efficient concatenation of C═C reduction, C-H bond activation, and C-C and C-N coupling reactions on Osmium: Assembly of two allylamines and an allene. Organometallics 2010, 29, 6298–6307. [Google Scholar] [CrossRef]
- Crespo, O.; Eguillor, B.; Esteruelas, M.A.; Fernández, I.; García-Raboso, J.; Gómez-Gallego, M.; Martín-Ortiz, M.; Oliva, M.; Sierra, M.A. Synthesis and characterisation of [6]-azaosmahelicenes: The first d4-heterometallahelicenes. Chem. Commun. 2012, 48, 5328–5330. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Fernández, I.; Herrera, A.; Martín-Ortiz, M.; Martínez-Álvarez, R.; Oliván, M.; Oñate, E.; Sierra, M.A.; Valencia, M. Multiple C-H bond activation of phenyl-substituted pyrimidines and triazines promoted by an Osmium polyhydride: Formation of osmapolycycles with three, five, and eight fused rings. Organometallics 2010, 29, 976–986. [Google Scholar] [CrossRef]
- Eguillor, B.; Esteruelas, M.A.; Lezáun, V.; Oliván, M.; Oñate, E.; Tsai, J.-Y.; Xia, C. A capped octahedral MHC6 compound of a Platinum group metal. Chem. A Eur. J. 2016, 22, 9106–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, T.; Stephenson, T.A. Synthesis of triple halide-bridged arene complexes of Ruthenium(II) and Osmium(II). J. Organomet. Chem. 1981, 208, 369–387. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Soukharev, V.S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. Low-potential cyclometalated Osmium(II) mediators of glucose oxidase. Inorg. Chem. 2003, 42, 6598–6600. [Google Scholar] [CrossRef]
- Abbenhuis, H.C.L.; Pfeffer, M.; Sutter, J.P.; de Cian, A.; Fischer, J.; Ji, H.L.; Nelson, J.H. Carbon-carbon and carbon-nitrogen bond formation mediated by Ruthenium(II) complexes: Synthesis of (1H)-isoquinolinium derivatives. Organometallics 1993, 12, 4464–4472. [Google Scholar] [CrossRef]
- Ferstl, W.; Sakodinskaya, I.K.; Beydoun-Sutter, N.; Le Borgne, G.; Pfeffer, M.; Ryabov, A.D. Mechanism of alkyne insertion into the Ru-C bonds of orthoruthenated compounds featuring similarity of the Ru(II) and Pd(II) reactions. Organometallics 1997, 16, 411–418. [Google Scholar] [CrossRef]
- Pfeffer, M.; Sutter, J.-P.; Urriolabeitia, E.P. Ruthenium mediated carbon-carbon and carbon-nitrogen bond formation: Parameters governing the reactivity of the metal centre. Bull. Soc. Chim. Fr. 1997, 134, 947–954. [Google Scholar]
- Fernandez, S.; Pfeffer, M.; Ritleng, V.; Sirlin, C. An effective route to cycloruthenated N-ligands under mild conditions. Organometallics 1999, 18, 2390–2394. [Google Scholar] [CrossRef]
- Cerón-Camacho, R.; Morales-Morales, D.; Hernandez, S.; Le Lagadec, R.; Ryabov, A.D. Easy access to bio-inspired Osmium(II) complexes through electrophilic intramolecular C(sp2)-H bond cyclometalation. Inorg. Chem. 2008, 47, 4988–4995. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Oñate, E.; Palacios, A.; Tsai, J.Y.; Xia, C. Preparation of capped octahedral OsHC6 complexes by sequential carbon-directed C-H bond activation reactions. Organometallics 2016, 35, 2532–2542. [Google Scholar] [CrossRef] [Green Version]
- Bolje, A.; Hohloch, S.; van der Meer, M.; Košmrlj, J.; Sarkar, B. RuII, OsII, and IrII complexes with chelating pyridyl-mesoionic carbene ligands: Structural characterization and applications in transfer hydrogenation catalysis. Chem. A Eur. J. 2015, 21, 6756–6764. [Google Scholar] [CrossRef]
- Cerón-Camacho, R.; Hernández, S.; Le Lagadec, R.; Ryabov, A.D. Cyclometalated [Os(C-N)x(N-N)3-x]m+ mimetics of tris(2,2′-bipyridine)Osmium(II): Covering a 2 V potential range by known (x = 0, 1) and new (x = 2, 3) species (C-N = o-2-phenylpyridinato). Chem. Commun. 2011, 47, 2823–2825. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Camacho, R.; Le Lagadec, R.; Kurnikov, I.V.; Ryabov, A.D. A glance at the reactivity of osma(II)cycles [Os(C-N)x(bpy)3-x]m+ (x=0-3) covering a 1.8 V potential range toward peroxidase through monte carlo simulations (-C-N = o-2-phenylpyridinato, bpy=2,2′-bipyridine). J. Inorg. Biochem. 2014, 134, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Gusev, D.G.; Dolgushin, F.M.; Antipin, M.Y. Cyclometalated Osmium complexes containing a tridentate PCP ligand. Organometallics 2001, 20, 1001–1007. [Google Scholar] [CrossRef]
- Gusev, D.G.; Lough, A.J. Double C-H activation on Osmium and Ruthenium centers: Carbene vs. olefin products. Organometallics 2002, 21, 2601–2603. [Google Scholar] [CrossRef]
- Bolaño, T.; Castarlenas, R.; Esteruelas, M.A.; Oñate, E. Assembly of an allenylidene ligand, a terminal alkyne, and an acetonitrile molecule: Formation of osmacyclopentapyrrole derivatives. J. Am. Chem. Soc. 2006, 128, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.-Y.; Zhong, Y.-W. Monometallic Osmium(II) complexes with bis(N-methylbenzimidazolyl)benzene or -pyridine: A comparison study with Ruthenium(II) analogues. Inorg. Chem. 2013, 52, 6464–6472. [Google Scholar] [CrossRef]
- Wen, T.B.; Zhou, Z.Y.; Jia, G. Coupling reaction of phenylacetylene with OsHn(PPh3)(2,6-(PPh2CH2)2C6H3) (n = 1, 3). Organometallics 2003, 22, 4947–4951. [Google Scholar] [CrossRef]
- Barrio, P.; Esteruelas, M.A.; Lledós, A.; Oñate, E.; Tomàs, J. Influence of the cis ligand on the H-H separation and the rotation barrier of the dihydrogen in Osmium-elongated dihydrogen complexes containing an ortho-metalated ketone. Organometallics 2004, 23, 3008–3015. [Google Scholar] [CrossRef]
- Gong, L.; Chen, Z.; Lin, Y.; He, X.; Wen, T.B.; Xu, X.; Xia, H. Osmabenzenes from Osmacycles containing an η2-coordinated olefin. Chem. A Eur. J. 2009, 15, 6258–6266. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, M.; Cannas, D.M.; Just-Baringo, X.; Vitorica-Yrezabal, I.J.; Larrosa, I. Cyclometallated Ruthenium catalyst enables late-stage directed arylation of pharmaceuticals. Nat. Chem. 2018, 10, 724–731. [Google Scholar] [CrossRef]
- Kapdi, A.; Maiti, D. (Eds.) Palladacycles: Catalysis and Beyond; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128155059. [Google Scholar]
- Liu, S.H.; Lo, S.T.; Wen, T.B.; Williams, I.D.; Zhou, Z.Y.; Lau, C.P.; Jia, G. Reactions of hydrogen with Ruthenium and Osmium complexes containing tridentate ligands Cy2PCH2CH(CH2)2PCy2 and 2,6-(Ph2PCH2)2C6H3. Inorg. Chim. Acta 2002, 334, 122–130. [Google Scholar] [CrossRef]
- Wen, T.B.; Zhou, Z.Y.; Jia, G. Osmium-mediated hexamerization of phenylacetylene. Angew. Chem. Int. Ed. 2006, 45, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Baratta, W.; Ballico, M.; Baldino, S.; Chelucci, G.; Herdtweck, E.; Siega, K.; Magnolia, S.; Rigo, P. New benzo[h]quinoline-based ligands and their Pincer Ru and Os complexes for efficient catalytic transfer hydrogenation of carbonyl compounds. Chem. A Eur. J. 2008, 14, 9148–9160. [Google Scholar] [CrossRef]
- Baratta, W.; Ballico, M.; Chelucci, G.; Siega, K.; Rigo, P. Osmium(II) CNN pincer complexes as efficient catalysts for both asymmetric transfer and H2 hydrogenation of ketones. Angew. Chem. Int. Ed. 2008, 47, 4362–4365. [Google Scholar] [CrossRef] [PubMed]
- Baratta, W.; Fanfoni, L.; Magnolia, S.; Siega, K.; Rigo, P. Benzo[h]quinoline pincer Ruthenium and Osmium catalysts for hydrogenation of ketones. Eur. J. Inorg. Chem. 2010, 1419–1423. [Google Scholar] [CrossRef]
- Baratta, W.; Benedetti, F.; Del Zotto, A.; Fanfoni, L.; Felluga, F.; Magnolia, S.; Putignano, E.; Rigo, P. Chiral pincer Ruthenium and Osmium complexes for the fast and efficient hydrogen transfer reduction of ketones. Organometallics 2010, 29, 3563–3570. [Google Scholar] [CrossRef]
- Baratta, W.; Bossi, G.; Putignano, E.; Rigo, P. Pincer and diamine Ru and Os diphosphane complexes as efficient catalysts for the dehydrogenation of alcohols to ketones. Chem. A Eur. J. 2011, 17, 3474–3481. [Google Scholar] [CrossRef]
- Bossi, G.; Putignano, E.; Rigo, P.; Baratta, W. Pincer Ru and Os complexes as efficient catalysts for racemization and deuteration of alcohols. Dalton Trans. 2011, 40, 8986–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruver, B.C.; Adams, J.J.; Arulsamy, N.; Roddick, D.M. Acceptor pincer chemistry of Osmium: Catalytic alkane dehydrogenation by (CF3 PCP)Os(cod)(H). Organometallics 2013, 32, 6468–6475. [Google Scholar] [CrossRef]
- Bolaño, T.; Esteruelas, M.A.; Fernández, I.; Oñate, E.; Palacios, A.; Tsai, J.Y.; Xia, C. Osmium(II)-bis(dihydrogen) complexes containing Caryl, CNHC-chelate ligands: Preparation, bonding situation, and acidity. Organometallics 2015, 34, 778–789. [Google Scholar] [CrossRef] [Green Version]
- Bolaño, T.; Esteruelas, M.A.; Gay, M.P.; Oñate, E.; Pastor, I.M.; Yus, M. An acyl-NHC Osmium cooperative system: Coordination of small molecules and heterolytic B-H and O-H bond activation. Organometallics 2015, 34, 3902–3908. [Google Scholar] [CrossRef] [Green Version]
- Solinas, M.; Sechi, B.; Baldino, S.; Baratta, W.; Chelucci, G. Hydrogenation of imines catalyzed by 2-(Aminomethyl)pyridine-based ruthenium and Osmium complexes. ChemistrySelect 2016, 1, 2492–2497. [Google Scholar] [CrossRef]
- Buil, M.L.; Esteruelas, M.A.; Izquierdo, S.; Nicasio, A.I.; Oñate, E. N-H and C-H bond activations of an isoindoline promoted by Iridium- and Osmium-polyhydride complexes: A noninnocent bridge ligand for acceptorless and Base-Free Dehydrogenation of secondary alcohols. Organometallics 2020, 39, 2719–2731. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, Z.; Shen, X.; Ivlev, S.; Meggers, E. Asymmetric catalysis with a chiral-at-Osmium complex. Chem. Commun. 2020, 56, 7714–7717. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, L.A.; Huo, H.; Shen, X.; Harms, K.; Gong, L.; Meggers, E. Asymmetric lewis acid catalysis directed by octahedral rhodium centrochirality. Chem. Sci. 2015, 6, 1094–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Tan, Y.; Harms, K.; Marsch, M.; Riedel, R.; Zhang, L.; Meggers, E. Octahedral Ruthenium complex with exclusive metal-centered chirality for highly effective asymmetric catalysis. J. Am. Chem. Soc. 2017, 139, 4322–4325. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, S.; Hong, Y.; Winterling, E.; Tan, Y.; Hemming, M.; Harms, K.; Houk, K.N.; Meggers, E. Non-C2-symmetric chiral-at-Ruthenium catalyst for highly efficient enantioselective intramolecular C(sp3)-H amidation. J. Am. Chem. Soc. 2019, 141, 19048–19057. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Lugo, C.; Le Lagadec, R.; Ryabov, A.D.; Valverde, G.C.; Morales, S.L.; Alexandrova, L. “Living” radical polymerization of styrene catalyzed by cyclometalated Ruthenium(II) complexes bearing nonlabile ligands. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 3814–3828. [Google Scholar] [CrossRef]
- Ryabov, A.D. Transition metal chemistry of glucose oxidase, horseradish peroxidase, and related enzymes. Adv. Inorg. Chem. 2004, 55, 201–269. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Sukbarev, V.S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. New synthesis and new bio-application of cyclometalated Ruthenium(II) complexes for fast mediated electron transfer with peroxidase and glucose oxidase. Inorg. Chem. 2001, 40, 6529–6532. [Google Scholar] [CrossRef]
- Le Lagadec, R.; Rubio, L.; Alexandrova, L.; Toscano, R.A.; Ivanova, E.V.; Meškys, R.; Laurinavičius, V.; Pfeffer, M.; Ryabov, A.D. Cyclometalated N,N-dimethylbenzylamine Ruthenium(II) complexes [Ru(C6HR1R2R3-o-CH2NMe2)(bpy)(RCN)2]PF6 for bioapplications: Synthesis, characterization, crystal structures, redox properties, and reactivity toward PQQ-dependent glucose dehydrogenase. J. Organomet. Chem. 2004, 689, 4820–4832. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Kurova, V.S.; Ivanova, E.V.; Le Lagadec, R.; Alexandrova, L. Redox mediation and photomechanical oscillations involving photosensitive cyclometalated Ru(II) complexes, glucose oxidase, and peroxidase. Anal. Chem. 2005, 77, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Saji, T.; Aoyagui, S. Polarographic studies on bipyridines complexes. II. Correlation between charge-transfer frequencies and oxidation potentials of tris (2,2′-bipyridine) complexes of iron, Ruthenium, Osmium, Cobalt and Chromium. J. Electroanal. Chem. 1975, 60, 1–10. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Cerón-Camacho, R.; Saavedra-Díaz, O.; Denardo, M.A.; Ghosh, A.; Le Lagadec, R.; Collins, T.J. TAML activator-based amperometric analytical devices as alternatives to peroxidase biosensors. Anal. Chem. 2012, 84, 9096–9100. [Google Scholar] [CrossRef] [PubMed]
- Demas, J.N.; DeGraff, Β.A. Design and applications of highly luminescent transition metal complexes. Anal. Chem. 1991, 63, 829A–837A. [Google Scholar] [CrossRef]
- Bhaumik, C.; Das, S.; Maity, D.; Baitalik, S. Luminescent bis-tridentate Ruthenium(II) and Osmium(II) complexes based on terpyridyl-imidazole ligand: Synthesis, structural characterization, photophysical, electrochemical, and solvent dependence studies. Dalton Trans. 2012, 41, 2427–2438. [Google Scholar] [CrossRef]
- Wilkinson, A.J.; Puschmann, H.; Howard, J.A.K.; Foster, C.E.; Williams, J.A.G. Luminescent complexes of Iridium(III) containing N∧C∧N-coordinating terdentate ligands. Inorg. Chem. 2006, 45, 8685–8699. [Google Scholar] [CrossRef] [PubMed]
- Wadman, S.H.; Lutz, M.; Tooke, D.M.; Spek, A.L.; František, H.; Havenith, R.W.A.; Van Klink, G.P.M.; Van Koten, G. Consequences of N, C, N′- and C, N, N′-coordination modes on electronic and photophysical properties of cyclometalated aryl ruthenium(II) complexes. Inorg. Chem. 2009, 48, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Demovič, L.; Kellö, V.; Urban, M. Role of relativity in energy pattern of low-lying terms of Fe, Ru and Os. Comput. Chem. 2016, 1084, 157–161. [Google Scholar] [CrossRef]
- Woźna, A.; Kapturkiewicz, A.; Angulo, G. Synthesis and characterization of heteroleptic cyclometalated divalent Osmium Os[P(C6H5)3]2(CO)(N∩C−)Cl complexes. Inorg. Chem. Commun. 2013, 37, 26–29. [Google Scholar] [CrossRef]
- Chung, L.H.; Lo, H.S.; Ng, S.W.; Ma, D.L.; Leung, C.H.; Wong, C.Y. Luminescent Iridium(III) complexes supported by N-heterocyclic carbene-based C^C^C-pincer ligands and aromatic diimines. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.H.; Chan, S.C.; Lee, W.C.; Wong, C.Y. Emissive Osmium(II) complexes supported by N-heterocyclic carbene-based C^C^C-pincer ligands and aromatic diimines. Inorg. Chem. 2012, 51, 8693–8703. [Google Scholar] [CrossRef]
- Alabau, R.G.; Eguillor, B.; Esler, J.; Esteruelas, M.A.; Oliván, M.; Oñate, E.; Tsai, J.-Y.; Xia, C. CCC-pincer-NHC Osmium complexes: New types of blue-green emissive neutral compounds for organic light-emitting devices (OLEDs). Organometallics 2014, 33, 5582–5596. [Google Scholar] [CrossRef]
- Alabau, R.G.; Esteruelas, M.A.; Oliván, M.; Oñate, E.; Palacios, A.U.; Tsai, J.Y.; Xia, C. Osmium(II) complexes containing a dianionic CCCC-donor tetradentate ligand. Organometallics 2016, 35, 3981–3995. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.-J.; Shao, J.-Y.; Gong, Z.-L.; Zhong, Y.-W. Cyclometalated Osmium-amine electronic communication through the p-oligophenylene Wire. Inorg. Chem. 2015, 54, 10776–10784. [Google Scholar] [CrossRef]
- Castro-Rodrigo, R.; Esteruelas, M.A.; Gómez-Bautista, D.; Lezáun, V.; López, A.M.; Oliván, M.; Oñate, E. Influence of the bite angle of dianionic C,N,C-pincer ligands on the chemical and photophysical properties of Iridium(III) and Osmium(IV) hydride complexes. Organometallics 2019, 38, 3707–3718. [Google Scholar] [CrossRef]
- Alabau, R.G.; Esteruelas, M.A.; Oliván, M.; Oñate, E. Preparation of phosphorescent Osmium(IV) complexes with N,N′,C- and C,N,C′-pincer ligands. Organometallics 2017, 36, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Zych, D.; Slodek, A.; Matuszczyk, D.; Golba, S. Comprehensive study of mononuclear Osmium complexes with various pyrene ligands. Eur. J. Inorg. Chem. 2018, 5117–5128. [Google Scholar] [CrossRef]
- Zych, D. 1,3-Di(hetero)aryl-7-substituted pyrenes—An undiscovered area of important pyrene derivatives. Proceedings 2019, 41, 28. [Google Scholar] [CrossRef] [Green Version]
- Zych, D.; Slodek, A.; Golba, S.; Krompiec, S. Cyclometalated Ruthenium, Osmium, and Iridium complexes bridged by an NCN-pyrene-NCN derivative-synthesis and comparison of optical, thermal, and electrochemical properties. Eur. J. Inorg. Chem. 2018, 1581–1588. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, H. Future potential of Osmium complexes as anticancer drug candidates, photosensitizers and organelle-targeted probes. Dalton Trans. 2018, 47, 14841–14854. [Google Scholar] [CrossRef] [PubMed]
- Omae, I. Applications of five-membered ring products of cyclometalation reactions as anticancer agents. Coord. Chem. Rev. 2014, 280, 84–95. [Google Scholar] [CrossRef]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Jia, P.; Ouyang, R.; Cao, P.; Tong, X.; Zhou, X.; Lei, T.; Zhao, Y.; Guo, N.; Chang, H.; Miao, Y.; et al. Review: Recent advances and future development of metal complexes as anticancer agents. J. Coord. Chem. 2017, 70, 2175–2201. [Google Scholar] [CrossRef]
- Alessio, E. (Ed.) Bioinorganic Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 9783527633104. [Google Scholar]
- Jaouen, G.; Salmain, M. (Eds.) Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging; John Wiley & Sons: Weinheim, Germany, 2015; ISBN1 3527335277. ISBN2 9783527335275. [Google Scholar]
- Casini, A.; Vessières, A.; Meier-Menches, S.M. (Eds.) Metal-Based Anticancer Agents; Royal Society of Chemistry: Cambridge, UK, 2019; ISBN 978-1-78801-406-9. [Google Scholar]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Alessio, E.; Messori, L. Anticancer drug candidates face-to-face: A case story in medicinal inorganic chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [Green Version]
- Browne, W.R.; Holder, A.A.; Lawrence, M.A.; Bullock, J.L., Jr.; Lilge, L. (Eds.) Ruthenium Complexes: Photochemical and Biomedical Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; ISBN1 9783527339570. ISBN2 9783527695225. [Google Scholar]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) compounds: Next-generation anticancer metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Gaiddon, C.; Pfeffer, M. The Fate of Cycloruthenated compounds: From C-H activation to innovative anticancer therapy. Eur. J. Inorg. Chem. 2017, 2017, 1639–1654. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Gong, L.; Kräling, K.; Gründler, K.; Frias, C.; Webster, R.D.; Meggers, E.; Prokop, A.; Xia, H. Unusual η2-allene osmacycle with apoptotic properties. ChemBioChem 2010, 11, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Boff, B.; Gaiddon, C.; Pfeffer, M. Cancer cell cytotoxicity of cyclometalated compounds obtained with Osmium(II) complexes. Inorg. Chem. 2013, 52, 2705–2715. [Google Scholar] [CrossRef]
- Licona, C.; Delhorme, J.-B.; Riegel, G.; Vidimar, V.; Cerón-Camacho, R.; Boff, B.; Venkatasamy, A.; Tomasetto, C.; Da Silva Figueiredo Celestino Gomes, P.; Rognan, D.; et al. Anticancer activity of Ruthenium and Osmium cyclometalated compounds: Identification of ABCB1 and EGFR as resistance mechanisms. Inorg. Chem. Front. 2020, 7, 678–688. [Google Scholar] [CrossRef]
- Riedl, C.A.; Flocke, L.S.; Hejl, M.; Roller, A.; Klose, M.H.M.M.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Introducing the 4-phenyl-1,2,3-triazole moiety as a versatile scaffold for the development of cytotoxic Ruthenium(II) and Osmium(II) arene cyclometalates. Inorg. Chem. 2017, 56, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Rono, C.K.; Chu, W.K.; Darkwa, J.; Meyer, D.; Makhubela, B.C.E. Triazolyl RuII, RhIII OsII, and IrIII complexes as potential anticancer agents: Synthesis, structure elucidation, cytotoxicity, and DNA model interaction studies. Organometallics 2019, 38, 3197–3211. [Google Scholar] [CrossRef]
- Ortega, E.; Yellol, J.G.; Rothemund, M.; Ballester, F.J.; Rodríguez, V.; Yellol, G.; Janiak, C.; Schobert, R.; Ruiz, J. A new C,N-cyclometalated Osmium(II) arene anticancer scaffold with a handle for functionalization and antioxidative properties. Chem. Commun. 2018, 54, 11120–11123. [Google Scholar] [CrossRef] [Green Version]
- Valencia, M.; Merinero, A.D.; Lorenzo-Aparicio, C.; Gómez-Gallego, M.; Sierra, M.A.; Eguillor, B.; Esteruelas, M.A.; Oliván, M.; Oñate, E. Osmium-promoted σ-bond activation reactions on nucleosides. Organometallics 2020, 39, 312–323. [Google Scholar] [CrossRef]
Figure 1.
Chelate coordination and cyclometalated complexes.

Figure 2.
Year progression of publications mentioning osmacycles (black) and ruthenacycles (grey) (retrieved December 2020, SciFinder with keywords: osmacycle, osmium AND cyclometalated, osmium AND cyclometallated, cyclometalated AND osmium, metalacycle AND osmium or ruthenacycle, ruthenium AND cyclometalated, cyclometallated AND ruthenium, cyclometalated AND ruthenium, metalacycle AND ruthenium).
Figure 2.
Year progression of publications mentioning osmacycles (black) and ruthenacycles (grey) (retrieved December 2020, SciFinder with keywords: osmacycle, osmium AND cyclometalated, osmium AND cyclometallated, cyclometalated AND osmium, metalacycle AND osmium or ruthenacycle, ruthenium AND cyclometalated, cyclometallated AND ruthenium, cyclometalated AND ruthenium, metalacycle AND ruthenium).

Figure 3.
Synthesis of C~O cyclometalated complexes.
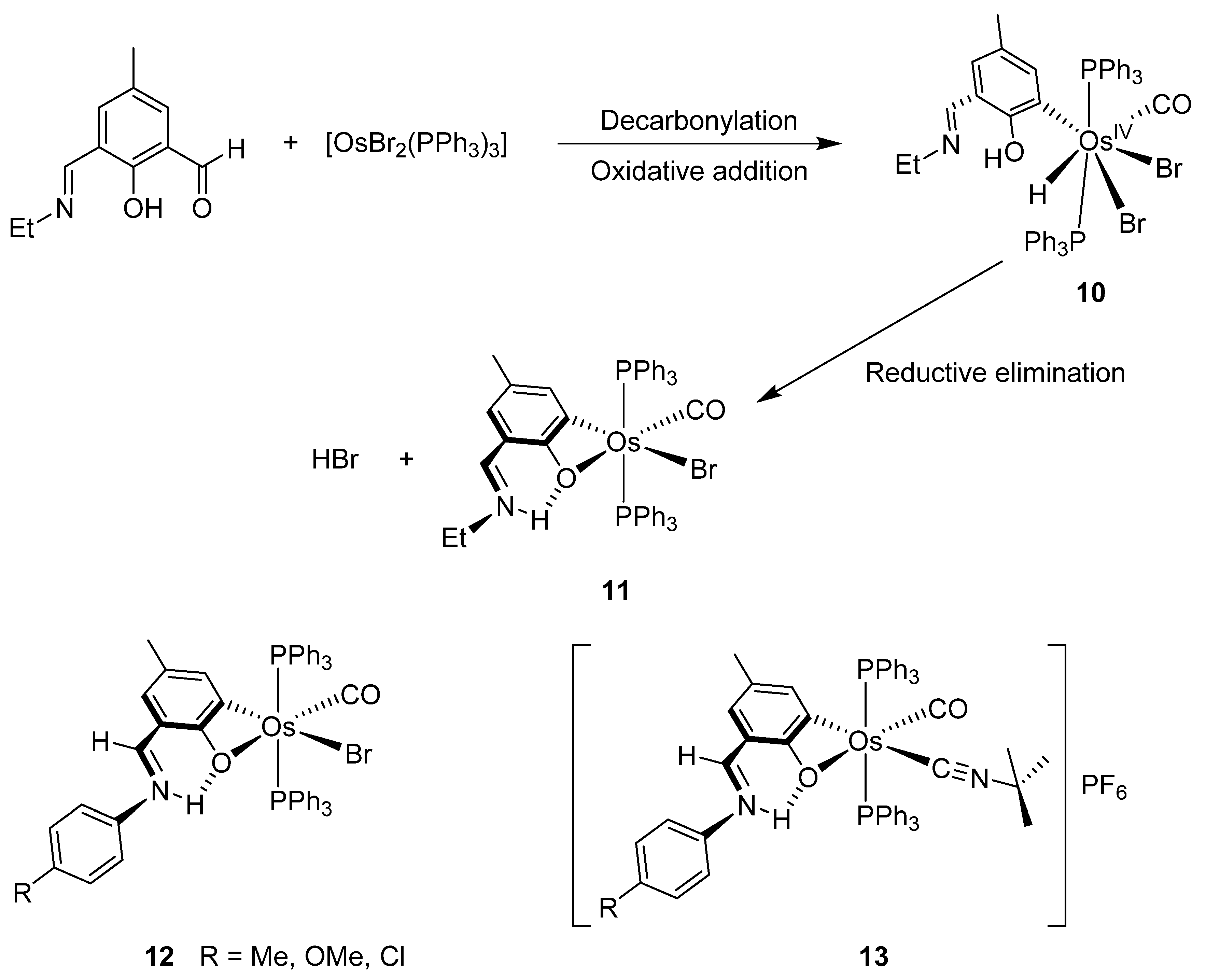
Figure 4.
Cyclometalated complexes derived from 2-vinylpyridine.

Figure 5.
Synthesis of C~N~O pincer complexes.

Figure 6.
Synthesis of P~C~P pincer complex from 1,3-(R2PCH2)2C6H4.
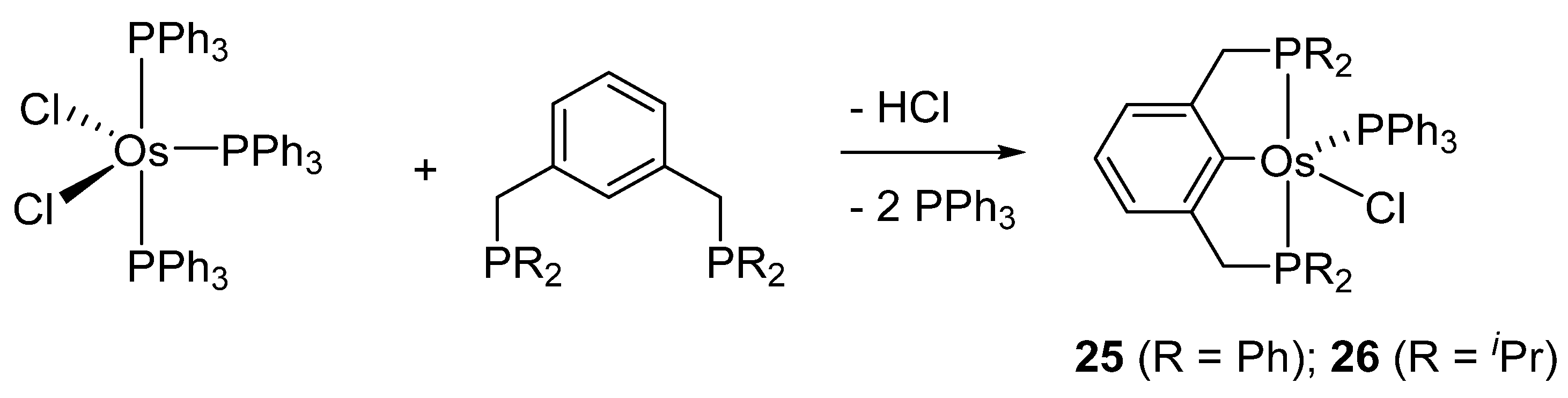
Figure 7.
Synthesis of P~C~P pincer complexes by cleavage of a C-C bond.
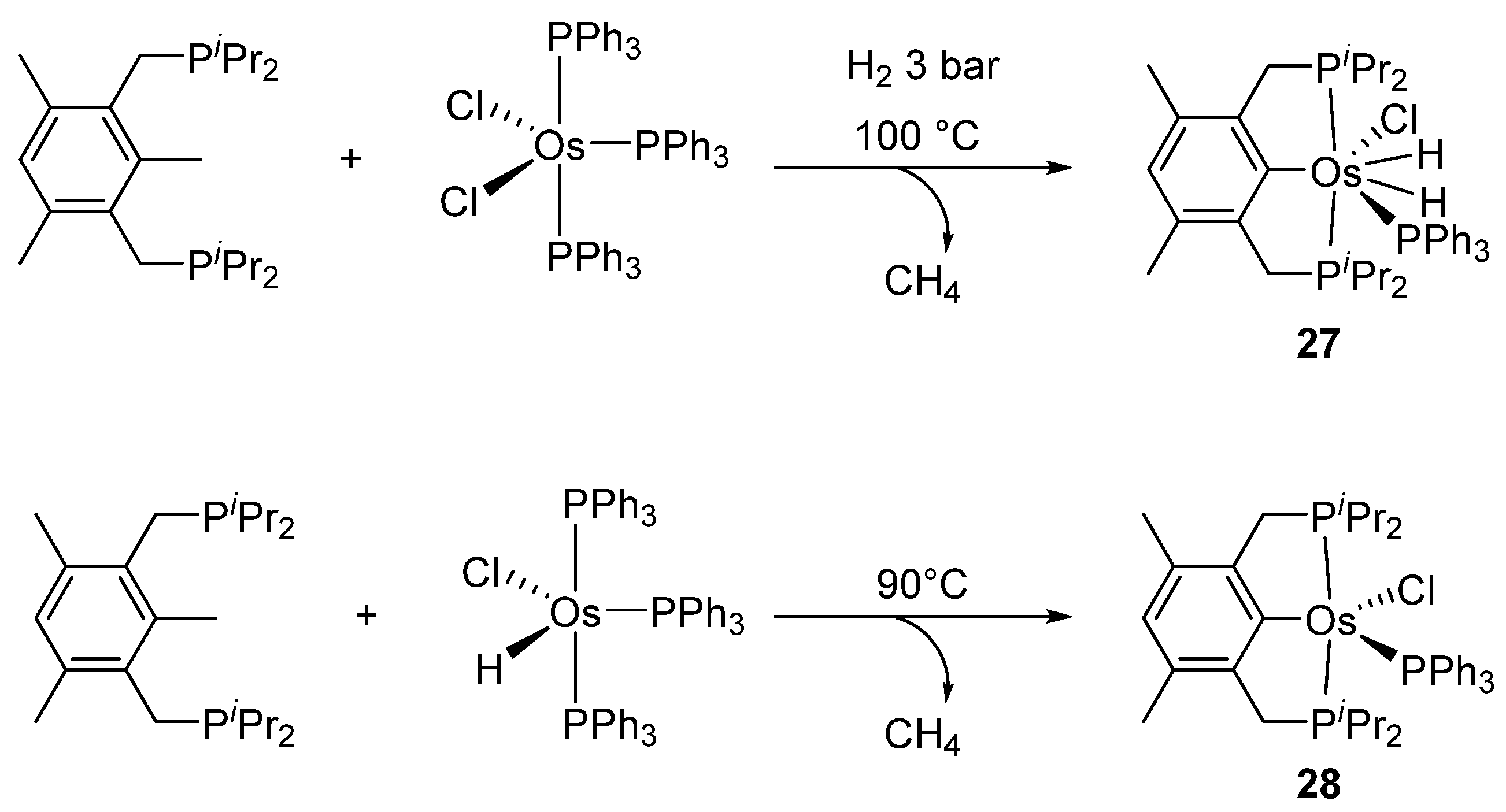
Figure 8.
Synthetic route for a series of P~C~P pincer complexes by a double C−H activation.

Figure 9.
Cyclometalated osmium complexes obtained by C–H bond activation of phenols and hydroxamic acids.
Figure 9.
Cyclometalated osmium complexes obtained by C–H bond activation of phenols and hydroxamic acids.

Figure 10.
Synthesis of bis-(P~C~P) pincer complexes from 1,3-bis(diphenyl-phosphinomethyl)benzene.
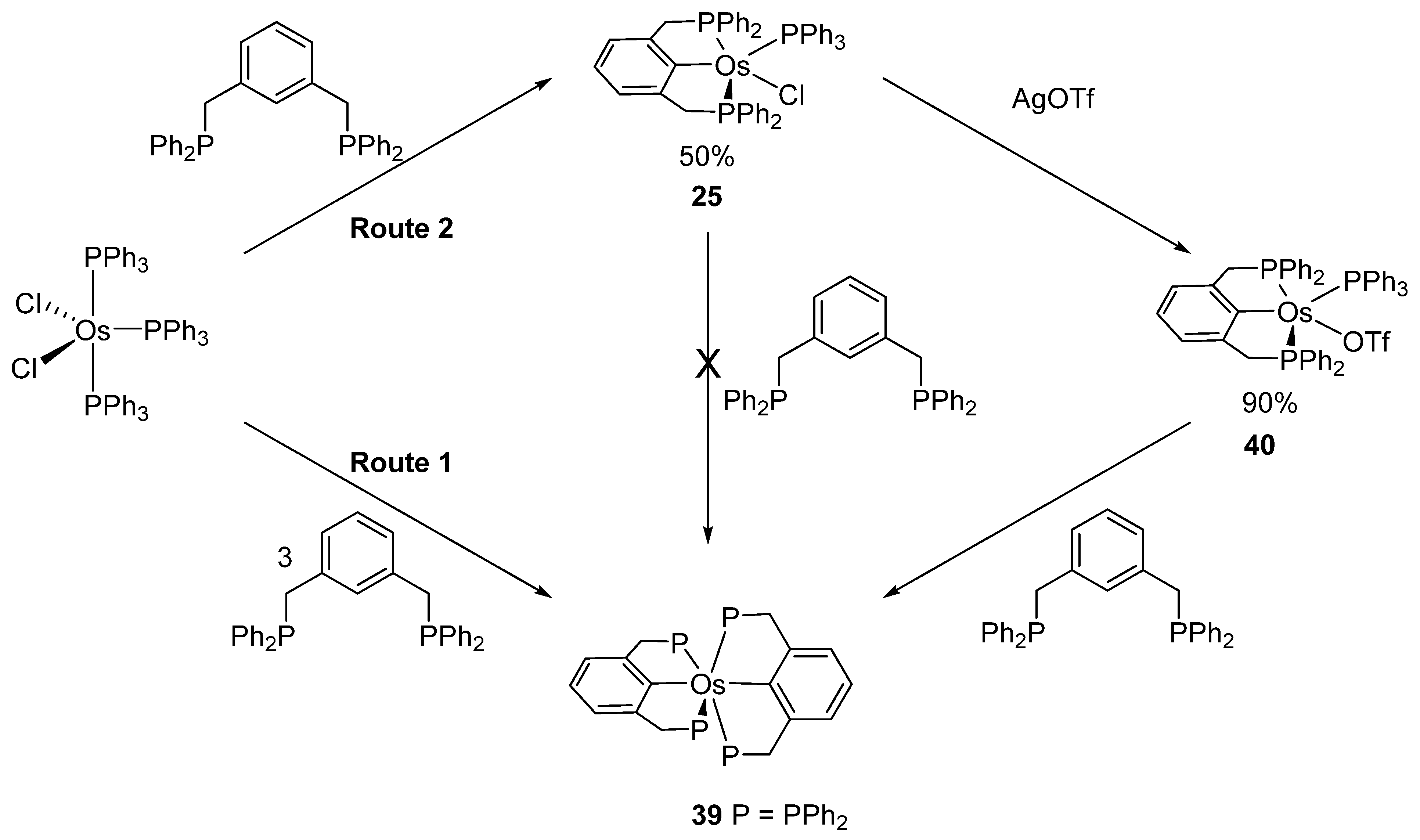
Figure 11.
Unusual osmacycles prepared by activation of alkynes.
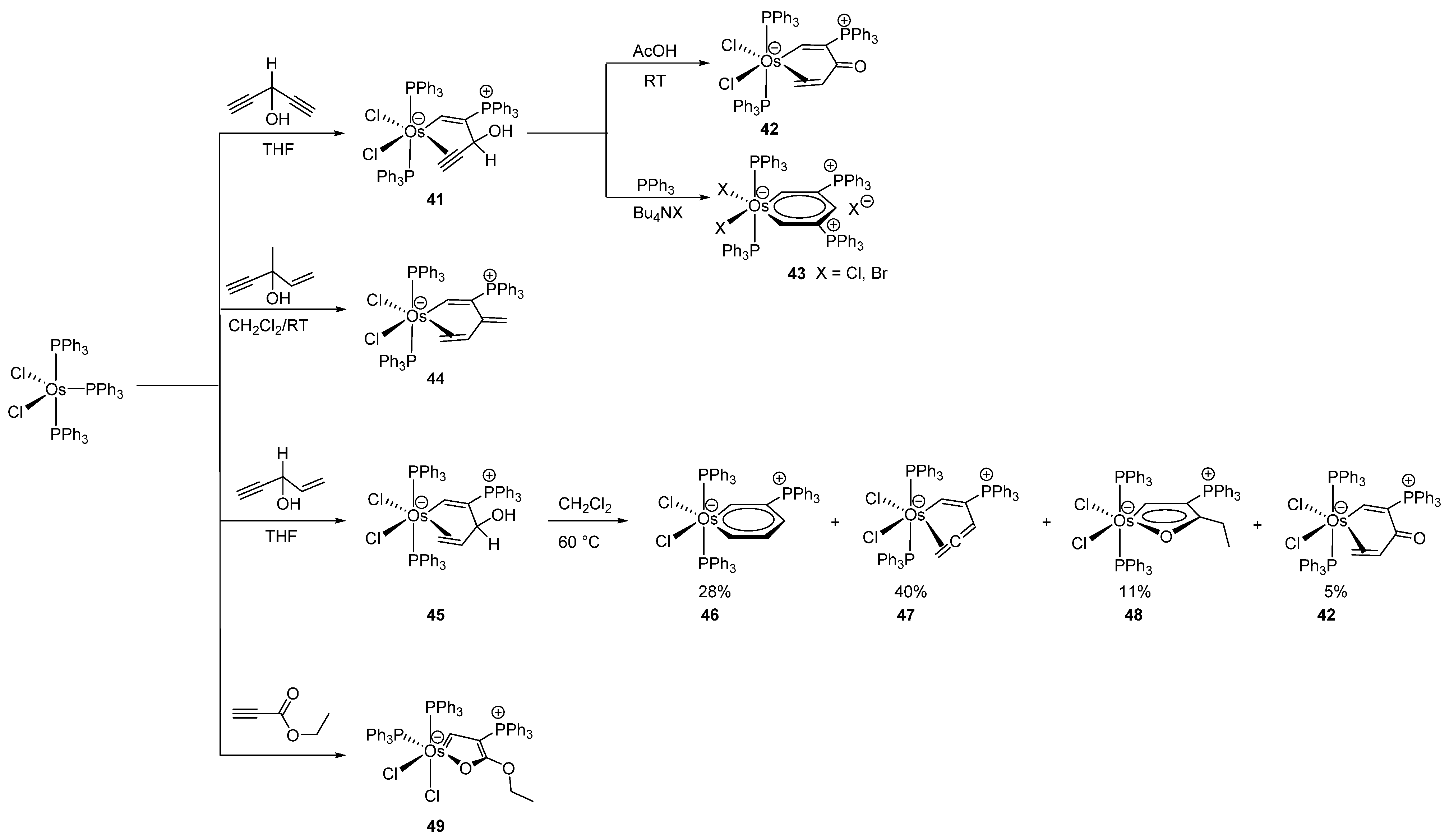
Figure 12.
Synthesis of cyclometalated complex by oxidative addition of the phenyl group of an imine and subsequent reductive elimination.
Figure 12.
Synthesis of cyclometalated complex by oxidative addition of the phenyl group of an imine and subsequent reductive elimination.

Figure 13.
Osmium(II) complexes with cyclometalated phenylpyridines.

Figure 14.
Use of organomercurial derivative to obtain cyclometalated complexes.
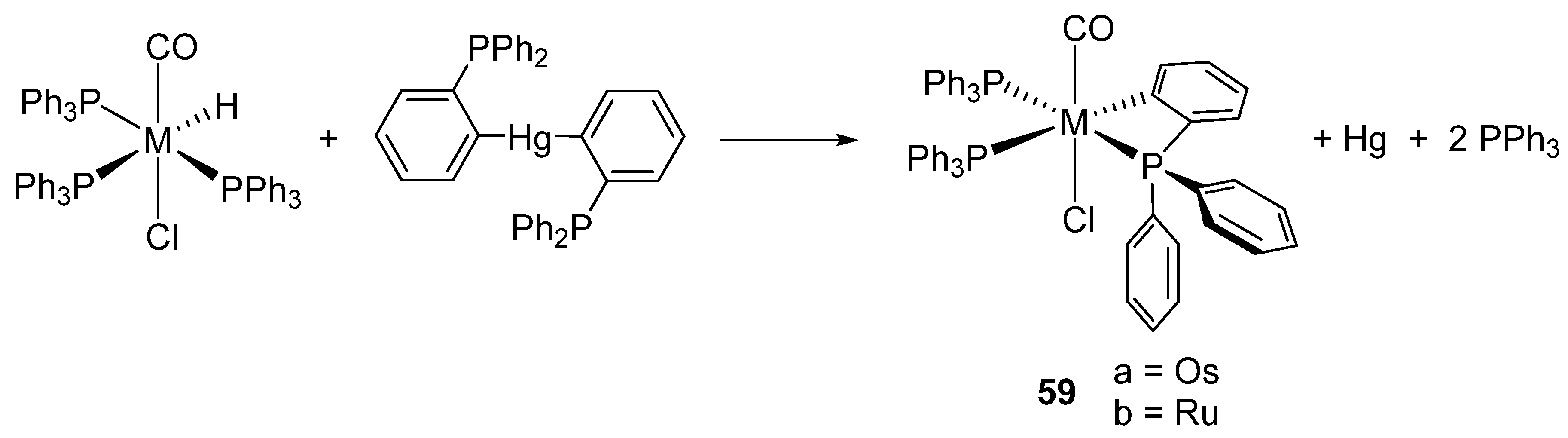
Figure 15.
Reaction between [OsH2Cl2(PiPr3)2] and allylamine.

Figure 16.
Cyclometalation via by C–H activation assisted by reductive elimination of hydride ligands.
Figure 16.
Cyclometalation via by C–H activation assisted by reductive elimination of hydride ligands.
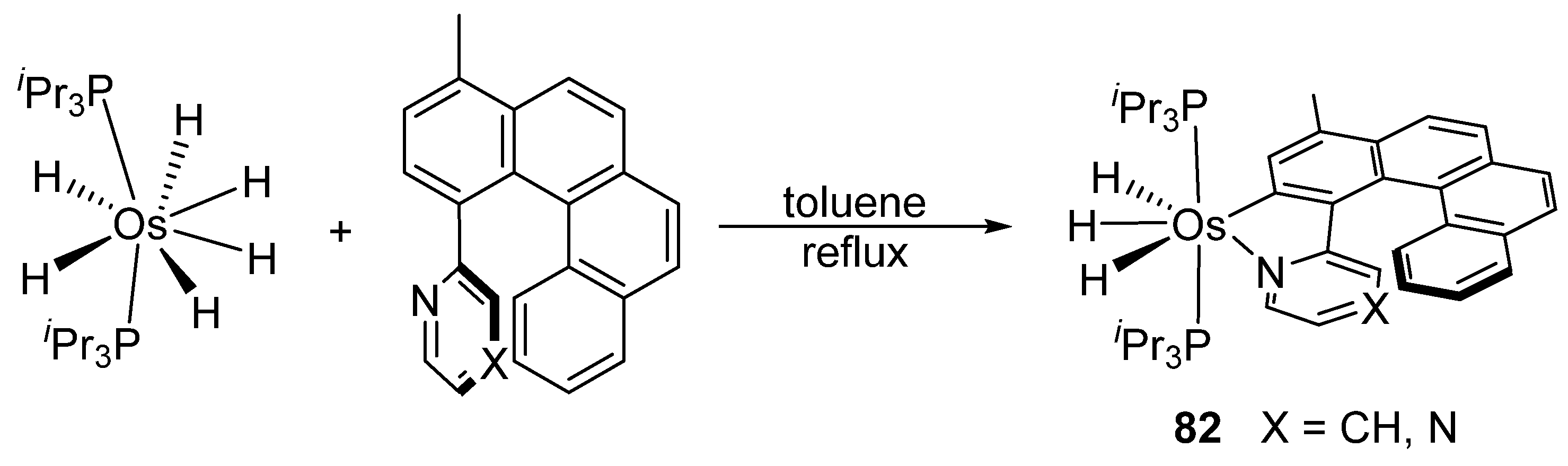
Figure 17.
Synthesis of bis-cyclometalated complexes from [OsH6(PiPr3)2].
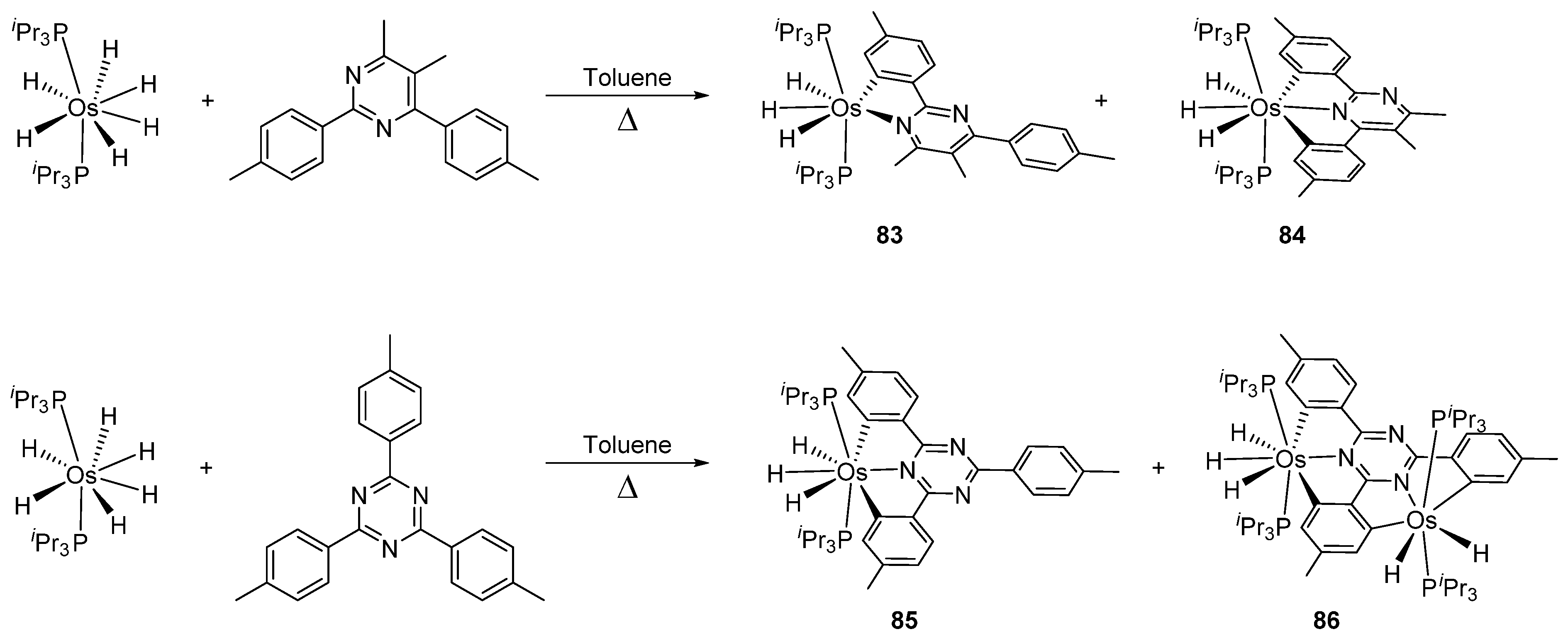
Figure 18.
Synthetic routes to tris-cyclometalated complexes from mono-cyclometalated derivatives.
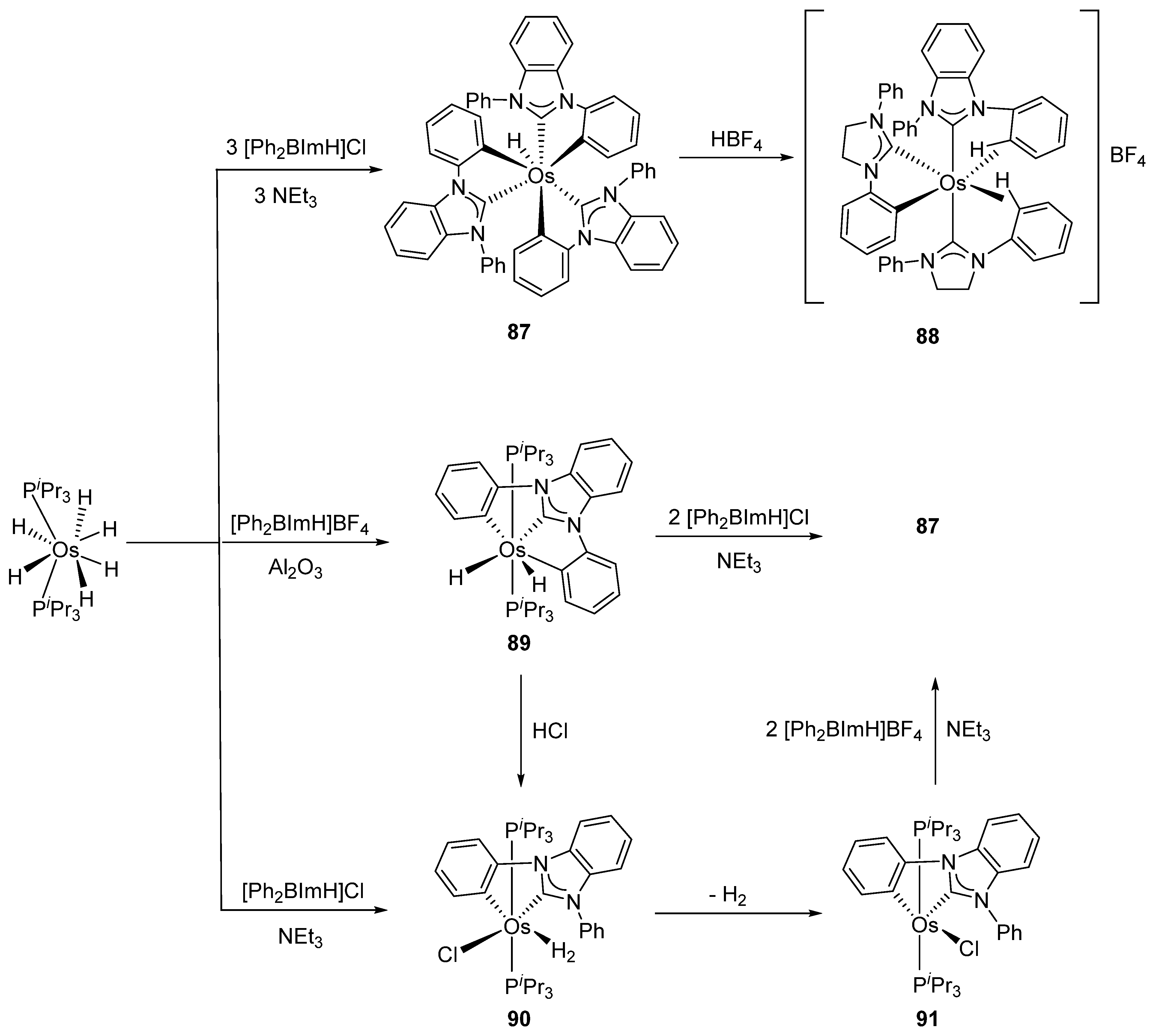
Figure 19.
Cyclometalated osmium complexes obtained from [Os(η6-C6H6)(μ-Cl)Cl]2.
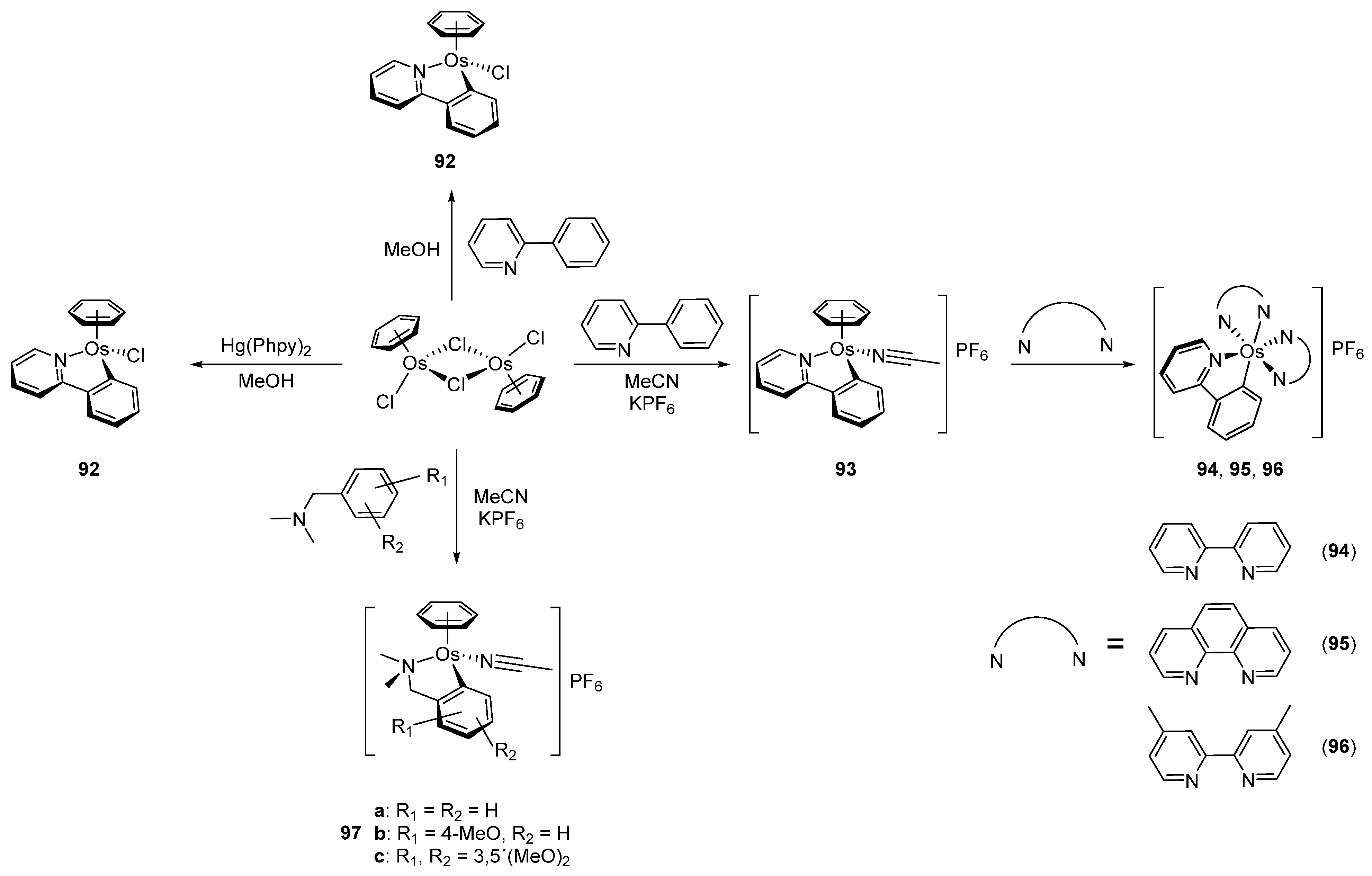
Figure 20.
Synthesis of mono-cyclometalated complexes through transmetalation reactions with silver(I) derivatives.
Figure 20.
Synthesis of mono-cyclometalated complexes through transmetalation reactions with silver(I) derivatives.
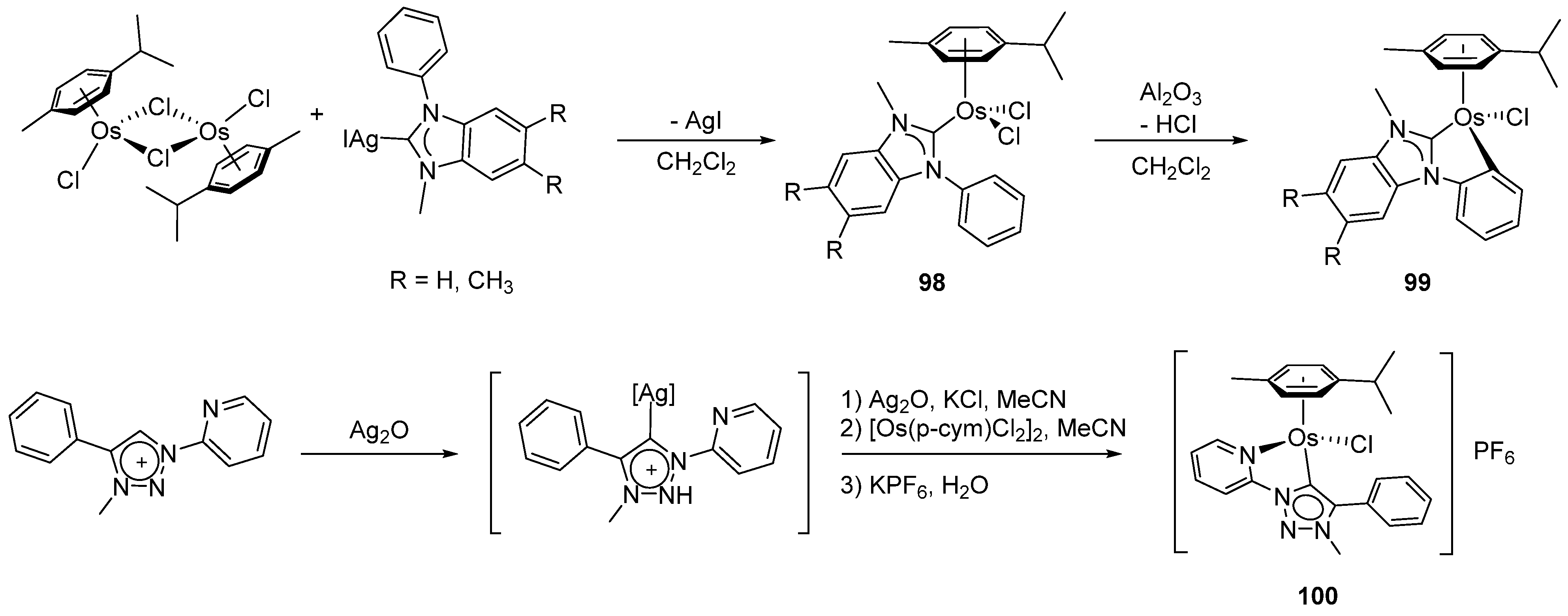
Figure 21.
Synthesis of bis- and tris-cyclometalated complexes (top) and cyclic voltammograms showing the influence of the number of Os−C bonds on the OsII/OsIII reduction potentials). Bottom: cyclic voltammograms of 103, 94, 101 and 102 (1 mM) in MeCN: 25 °C, scan rate 0.1 V s−1, 0.1 M nBu4NPF6, glassy carbon electrode. Inset shows a plot of the formal reduction potential vs. the number of C donors.
Figure 21.
Synthesis of bis- and tris-cyclometalated complexes (top) and cyclic voltammograms showing the influence of the number of Os−C bonds on the OsII/OsIII reduction potentials). Bottom: cyclic voltammograms of 103, 94, 101 and 102 (1 mM) in MeCN: 25 °C, scan rate 0.1 V s−1, 0.1 M nBu4NPF6, glassy carbon electrode. Inset shows a plot of the formal reduction potential vs. the number of C donors.

Figure 22.
Synthesis tris-cyclometalated complex from the osmium dimer precursor.

Figure 23.
Synthesis of osmium P~C~P pincer using [Et4N]2[OsCl6].

Figure 24.
Osmium P~C~P pincer complex obtained by double C−H bond activation.
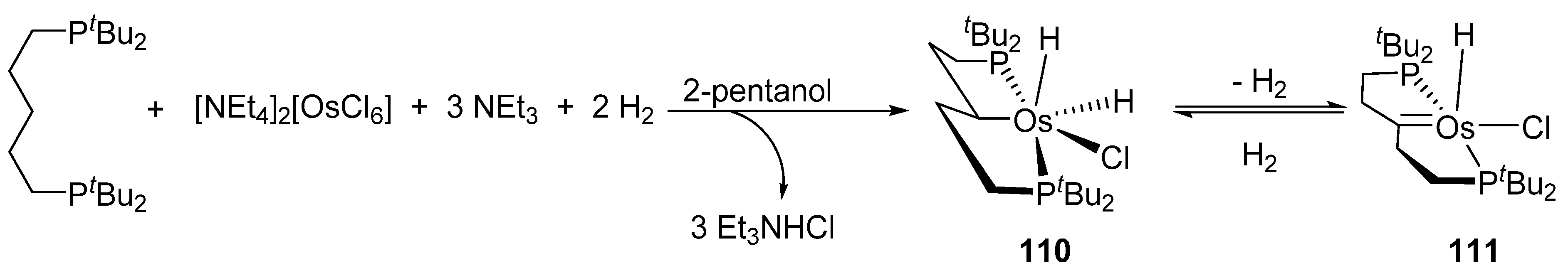
Figure 25.
Synthesis of an osmacyclopentapyrrole complex from allenylidene derivative.
Figure 26.
Synthesis of bis-pincer complexes.

Figure 27.
Reactions of four-membered metalacycles.
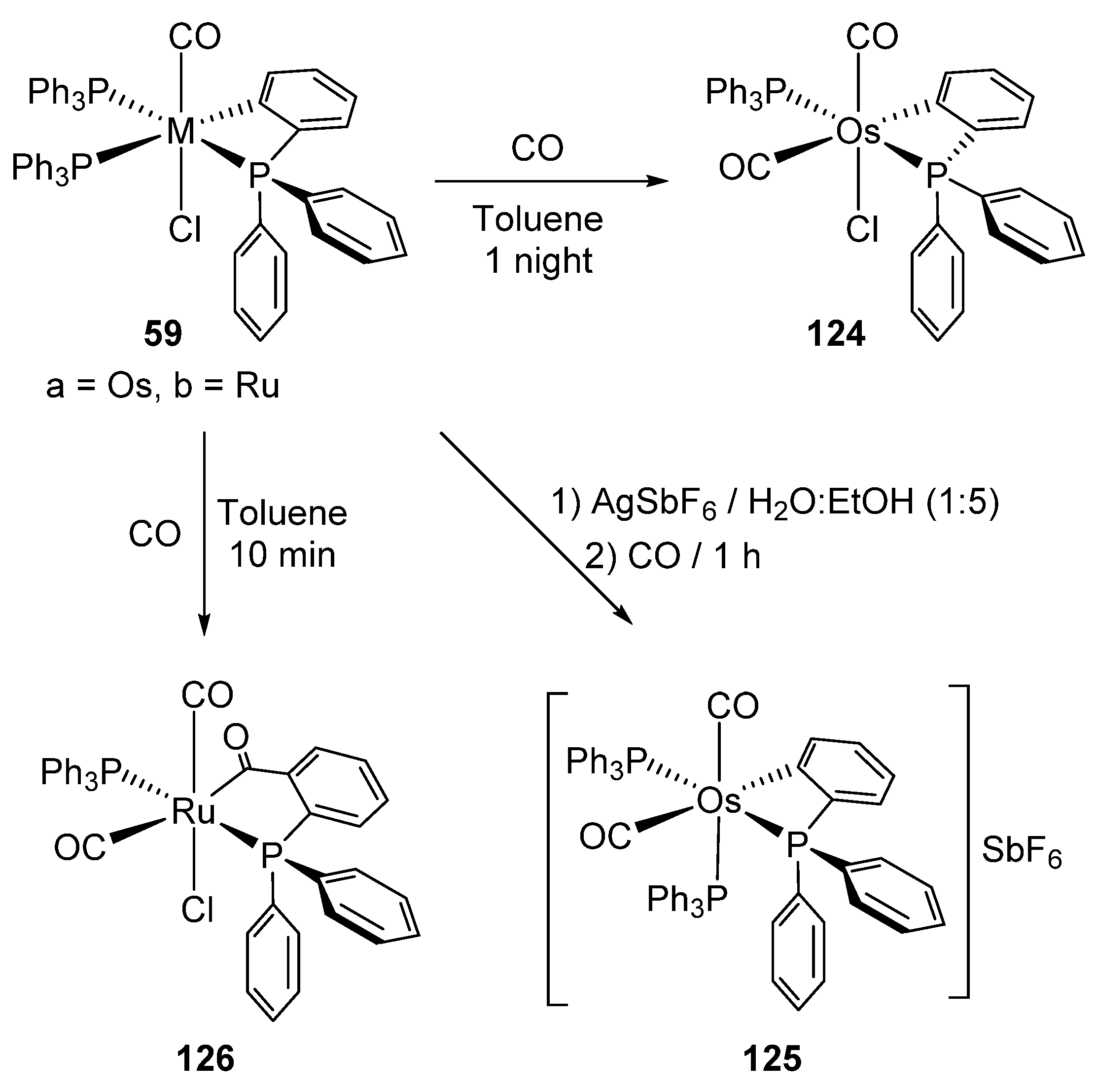
Figure 28.
Electrophilic substitution reactions at the phenyl ring of cyclometalated complex 51.

Figure 29.
Reactivity of P~C~P pincer complexes 108 and 109.
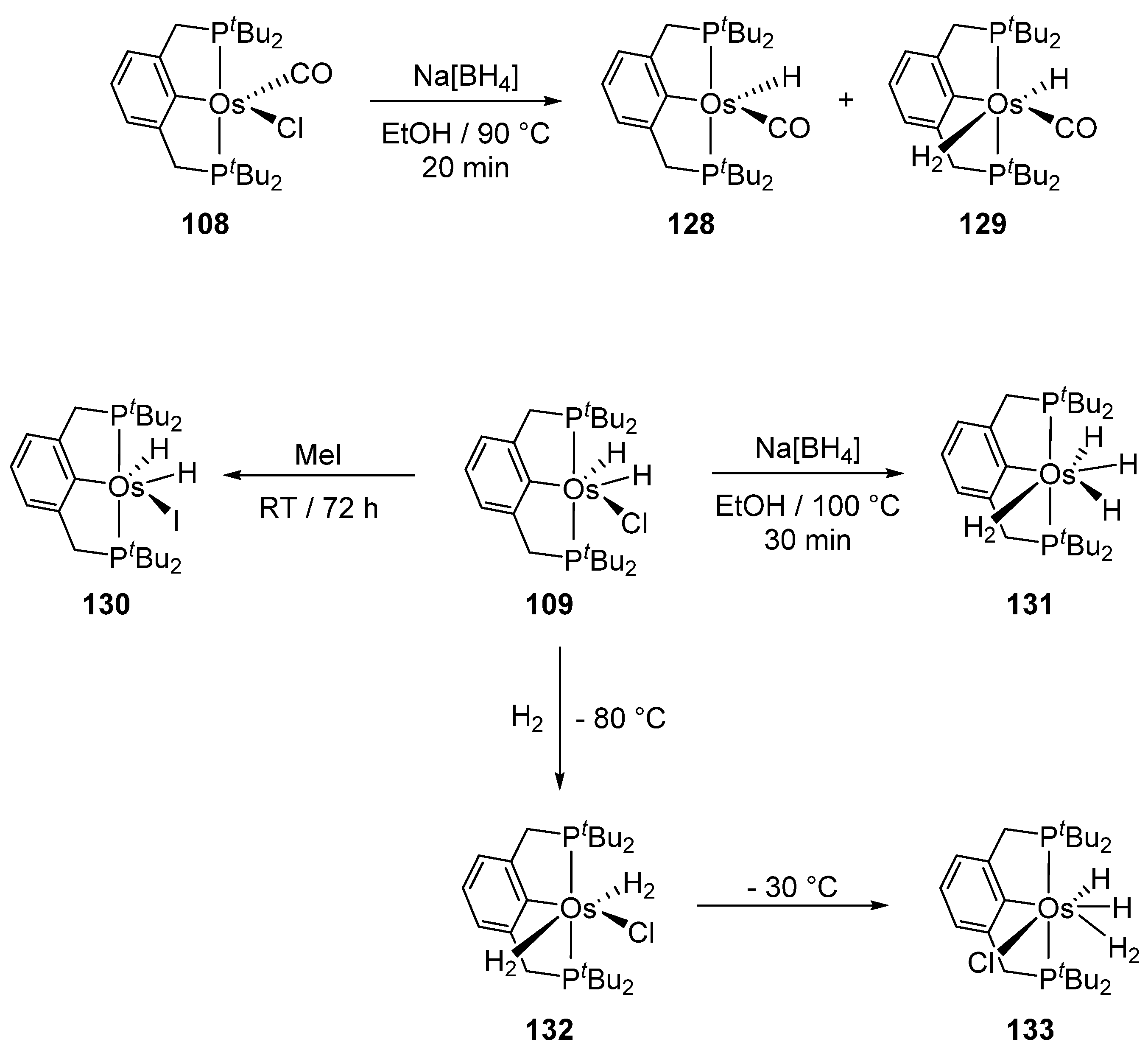
Figure 30.
Reactions between osmium P~C~P pincer complex 25 with alkynes.
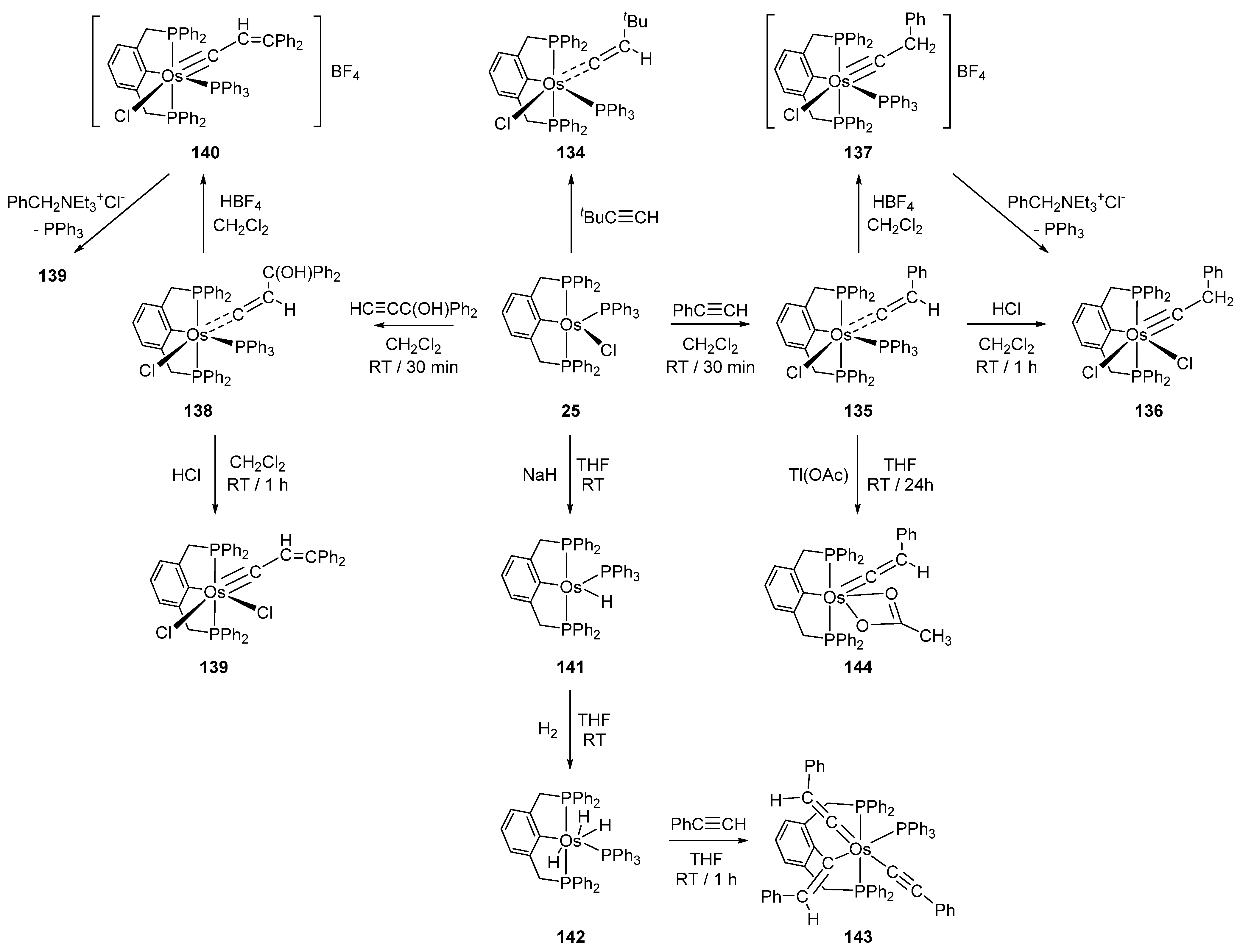
Figure 31.
Reactions of P~C~P pincer complex 26 with H2 and CO.
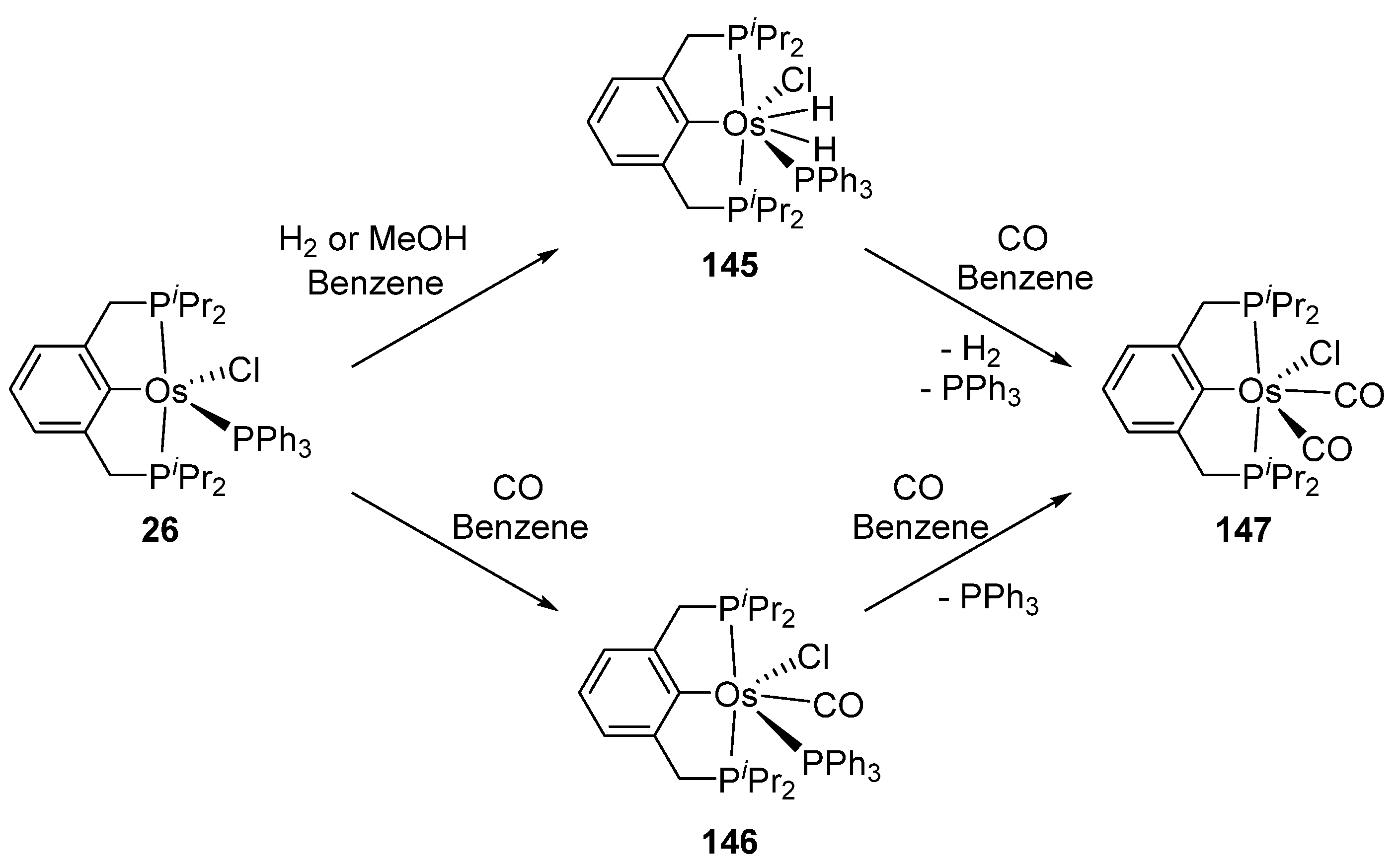
Figure 32.
Protonation of the cyclometalated fragment by HBF4.

Figure 33.
Reactions of hydride complexes with HBF4.
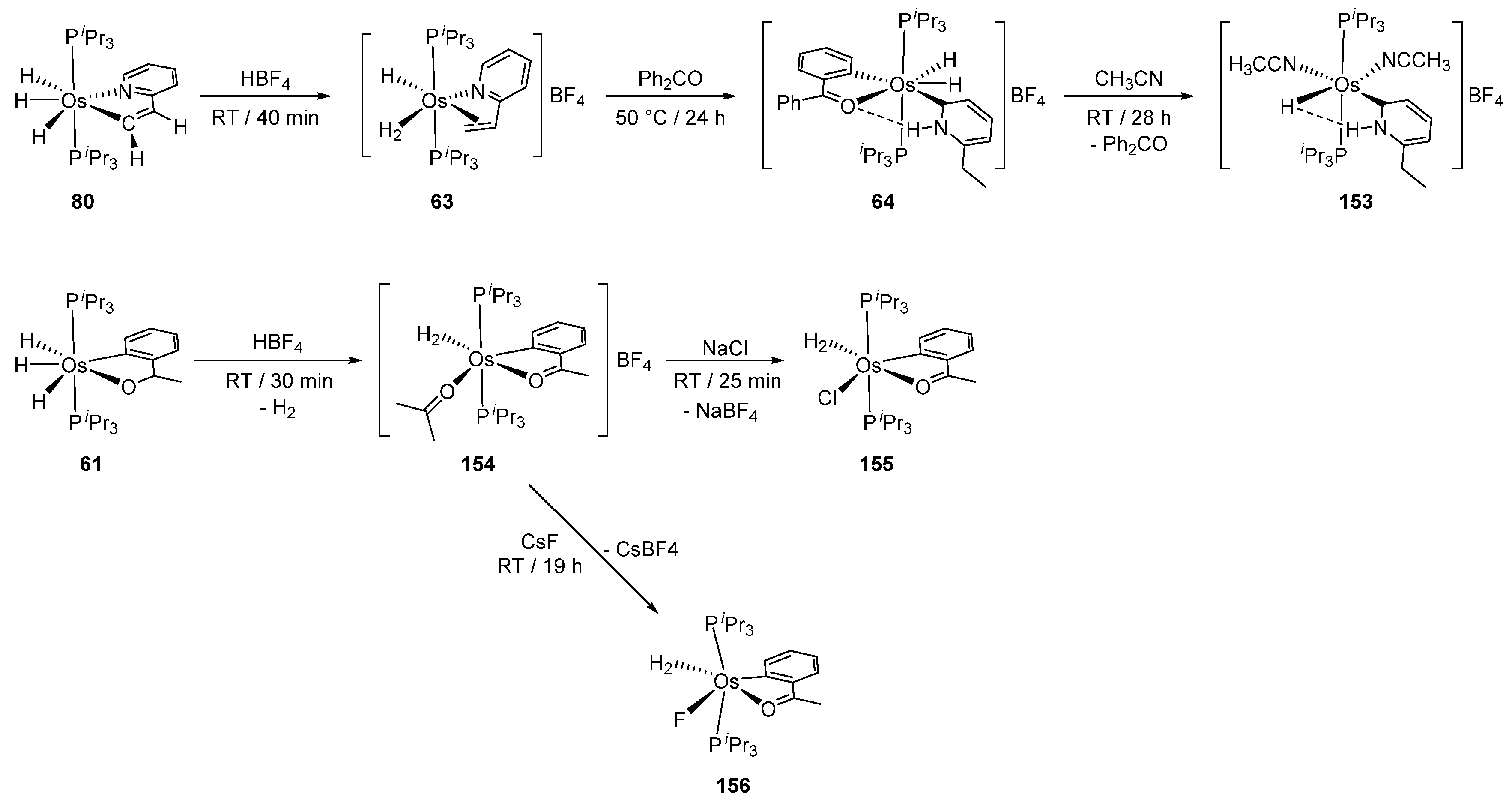
Figure 34.
Synthetic routes to octahedral cyclometalated derivatives by ligand substitution starting from piano-stool complex 93.
Figure 34.
Synthetic routes to octahedral cyclometalated derivatives by ligand substitution starting from piano-stool complex 93.
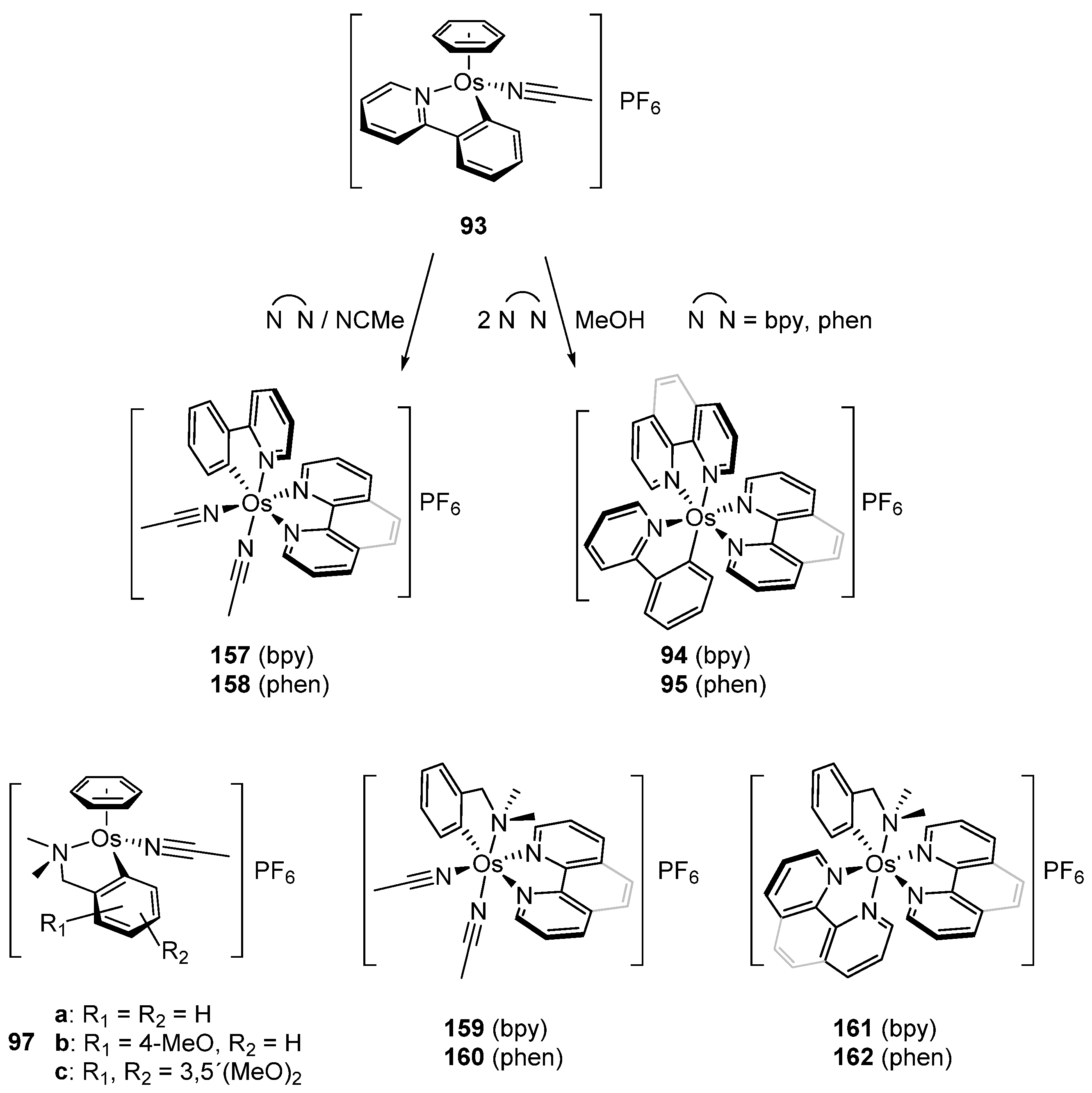
Figure 35.
Studies of reactivity of complex 44.

Figure 36.
Ring-expansion reaction from a five-membered to a nine-membered osmacycle.

Figure 37.
Coordination of dihydrogen to a pincer complex.
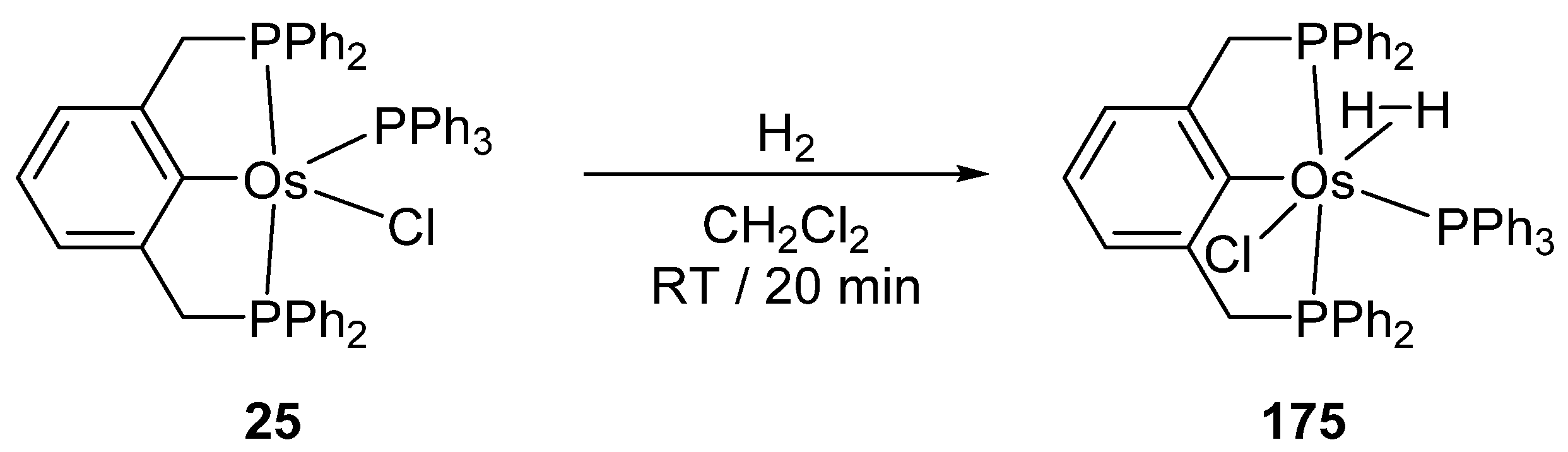
Figure 38.
Hexamerization of phenylacetylene at osmium pincer complex.
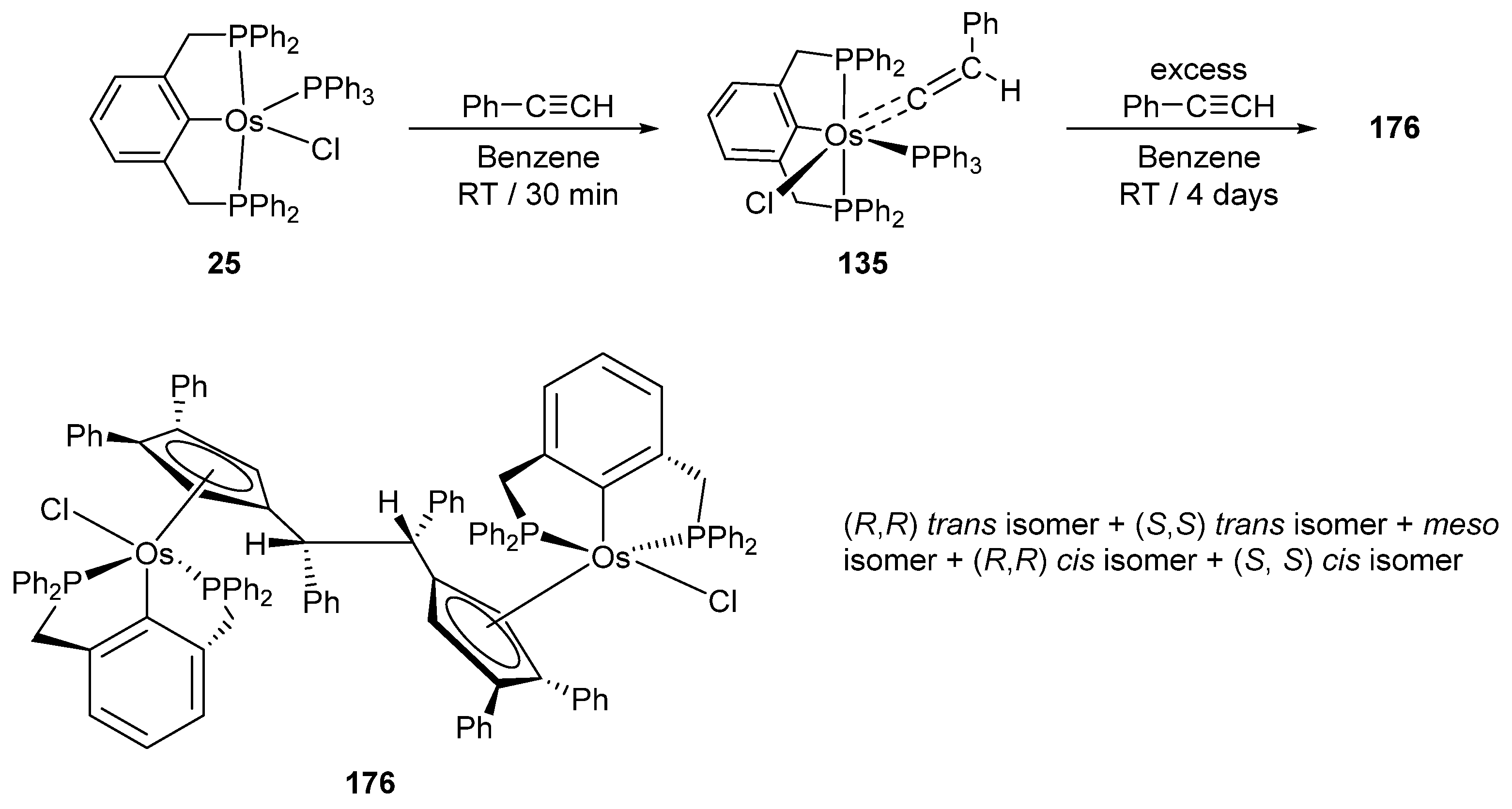
Figure 39.
Hydrogenation reactions catalyzed by osmium pincer complexes.
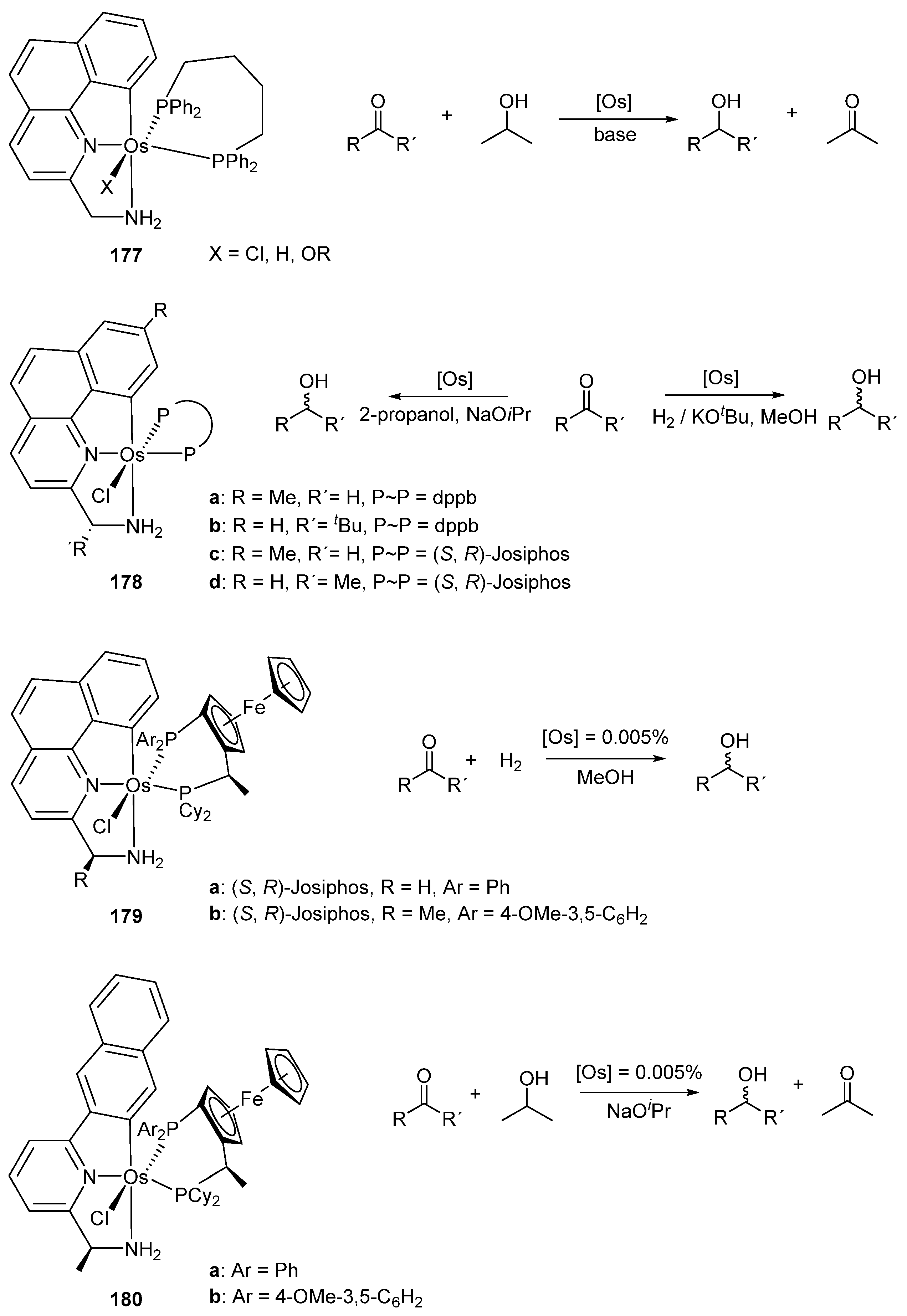
Figure 40.
C~N~N pincer complexes used for racemization of chiral alcohols.
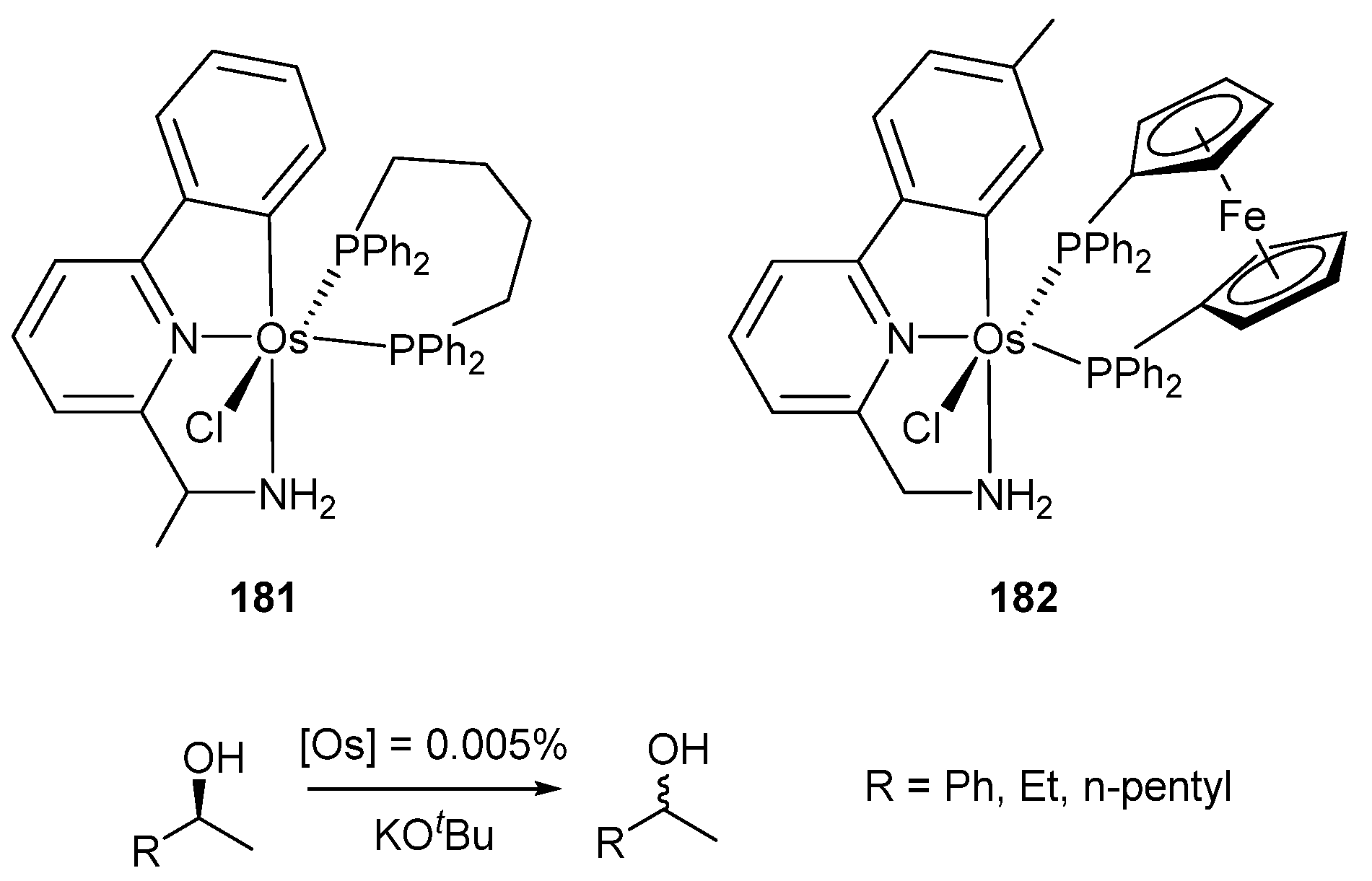
Figure 41.
Alkane dehydrogenation catalyzed by a fluorinated P~C~P pincer complex.
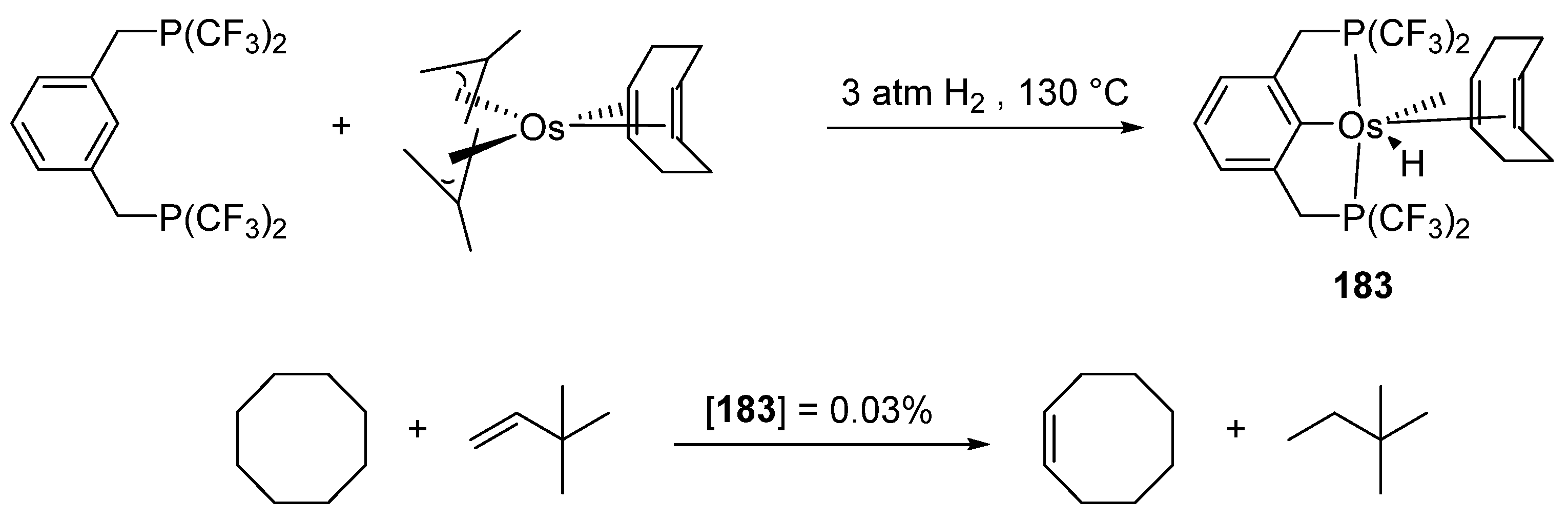
Figure 42.
Examples of hydride complexes used in the alcoholysis and hydrolysis of pinacolborane.

Figure 43.
Hydrogenation of imines catalyzed by C~N~N pincer complexes.
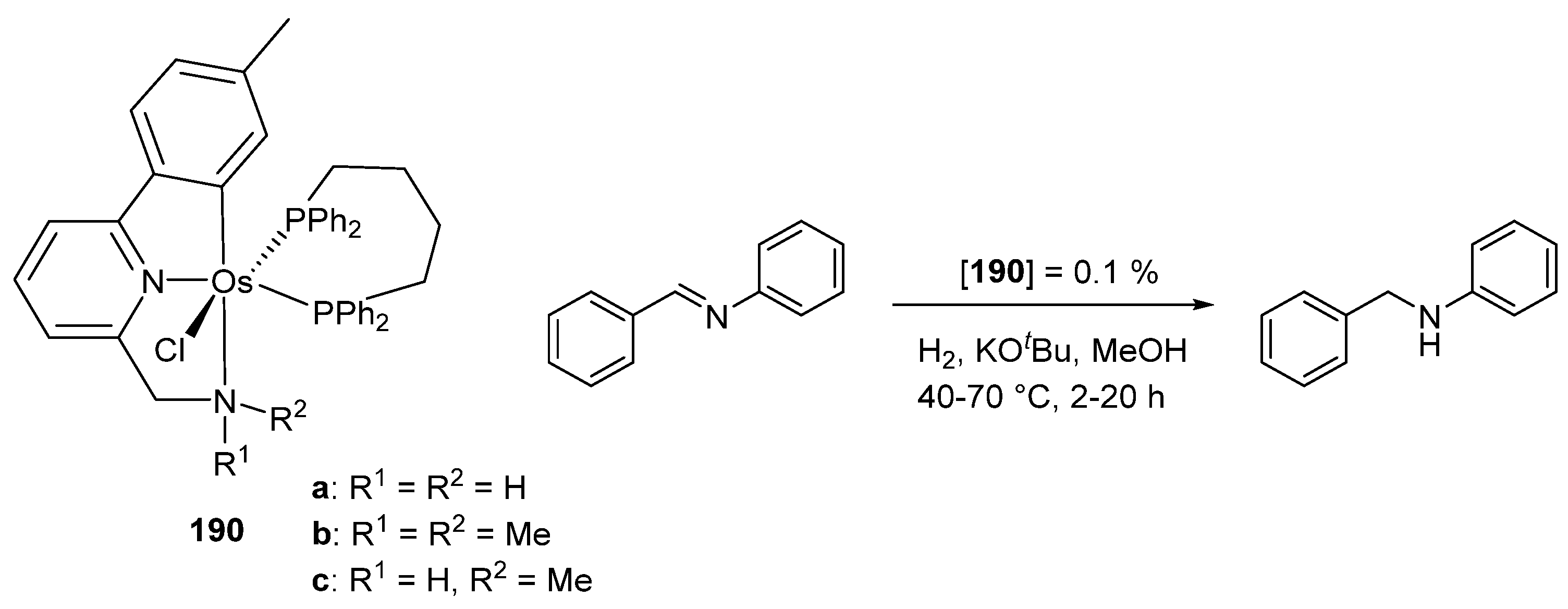
Figure 44.
Heterobimetallic complex used in dehydrogenation of secondary alcohols.
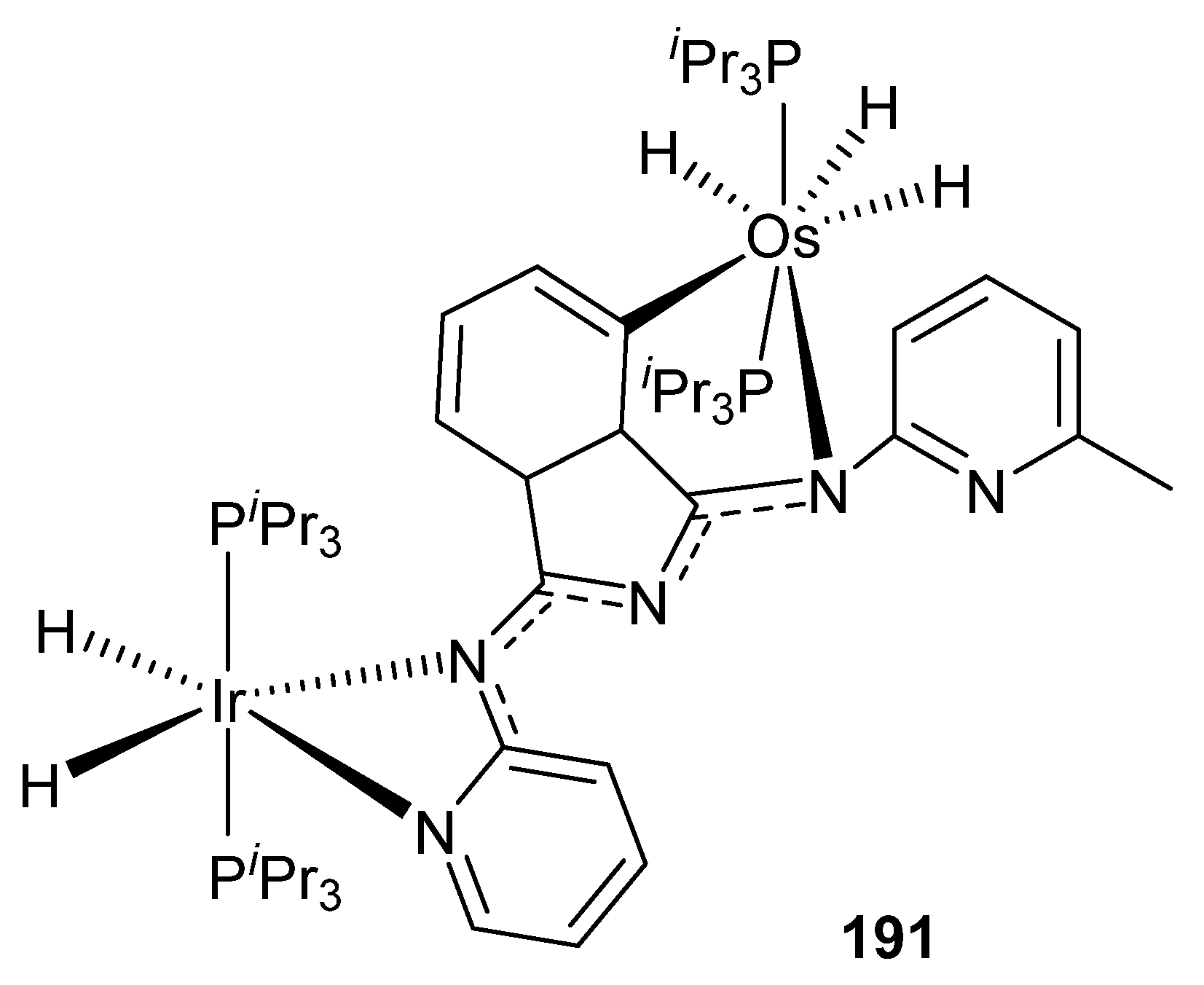
Figure 45.
Asymmetric catalysis by cyclometalated osmium complexes with central chirality.
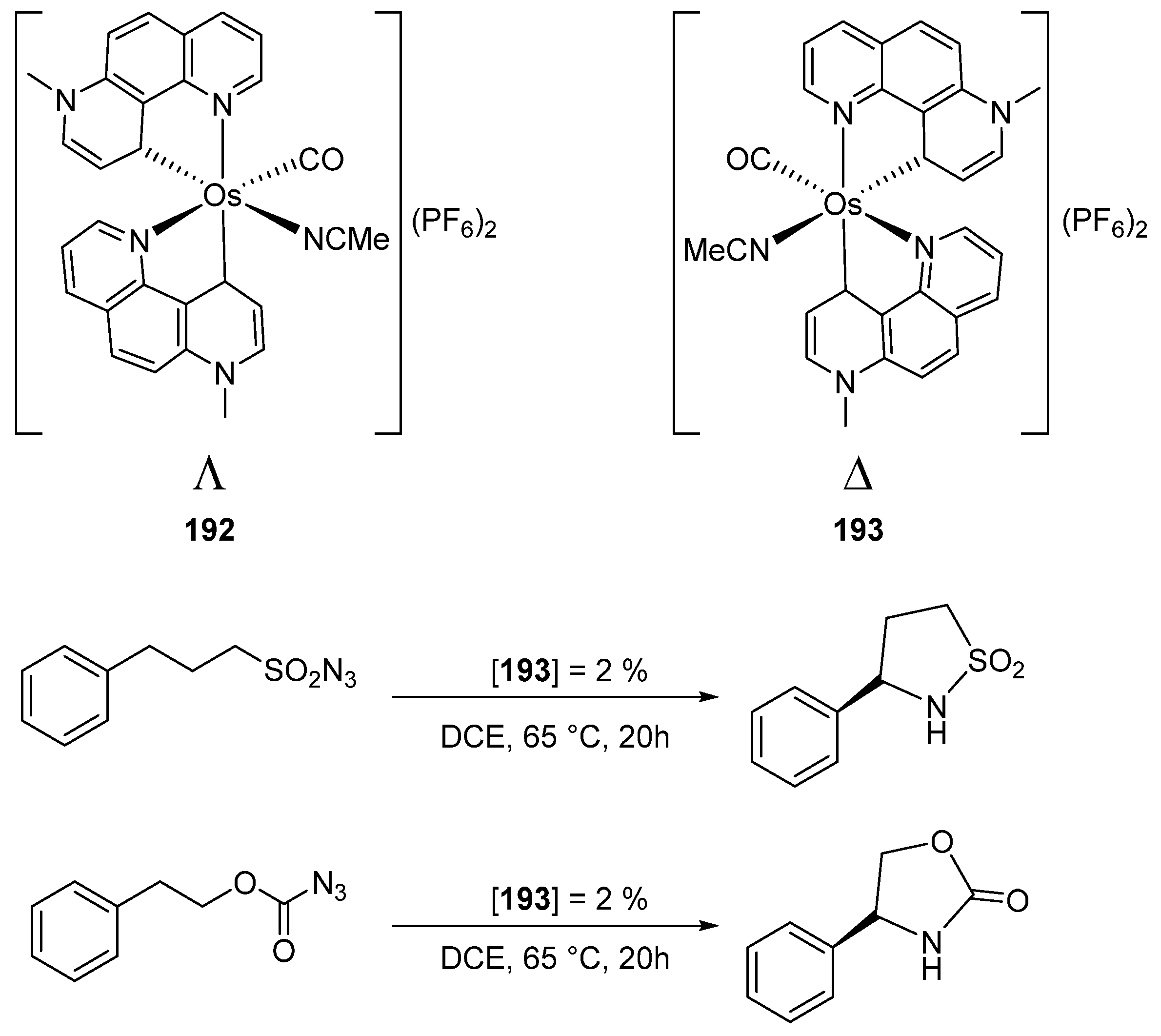
Figure 46.
Electron transfer pathway between osmium shuttle and glucose oxidase (GO).
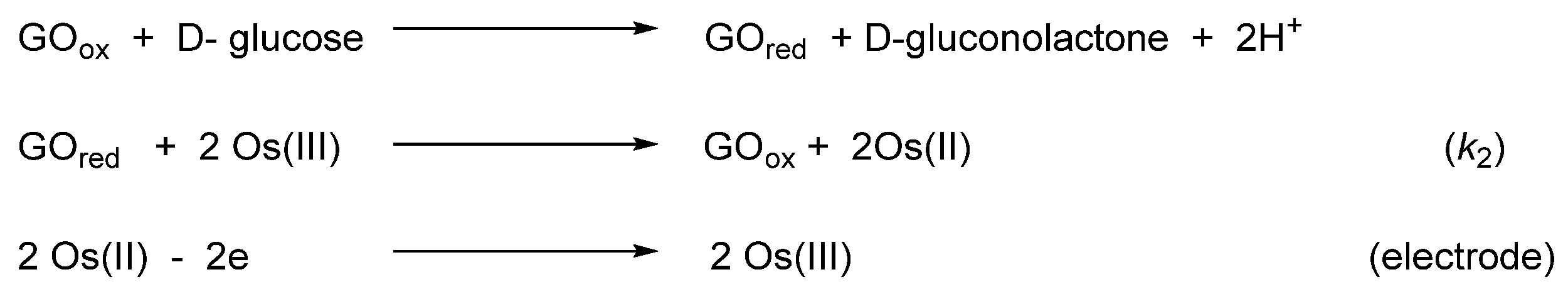
Figure 47.
Mono-, bis-, and tris-cyclometalated complexes used as electron shuttles and electron transfer pathway with horseradish peroxidase (HRP).
Figure 47.
Mono-, bis-, and tris-cyclometalated complexes used as electron shuttles and electron transfer pathway with horseradish peroxidase (HRP).
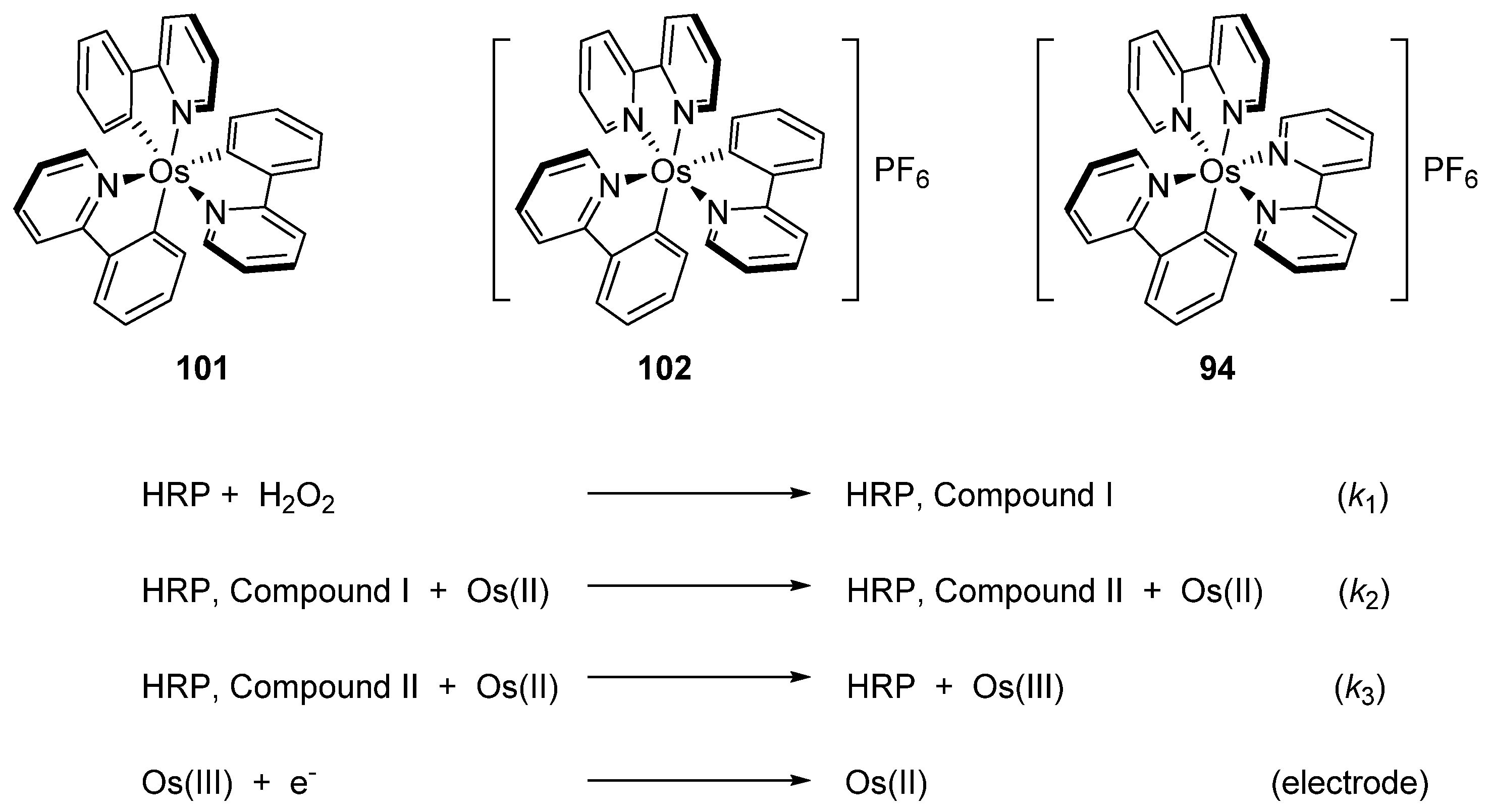
Figure 48.
Conceptual principle for a sensor using an iron(III)-TAML catalyst and the bis- osmacycle as a mediator.
Figure 48.
Conceptual principle for a sensor using an iron(III)-TAML catalyst and the bis- osmacycle as a mediator.
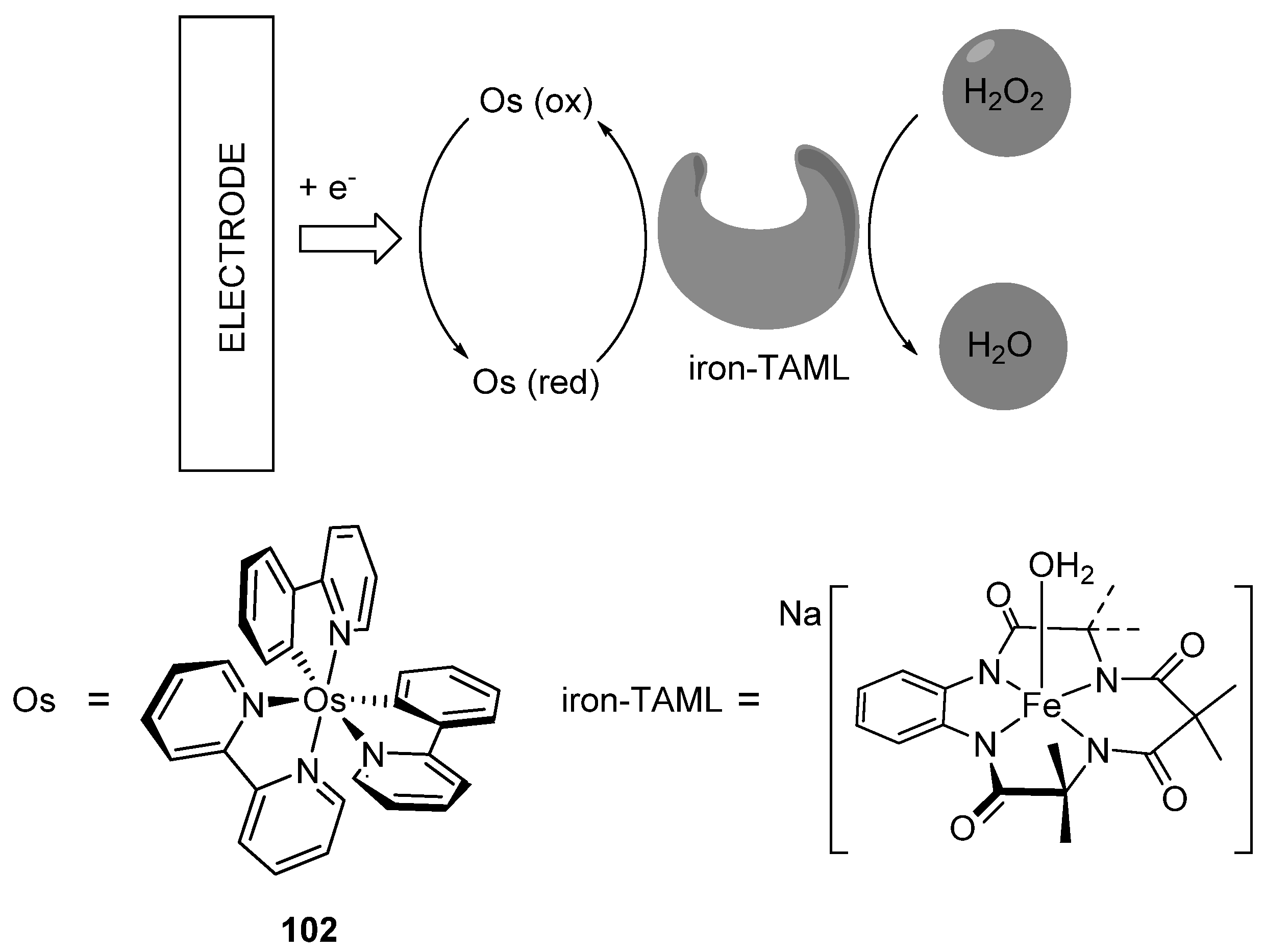
Figure 49.
Bimetallic luminescent complexes.

Figure 50.
Luminescent [OsCl(N~C)(PPh3)2(CO)] complexes.

Figure 51.
Luminescent carbene-based pincer complexes.

Figure 52.
Bis-tridentate cyclometalated osmium complexes studied as molecular wires.
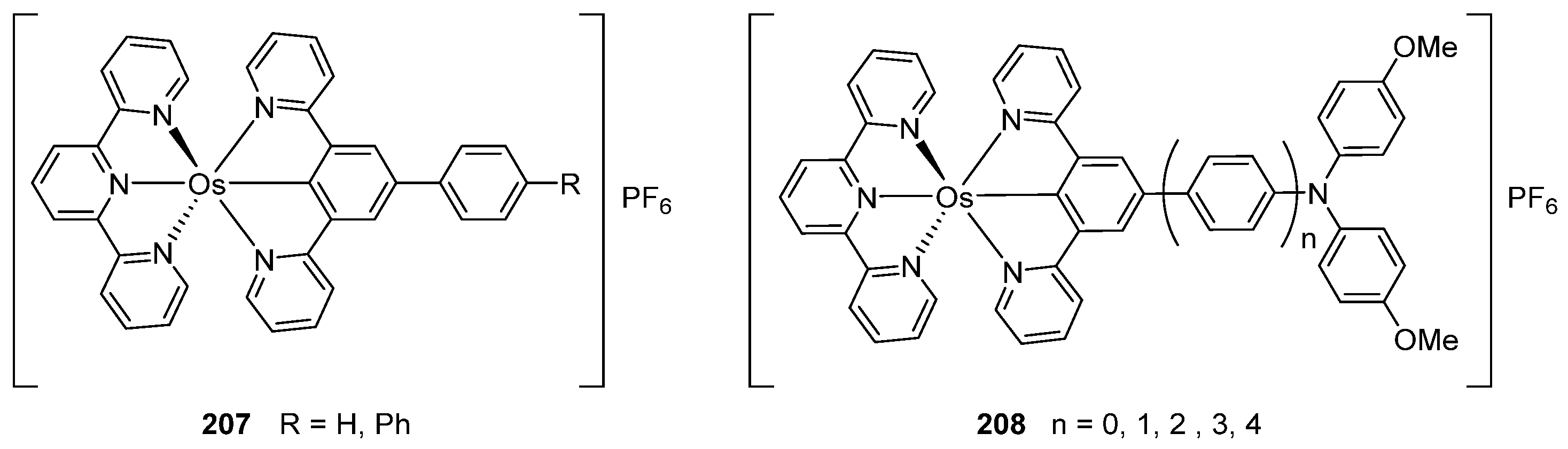
Figure 53.
Phosphorescent C~N~N and C~N~C osmium pincer complexes.
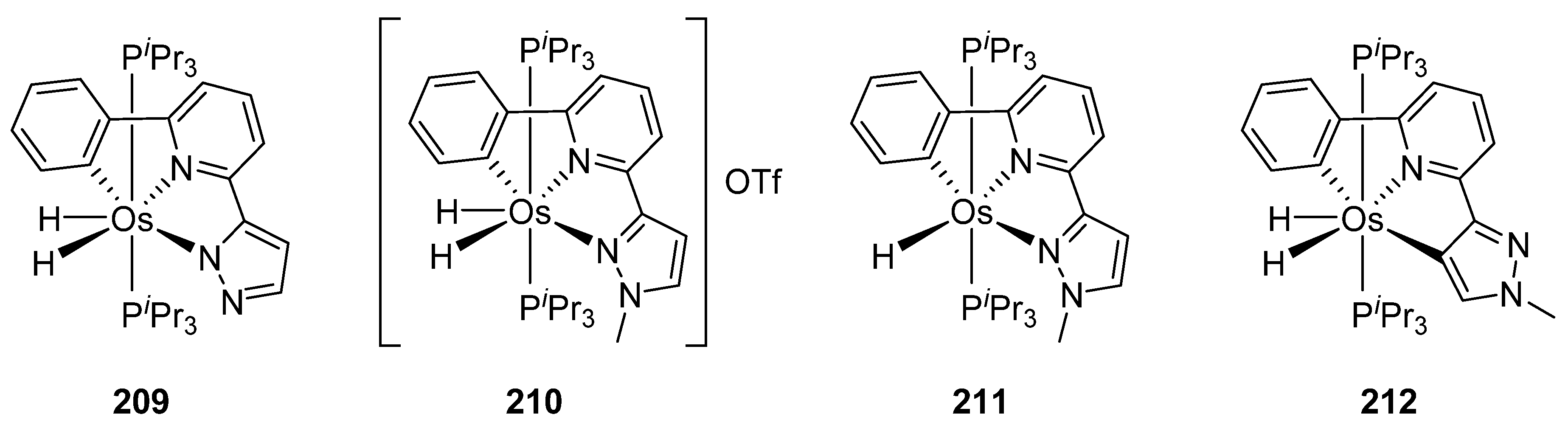
Figure 54.
Luminescent bis-pincer complexes with cyclometalated pyrene-type ligands.
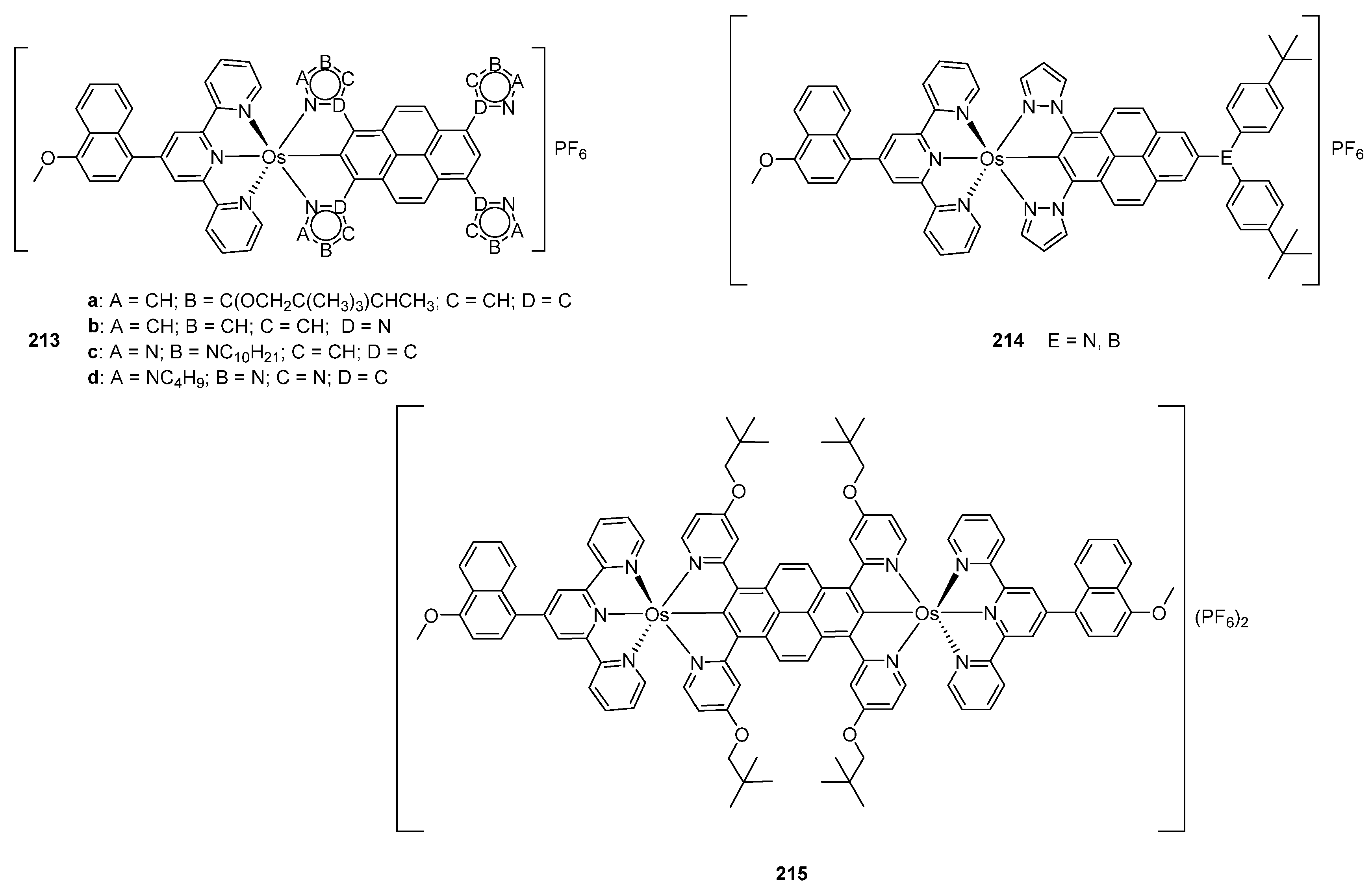
Figure 55.
η2-Allene complex with promising cytotoxic activity.
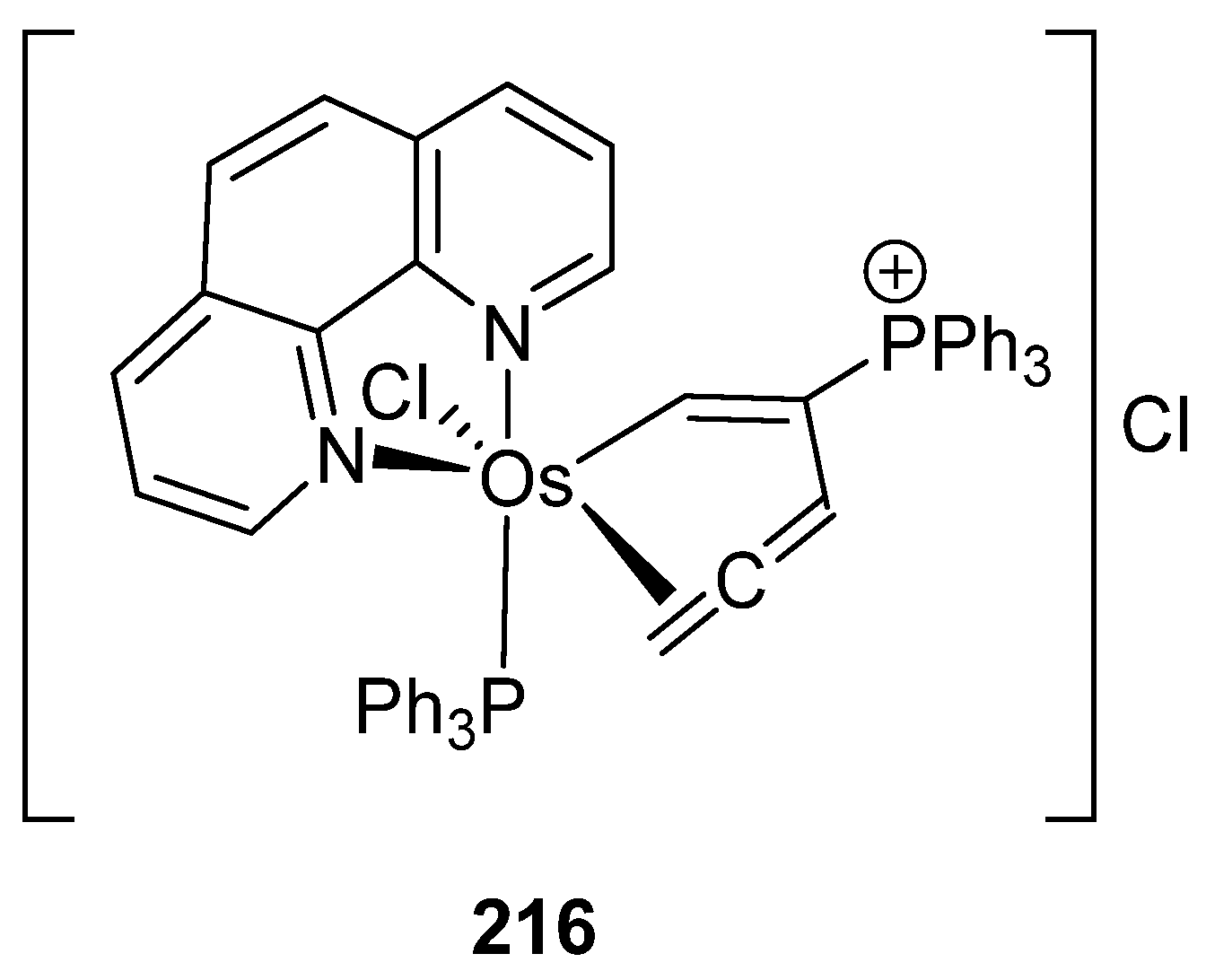
Figure 56.
Series of cyclometalated osmium complexes tested in the in vitro cell growth inhibition in A172 glioblastoma cell line.
Figure 56.
Series of cyclometalated osmium complexes tested in the in vitro cell growth inhibition in A172 glioblastoma cell line.
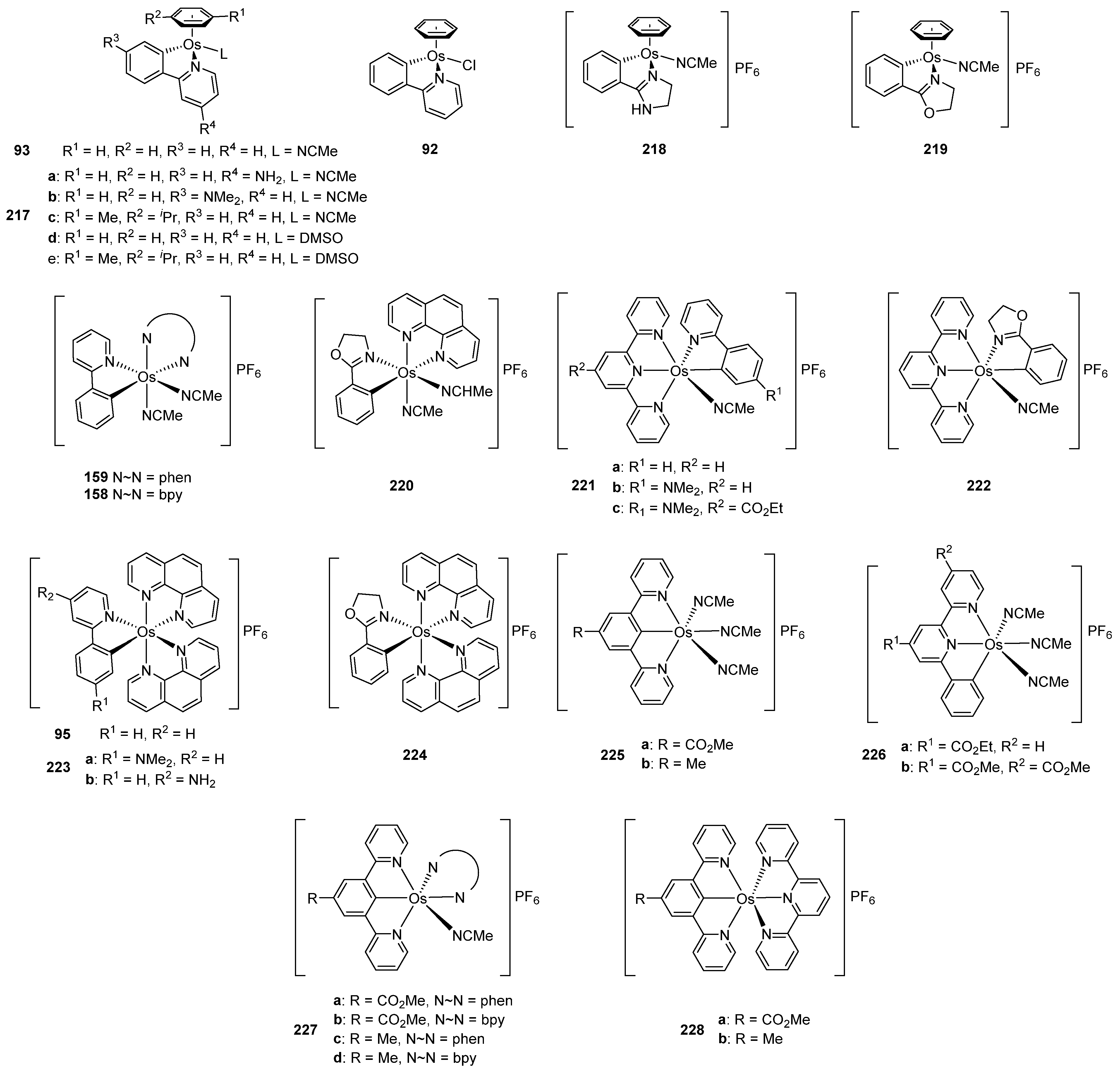
Figure 57.
Complexes tested against various human cancer cell lines.
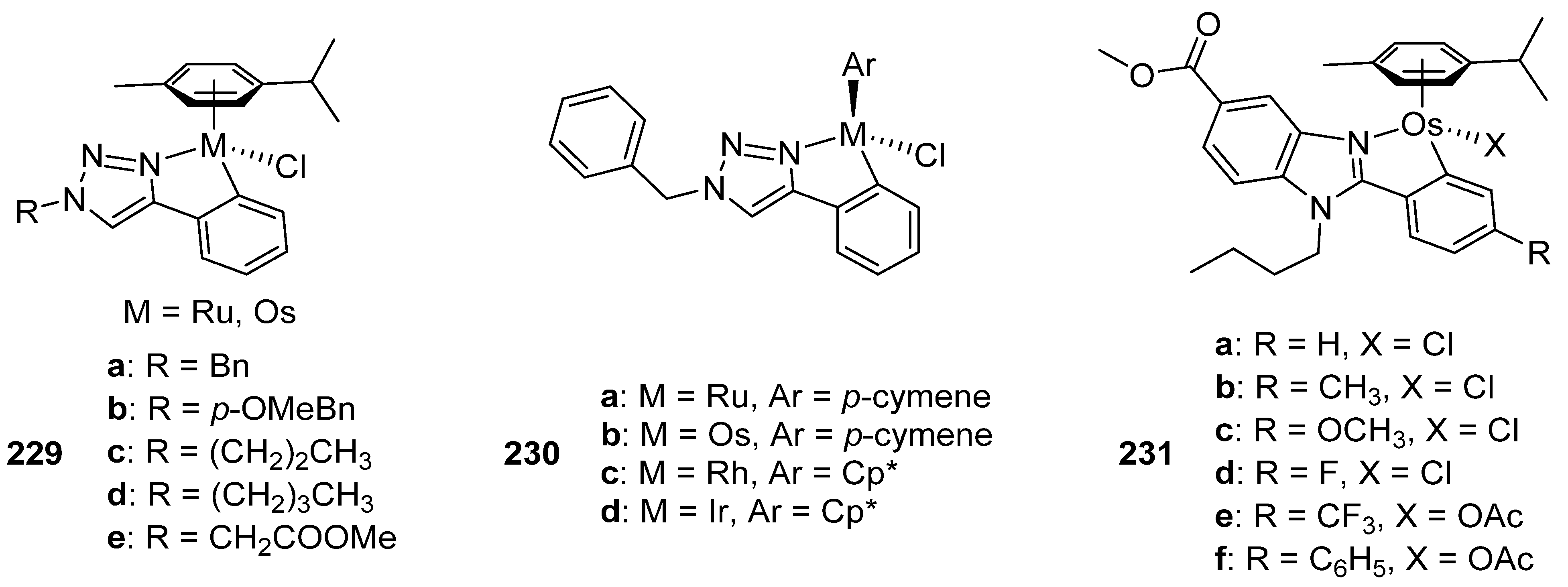
Figure 58.
Osmium complexes using nucleosides as cyclometalating ligands.
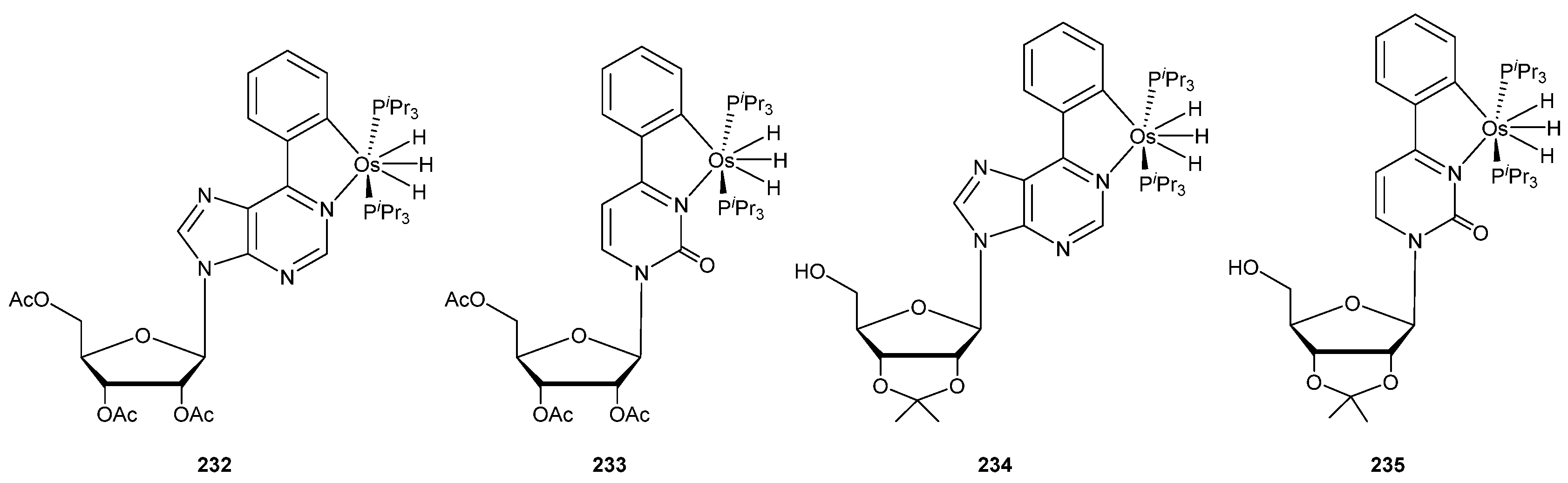

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
First osmacycles prepared between 1973 and 1997.
| Precursor | Cyclometalated Ligand | Osmacycle | Ref. |
|---|---|---|---|
| [Os3(CO)12] |  L1 |  1 speculative mixture of cis or trans | [28] |
| [OsH(O2CCF3)(CO)(PPh3)2] |  L2 |  2 | [29] |
| cis-[Os(PMe3)4H(CH2C(Me)3)] | PMe3 L3 |  3 | [30] |
| [Os(p-cymene)Me(Cl)(Me2SO)] prepared from [Os(p-cymene)Cl2]2 |  L4 | 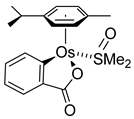 4 | [31] |
| [Os(tterpy)(O)2(OH)(NO3)] prepared from K2[Os(O)2(OH)4] | 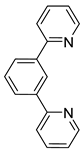 L5 | 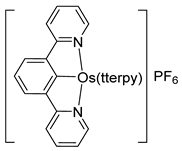 5 | [32] |
| [Os(tterpy)(O)2(OH)(NO3)] prepared from K2[Os(O)2(OH)4] |  L6 |  6 | [32] |
| [Os(tterpy)C13] prepared from [OsCl3] |  L6 | 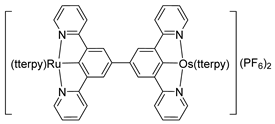 7 * | [33] |
| [Os(Cp)(PPh3)2]OTf |  L7 | 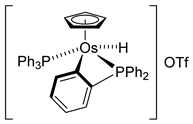 8 | [34] |
[Os(N~N~N)Cl3] prepared from [OsCl3] |  L8 | 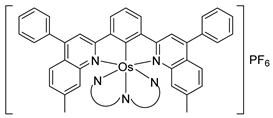 9 | [35] |
* tterpy = 4′-(4-tolyl)-2,2′:6′,2″-terpyridine.
Table 2.
Osmacycles obtained from [OsH6(PiPr3)2].
| Cyclometalated Ligand | Osmacycle | Yield | Ref. | ||
|---|---|---|---|---|---|
 L9 | 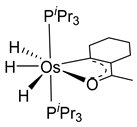 60 | 62% | [55] | ||
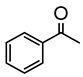 L10 | 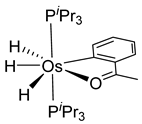 61 | 61% | [54] | ||
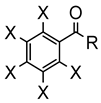 X = H, F; R = Ph, CH3 L11 | 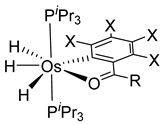 62 | 62% (X = H, R = Ph) 74% (X = F, R = CH3) 60% (X = F, R = Ph) | [54] | ||
 L12 |  63 | 92% | [56] | ||
 L13 | 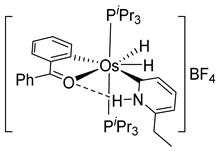 64 | 55% | [56] | ||
 L14 | 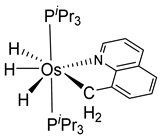 65 | 97% | [57] | ||
  L15, L16 | 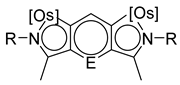 66 | 23% (E = N) 45% (E = CH) | [58] | ||
 L17 |  67 | 77% | [59] | ||
 L18 | 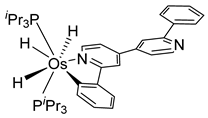 68 | 50% | [59] | ||
 L18 |  69 | 91% | [59] | ||
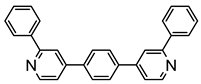 L19 | 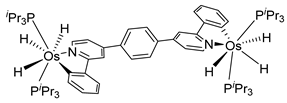 70 | 99% | [59] | ||
 L20 | 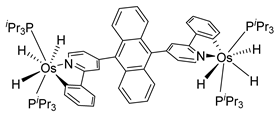 71 | 90% | [59] | ||
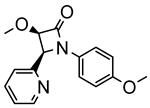 L21 | 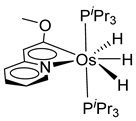 72 | 60% | [60] | ||
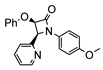 L22 | 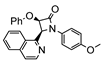 L23 |  L24 | 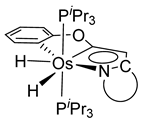 73 | 71% (L22) 60% (L23) 63% (L24) | [61] |
 L25 | 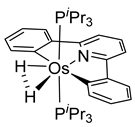 74 | 64% | [62] | ||
 L26 | 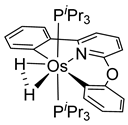 75 | 80% | [62] | ||
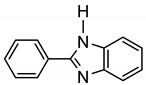 L27 |  76 | 53% | [63] | ||
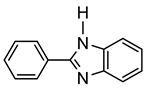 L27 | 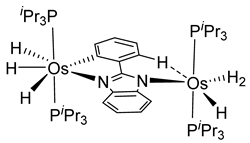 77 | 60% | [63] | ||
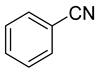 L28 |  78 | 73% (R = iPr3) 74% (R = Ph) R = Ph prepared from [OsH6(PiPr3)(PPh3)] | [64] | ||
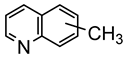 L29 | 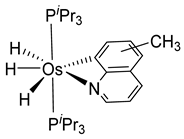 79 | 60–75% | [65] | ||
 L12 | 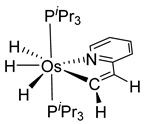 80 | 83% | [56] | ||
Table 3.
Reduction potential (OsII/OsIII) and rate constants k2 for the electron transfer between osmium complexes and the active site of glucose oxidase (GO). Reference electrode Ag/AgCl. [Os] 20 μM, GO 1.0 × 10−6 M, in phosphates buffer pH 7.0. Scan rate10 mV/s [76].
Table 3.
Reduction potential (OsII/OsIII) and rate constants k2 for the electron transfer between osmium complexes and the active site of glucose oxidase (GO). Reference electrode Ag/AgCl. [Os] 20 μM, GO 1.0 × 10−6 M, in phosphates buffer pH 7.0. Scan rate10 mV/s [76].
| Complex | E° (mV) | k2 (M−1s−1) |
|---|---|---|
| 162 | −51 | 0.67 × 106 |
| 163 | 13 | 4.80 × 106 |
| 161 | 32 | 2.00 × 106 |
| 94 | 84 | 2.90 × 106 |
| 95 | 31 | 1.80 × 106 |
| 159 | 109 | 2.90 × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cerón-Camacho, R.; Roque-Ramires, M.A.; Ryabov, A.D.; Le Lagadec, R. Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications. Molecules 2021, 26, 1563. https://doi.org/10.3390/molecules26061563
AMA Style
Cerón-Camacho R, Roque-Ramires MA, Ryabov AD, Le Lagadec R. Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications. Molecules. 2021; 26(6):1563. https://doi.org/10.3390/molecules26061563
Chicago/Turabian StyleCerón-Camacho, Ricardo, Manuel A. Roque-Ramires, Alexander D. Ryabov, and Ronan Le Lagadec. 2021. "Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications" Molecules 26, no. 6: 1563. https://doi.org/10.3390/molecules26061563