
Synthesis and Rational Design of New Appended 1,2,3-Triazole-uracil Ensembles as Promising Anti-Tumor Agents via In Silico VEGFR-2 Transferase Inhibition

Abstract
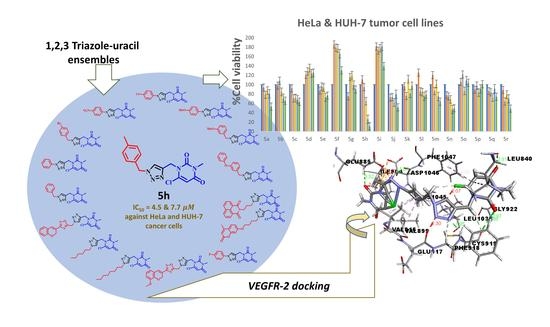
:
1. Introduction
2. Results and Discussion
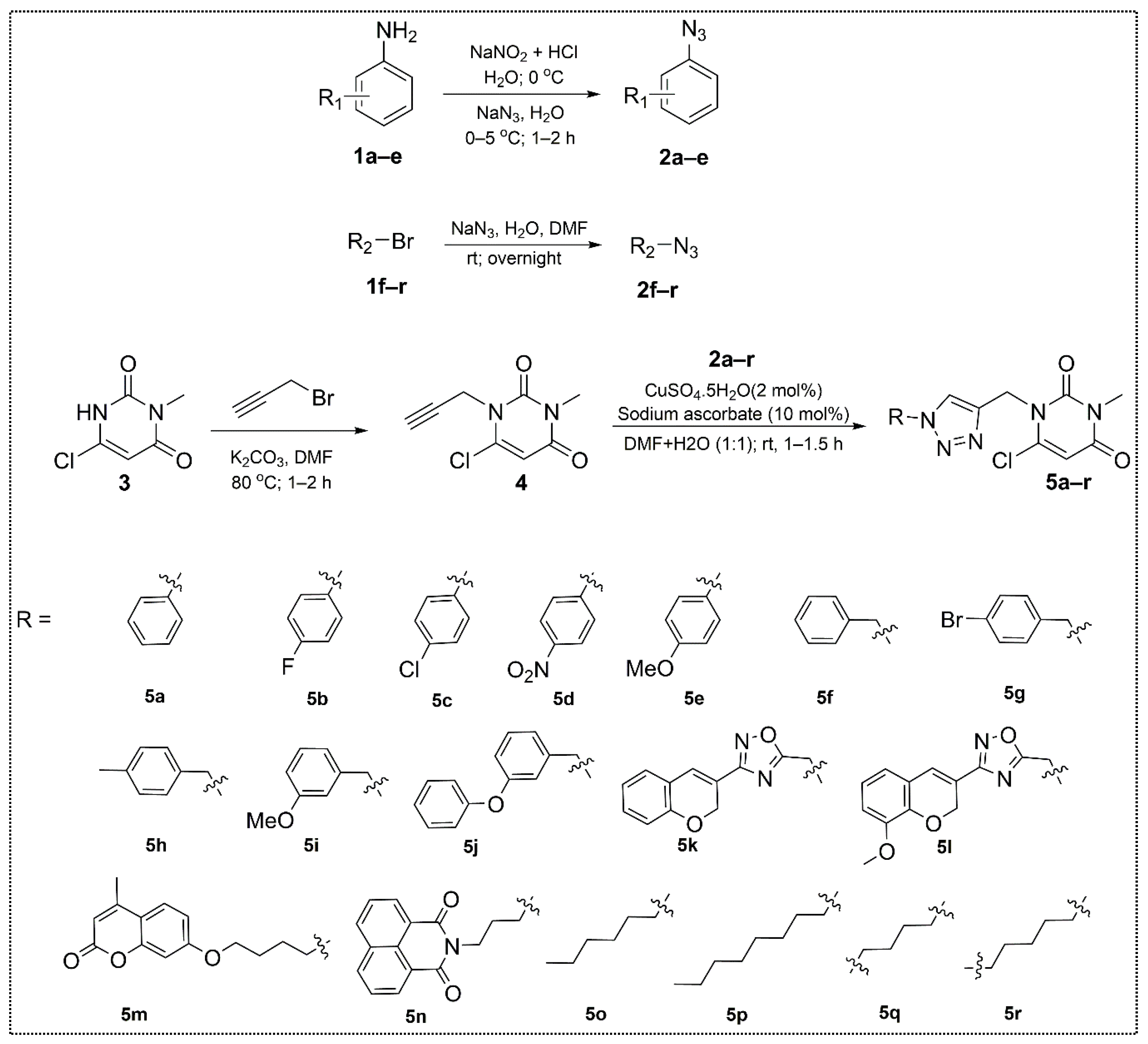
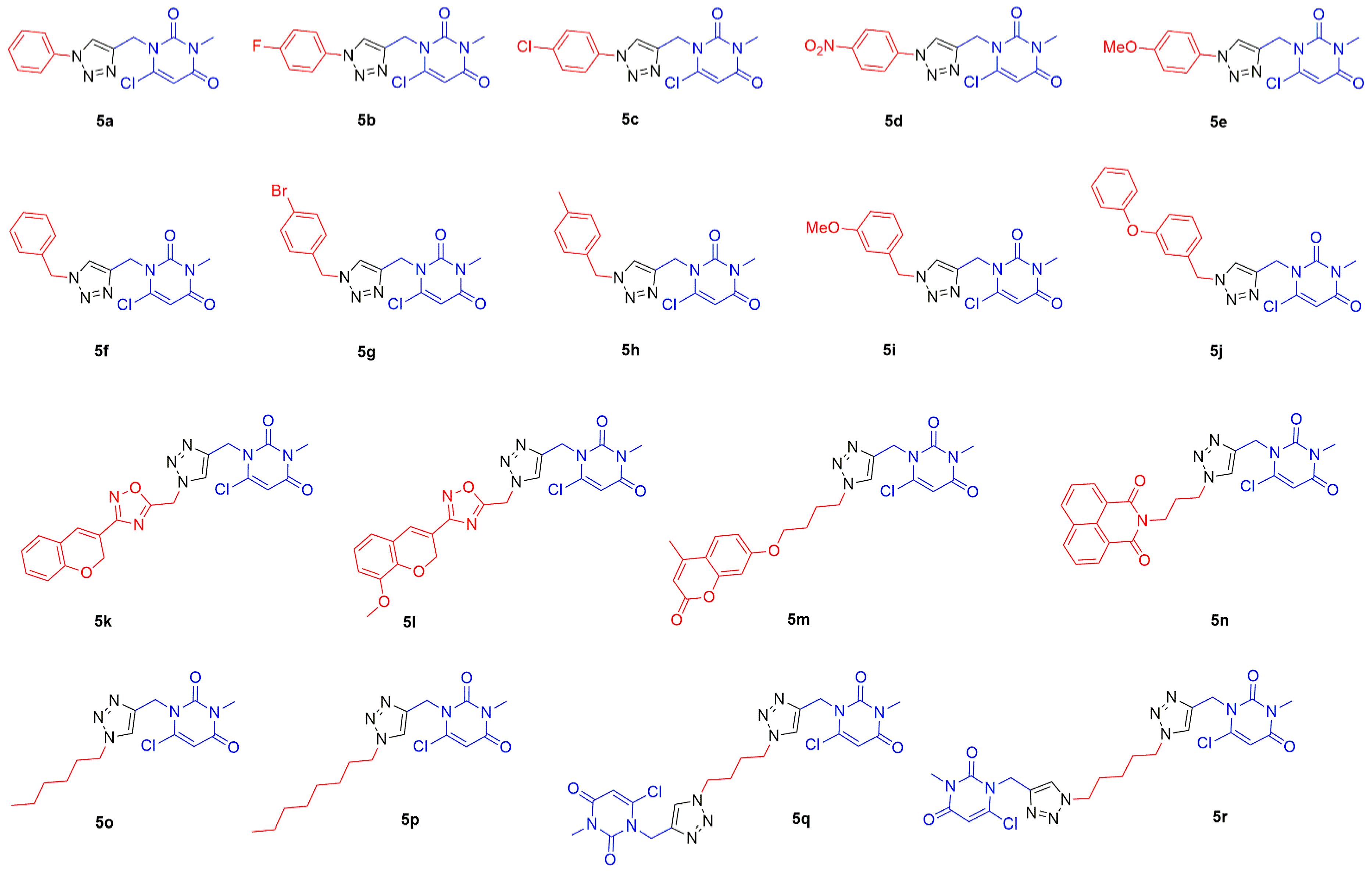
2.1. Chemistry
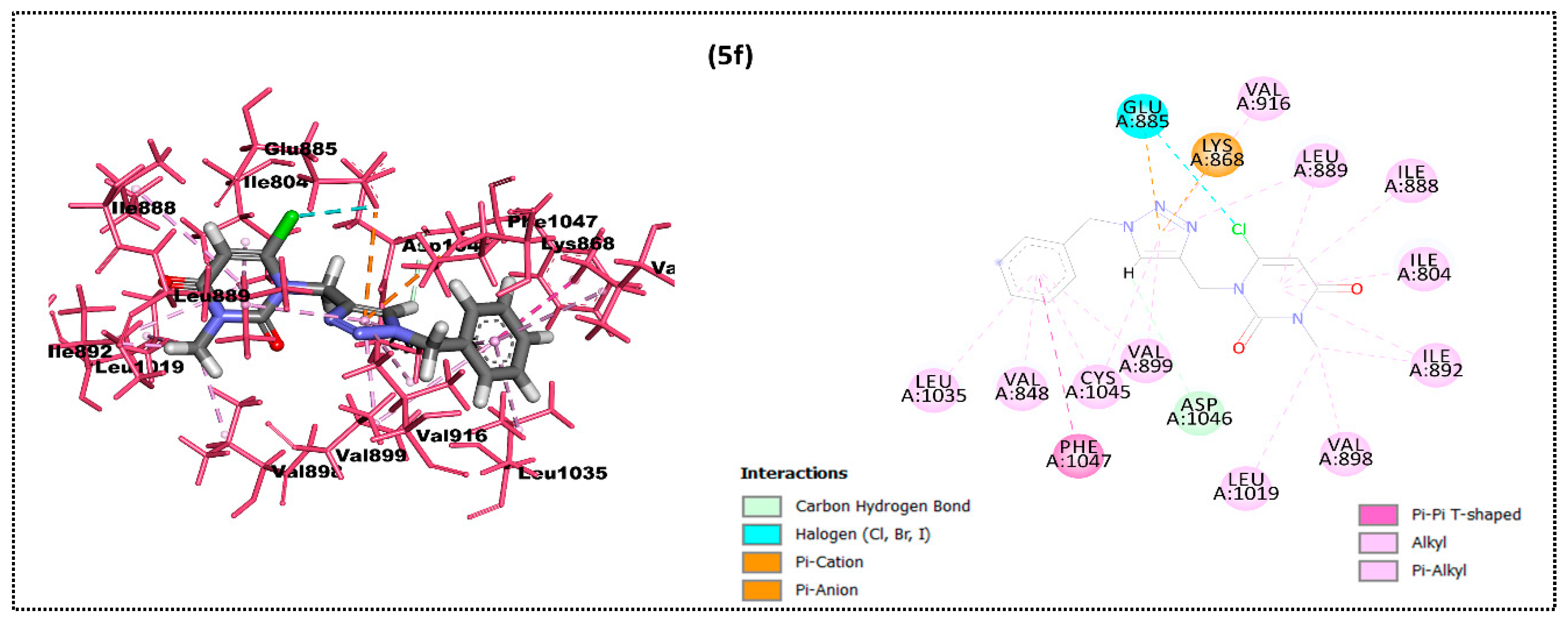
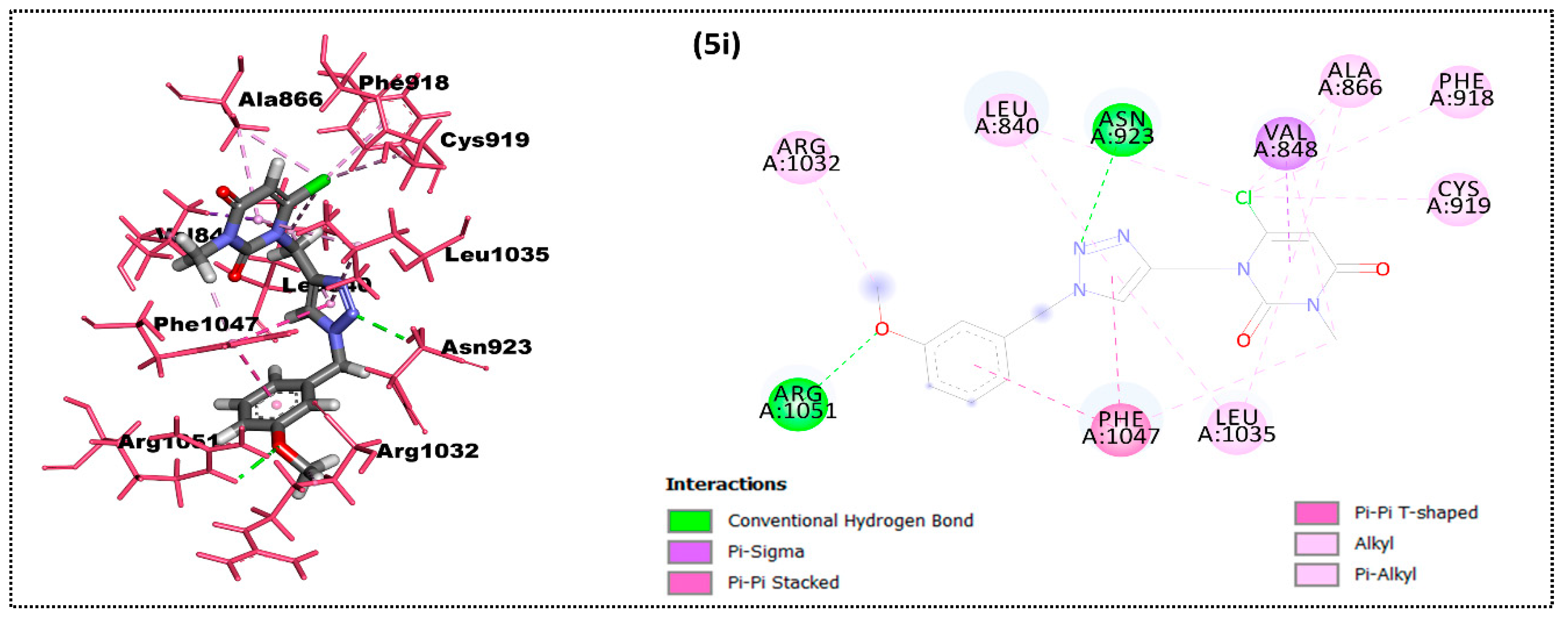
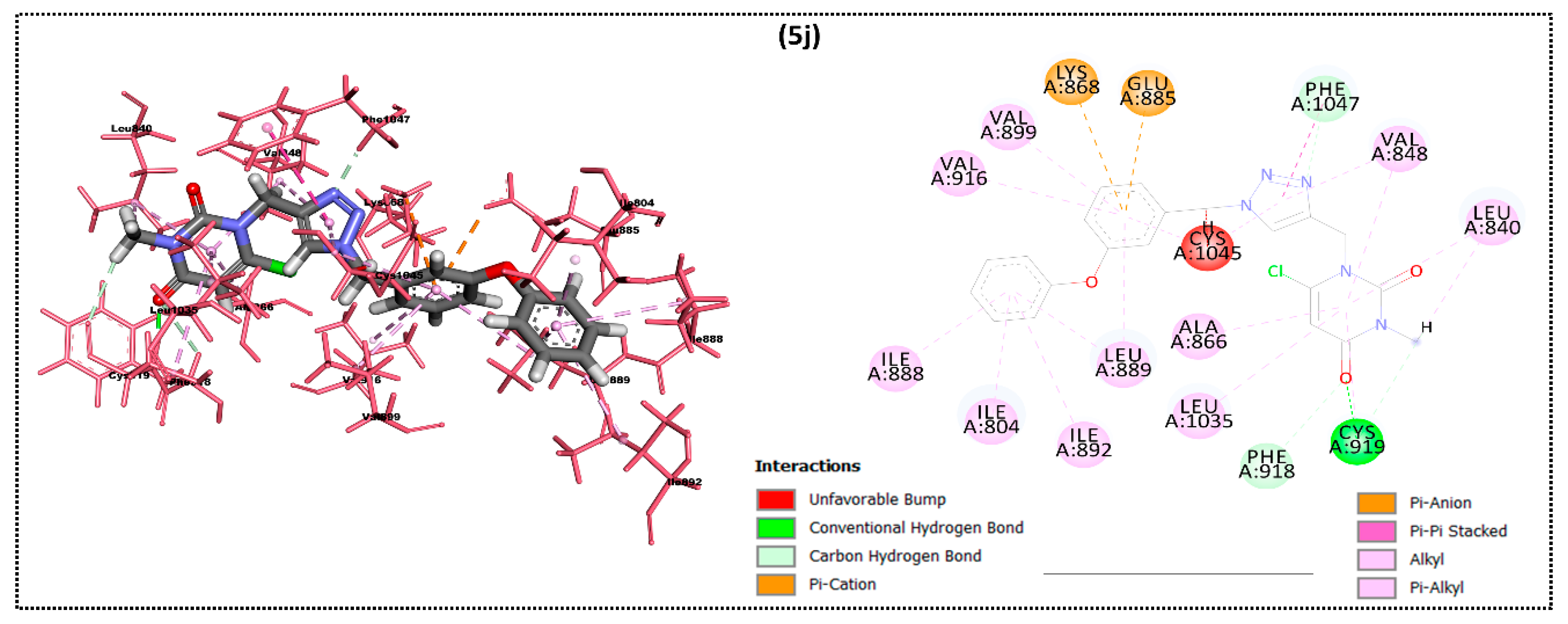
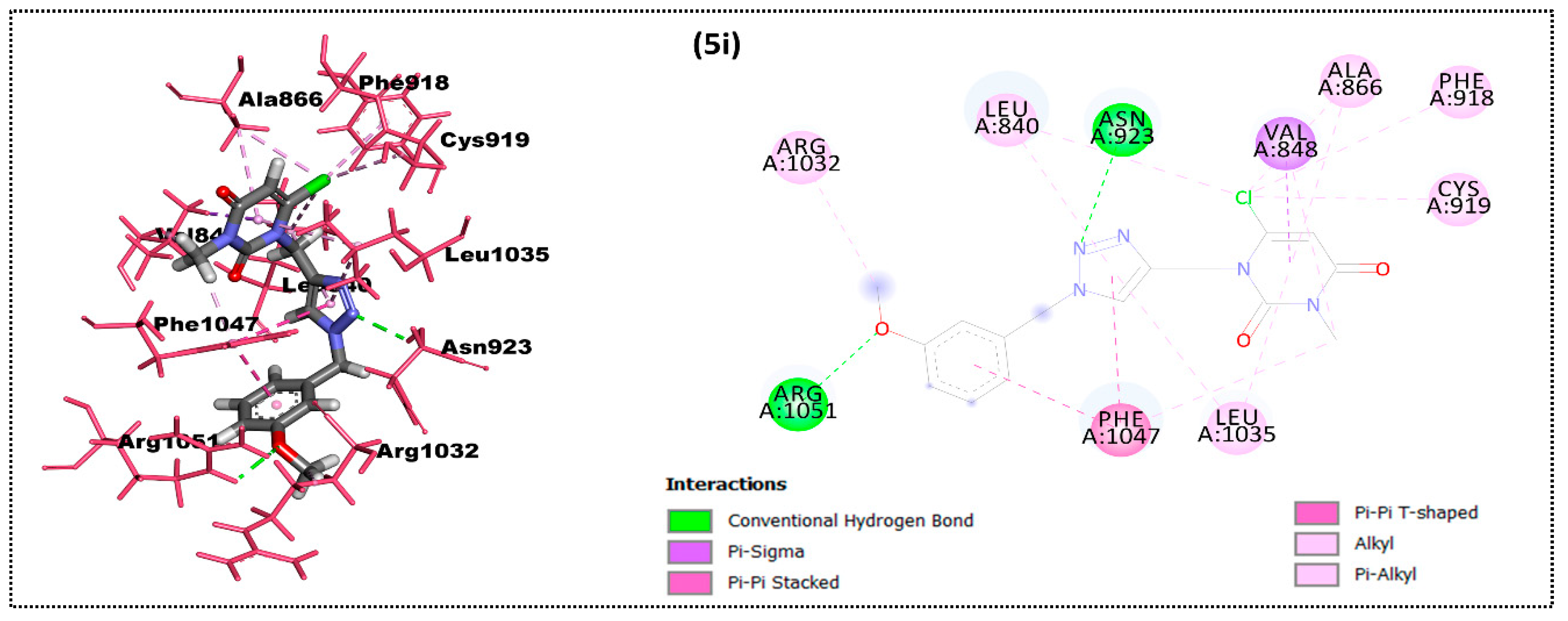
2.2. In Silico Docking Study
2.3. ADME Properties
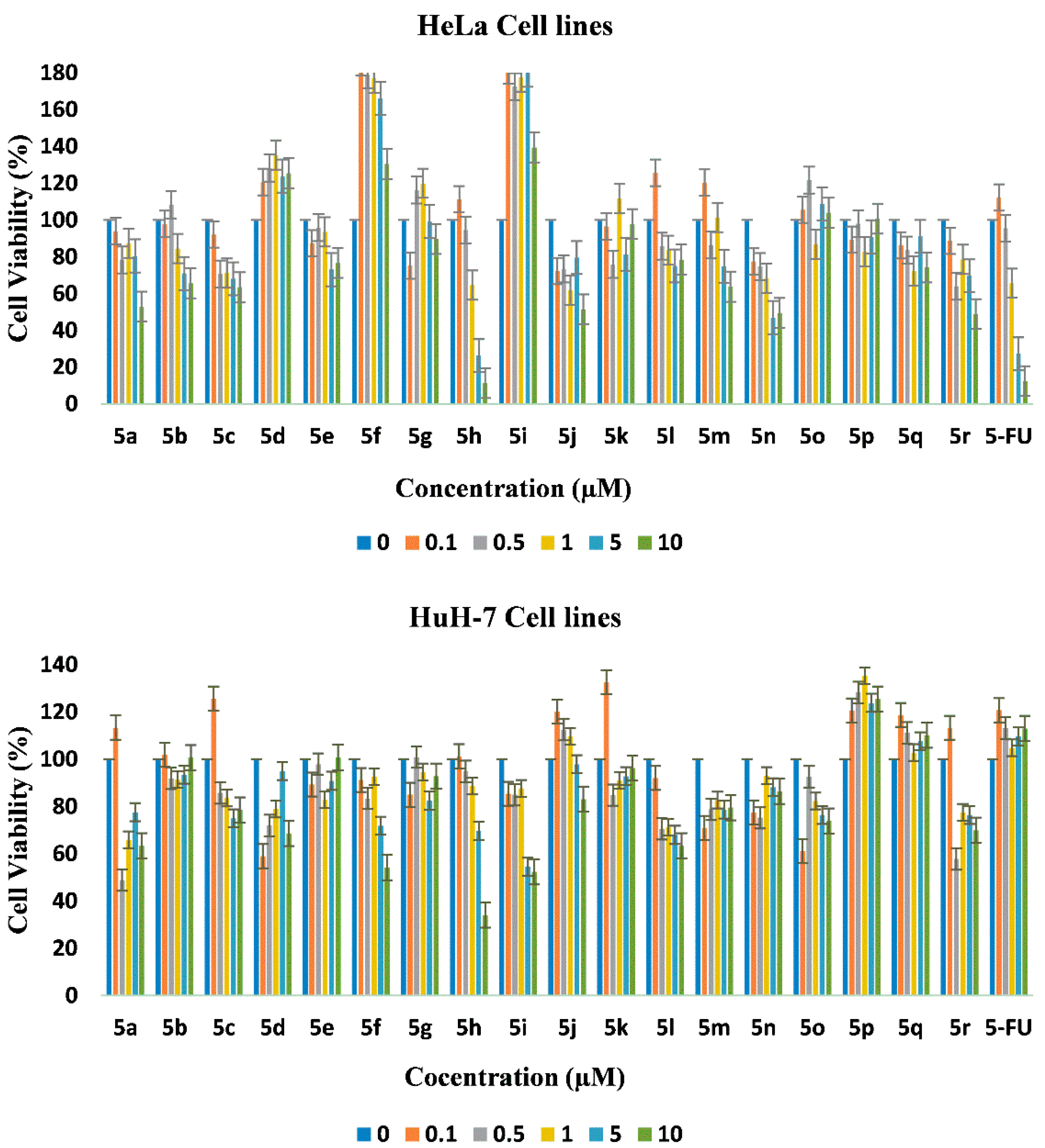
2.4. In Vitro Cytotoxicity
3. Materials and Methods
3.1. General Information
3.2. Syntheses
3.2.1. Synthesis of 6-Chloro-3-methyl-1-(prop-2-yn-1-yl)pyrimidine-2,4(1H,3H)-dione (4)
3.2.2. Synthesis of Substituted 1,2,3-Triazole-uracil Scaffolds 5a–r
3.3. In Silico Docking Simulations
3.3.1. Preparation of Ligands and Target Protein
3.3.2. Molecular Docking
3.4. ADME Profiles
3.5. Softwares, Suites and Webservers
3.6. In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, M. Amphiphilic glycoconjugates as potential anti-cancer chemotherapeutics. Eur. J. Med. Chem. 2018, 143, 1208–1253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. Tetrazole hybrids with potential anticancer activity. Eur. J. Med. Chem. 2019, 178, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2010, 26, 1–21. [Google Scholar] [CrossRef]
- Kaoukabi, H.; Kabri, Y.; Curti, C.; Taourirte, M.; Rodriguez-Ubis, J.C.; Snoeck, R.; Andrei, G.; Vanelle, P.; Lazrek, H.B. Dihydropyrimidinone/1,2,3-triazole hybrid molecules: Synthesis and anti-varicella-zoster virus (VZV) evaluation. Eur. J. Med. Chem. 2018, 155, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Ghobadi, E.; Saednia, S.; Hashemi, S.M. Current advances of triazole alcohols derived from fluconazole: Design, in vitro and in silico studies. Eur. J. Med. Chem. 2019, 170, 173–194. [Google Scholar] [CrossRef]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Yar, M.S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorganic Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Khurshed, B.; Jiangyu, Z.; Haji, A.A. 1,2,3-Triazole containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar]
- Kashmiri, L.; Pinki, Y. Recent advancements in 1,4-disubstituted 1H-1,2,3-Triazoles as potential anti-cancer agents. Anti-Cancer Agents Med. Chem. 2018, 18, 21–37. [Google Scholar]
- Pałasz, A.; Cież, D. In search of uracil derivatives as bioactive agents. Uracils and fused uracils: Synthesis, biological activity and applications. Eur. J. Med. Chem. 2015, 97, 582–611. [Google Scholar] [CrossRef]
- Senka, D.; Ljubica, G.O.; Jasmina, L.; Silvija, M.; Juraj, K.; Marija, K.; Marijana, J.; Silvana, R.M. Synthesis and biological evoluation of mono and bis ferrocene uracil derivatives. Appl. Organomet. Chem. 2020, 35, e6052. [Google Scholar]
- Valdenizia, R.S.; Rodrigo, S.C.; Luciano de Souza, S.; Milena, B.P.S.; Alzir, A.B.; Daniel, P.B. A ruthenium based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci. Rep. 2018, 8, 288. [Google Scholar]
- Rene, T.; Mikael, H.C. Moldock: A new technique for high accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Walter, F.D.A.J. Moldock applied to structure-based virtual screening. Curr. Drug Targets 2010, 11, 327–334. [Google Scholar]
- El-Adl, K.; Sakr, H.; Nasser, M.; Alswah, M.; Shoman, F.M.A. 5-(4-Methoxybenzylidene)thiazolidine-2,4-dione-derived VEGFR-2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluations. Arch. Pharm. 2020, 353, e2000079. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nat. Cell Biol. 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy. Int. J. Mol. Sci. 2014, 15, 15994–16011. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, M.; Wang, Y.; Fan, Y.; Chen, X.; Yang, Y.; Hua, Y.; Xie, W.; Lu, T.; Tang, W.; et al. Protein–ligand interaction-guided discovery of novel VEGFR-2 inhibitors. J. Biomol. Struct. Dyn. 2019, 38, 2559–2574. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Helby, A.-G.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorganic Chem. 2020, 105, 104380. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sharma, M.; Rahman, Q.I.; Akhtar, S.; Muddassir, M. Quantitative structure activity relationship and molecular simulations for the exploration of natural potent VEGFR-2 inhibitors: An in silico anti-angiogenic study. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, H.; You, Y.-Y.; Wang, X.-Y.; Lv, B.-B.; Cao, L.-Q.; Xue, J.-Y.; Xu, Y.-G.; Shi, L. Discovery of novel VEGFR-2 inhibitors embedding 6,7-dimethoxyquinazoline and diarylamide fragments. Bioorganic Med. Chem. Lett. 2021, 36, 127788. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Abdullaziz, M.A.; El Kerdawy, A.M.; Ragab, F.A.F.; Flanagan, K.J.; Mahmoud, A.E.E.; Ali, M.M.; El Diwani, H.I.; Senge, M.O. Targeting Receptor Tyrosine Kinase VEGFR-2 in Hepatocellular Cancer: Rational Design, Synthesis and Biological Evaluation of 1,2-Disubstituted Benzimidazoles. Molecules 2020, 25, 770. [Google Scholar] [CrossRef] [Green Version]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Eissa, I.H.; El-Hddad, S.S.; Shoman, F.M. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme. Bioorganic Chem. 2020, 102, 104059. [Google Scholar] [CrossRef]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; El-Gamal, K.M.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; El Sohly, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorganic Chem. 2020, 94, 103422. [Google Scholar] [CrossRef]
- El-Helby, A.A.; Sakr, H.; Eissa, I.H.; Abulkhair, H.; Al-Karmalawy, A.A.; El-Adl, K. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch. Pharm. 2019, 352, e1900113. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide derived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef]
- Mahmoud, H.K.; Farghaly, T.A.; Abdulwahab, H.G.; Al-Qurashi, N.T.; Shaaban, M.R. Novel 2-indolinone thiazole hybrids as sunitinib analogues: Design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity. Eur. J. Med. Chem. 2020, 208, 112752. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abou-Seri, S.M.; El Kerdawy, A.M.; Ayyad, R.R.; Hamdy, A.M.; Ghabbour, H.A.; Ali, M.M.; El Ella, D.A.A. Increasing the binding affinity of VEGFR-2 inhibitors by extending their hydrophobic interaction with the active site: Design, synthesis and biological evaluation of 1-substituted-4-(4-methoxybenzyl)phthalazine derivatives. Eur. J. Med. Chem. 2016, 113, 50–62. [Google Scholar] [CrossRef]
- Ahmed, E.Y.; Latif, N.A.A.; El-Mansy, M.F.; Elserwy, W.S.; Abdelhafez, O.M. VEGFR-2 inhibiting effect and molecular modeling of newly synthesized coumarin derivatives as anti-breast cancer agents. Bioorganic Med. Chem. 2020, 28, 115328. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.M.; El-Gazzar, M.G.; Aly, H.M.; Othman, R.A. Novel Anticancer Fused Pyrazole Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors. Front. Chem. 2020, 7, 917. [Google Scholar] [CrossRef]
- Ahmed, M.F.; Santali, E.Y.; El-Haggar, R. Novel piperazine–chalcone hybrids and related pyrazoline analogues targeting VEGFR-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J. Enzym. Inhib. Med. Chem. 2021, 36, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Faraji, A.; Bakhshaiesh, T.O.; Hasanvand, Z.; Motahari, R.; Nazeri, E.; Boshagh, M.A.; Firoozpour, L.; Mehrabi, H.; Khalaj, A.; Esmaeili, R.; et al. Design, synthesis and evaluation of novel thienopyrimidine-based agents bearing diaryl urea functionality as potential inhibitors of angiogenesis. Eur. J. Med. Chem. 2021, 209, 112942. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; El-Helby, A.-G.A.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; et al. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors. Bioorganic Med. Chem. 2021, 29, 115872. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorganic Chem. 2021, 107, 104532. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.-O.; Araceli, E.-V.; Guillermo, E.N.-S.; Manuel, E.P.-P.; Mario, A.R.-R.; Rosa, S. Multicomponent click synthesis of new 1,2,3-Triazole derivatives of pyrimidine nucleobases: Promising acidic corrosion inhibitors for steel. Molecules 2013, 18, 15064. [Google Scholar]
- Hanane, E.; Mustapha, A.A.; Ahmad, M.; Hasan, B.L. Nanocrystalline CuO: Synthesis and application as an efficient catalyst for the preparation of 1,2,3-triazole acyclic nucleosides via 1,3-dipolar cycloaddition. Catal. Commun. 2012, 26, 155. [Google Scholar]
- Kang, D.; Pang, X.; Lian, W.; Xu, L.; Wang, J.; Jia, H.; Zhang, B.; Liu, A.-L.; Du, G.-H. Discovery of VEGFR2 inhibitors by integrating naïve Bayesian classification, molecular docking and drug screening approaches. RSC Adv. 2018, 8, 5286–5297. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Molsoft, L.L.C. Drug-Likeness and Molecular Property Prediction. Available online: https://molsoft.com/mprop/ (accessed on 24 February 2021).
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Adnane, J.; Trail, P.A.; Levy, J.; Henderson, A.; Xue, D.; Bortolon, E.; Ichetovkin, M.; Chen, C.; McNabola, A. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xeno-graft models. Cancer Chemother. Pharmacol. 2007, 59, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-C.; Ou, D.-L.; Hsu, C.; Lin, K.-L.; Chang, C.-Y.; Lin, C.-Y.; Liu, S.-H.; Cheng, A.-L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2012, 108, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Alaparthi, M.D.; Gopinath, G.; Bandaru, S.; Sankeshi, V.; Mangalarapu, M.; Nagamalla, S.S.; Sudhakar, K.; Rani, A.R.; Sagurthi, S.R. Virtual screening of RAGE inhibitors using molecular docking. Bioinformation 2016, 12, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Torktaz, I.; Mohamadhashem, F.; Esmaeili, A.; Behjati, M.; Sharifzadeh, S. Virtual Screening and Pharmacophore Design for a Novel Theoretical Inhibitor of Macrophage Stimulating Factor as a Metastatic Agent. BioImpacts 2013, 3, 141–144. [Google Scholar]
- Brown, A.; Long, F.; Nicholls, R.A.; Toots, J.; Emsley, P.; Murshudov, G. Tools for macromolecular model building and re-finement into electron cryo-microscopy reconstructions. Acta Cryst. D 2015, 71, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Hsieh, Y.-J.; Huang, H.-S.; Leu, Y.-L.; Peng, K.-C.; Chang, C.-J.; Chang, M.-Y. Anticancer activity of Kalanchoe tubiflora extract against human lung cancer cells in vitro and in vivo. Environ. Toxicol. 2015, 31, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Gao, E.; Sun, Y.; Liu, Q.; Duan, L. An anticancer metallobenzylmalonate: Crystal structure and anticancer activity of a palla-dium complex of 2, 2′-bipyridine and benzylmalonate. J. Coord. Chem. 2006, 59, 1295–1300. [Google Scholar] [CrossRef]
- Ferrari, M.; Fornasiero, M.C.; Isetta, A.M. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [Google Scholar] [CrossRef]
- Rey, N.A.; Neves, A.; Silva, P.P.; Paula, F.C.; Silveira, J.N.; Botelho, F.V.; Vieira, L.Q.; Pich, C.T.; Terenzi, H.; Pereira-Maia, E.C. A synthetic dinuclear copper (II) hydrolase and its potential as antitumoral: Cytotoxicity, cellular uptake, and DNA cleavage. J. Inorg. Biochem. 2009, 103, 1323–1330. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Moldock Score [Grid] (Kcal/mol) | Moldock Score | Rerank Score | RMSD |
|---|---|---|---|---|
| 5a | −108.00 | −111.498 | −91.02 | 43.22 |
| 5b | −118.04 | −121.41 | −48.81 | 43.58 |
| 5c | −113.87 | −117.85 | −97.03 | 43.51 |
| 5d | −126.20 | −129.04 | −71.39 | 46.09 |
| 5e | −118.97 | −118.2 | −53.30 | 42.54 |
| 5f | −112.05 | −113.68 | −75.16 | 46.45 |
| 5g | −122.27 | −117.88 | −95.98 | 50.76 |
| 5h | −124.79 | −126.60 | −100.77 | 43.38 |
| 5i | −118.52 | −119.90 | −99.87 | 51.82 |
| 5j | −152.32 | −155.11 | −44.55 | 48.89 |
| 5k | −141.58 | −147.40 | −123.80 | 52.33 |
| 5l | −151.01 | −155.75 | −113.42 | 41.92 |
| 5m | −124.53 | −124.14 | −92.41 | 54.27 |
| 5n | −125.00 | −112.56 | −59.54 | 51.16 |
| 5o | −110.97 | −111.43 | −64.34 | 47.66 |
| 5p | −129.45 | −126.51 | −48.51 | 47.25 |
| 5q | −142.92 | −128.58 | −42.75 | 54.60 |
| 5r | −198.07 | −204.56 | −137.60 | 51.45 |
| 5-FU | −65.73 | −65.92 | −59.28 | 40.95 |
| C 1 | Gpcr Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor | milogP 2 | TPSA (A2) 3 | n Violation 4 | M.wt 5 | nO N 6 | nOHNH 7 | %ABS | MV 8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rule | ≤5 | — | ≤1 | <500 | <10 | <5 | ||||||||
| 5a | 0.11 | −0.15 | −0.21 | −0.22 | −0.28 | 0.09 | 1.28 | 74.73 | 0 | 317.74 | 7 | 0 | 83.21 | 259.51 |
| 5b | 0.13 | −0.16 | −0.15 | −0.17 | −0.27 | 0.06 | 1.45 | 74.73 | 0 | 335.73 | 7 | 0 | 83.21 | 264.45 |
| 5c | 0.12 | −0.15 | −0.20 | −0.21 | −0.27 | 0.06 | 1.96 | 74.73 | 0 | 352.18 | 7 | 0 | 83.21 | 273.05 |
| 5d | −0.04 | −0.19 | −0.32 | −0.28 | −0.35 | −0.05 | 1.24 | 120.55 | 0 | 362.73 | 10 | 0 | 67.41 | 282.85 |
| 5e | 0.07 | −0.21 | −0.21 | −0.19 | −0.27 | 0.03 | 1.34 | 83.96 | 0 | 347.76 | 8 | 0 | 80.03 | 285.06 |
| 5f | 0.07 | −0.19 | −0.21 | −0.26 | −0.21 | 0.07 | 1.60 | 74.73 | 0 | 331.76 | 7 | 0 | 83.21 | 276.32 |
| 5g | −0.04 | −0.26 | −0.24 | −0.36 | −0.31 | −0.01 | 2.41 | 74.73 | 0 | 410.66 | 7 | 0 | 83.21 | 294.20 |
| 5h | 0.02 | −0.25 | −0.24 | −0.28 | −0.24 | 0.01 | 2.05 | 74.73 | 0 | 345.79 | 7 | 0 | 83.21 | 292.88 |
| 5i | 0.07 | −0.15 | −0.11 | −0.10 | −0.24 | 0.01 | 1.64 | 83.96 | 0 | 361.79 | 8 | 0 | 80.03 | 301.86 |
| 5j | 0.07 | −0.15 | −0.11 | −0.10 | −0.10 | 0.07 | 3.33 | 83.96 | 0 | 423.86 | 8 | 0 | 80.03 | 356.71 |
| 5k | 0.06 | −0.24 | −0.34 | −0.15 | −0.19 | 0.01 | 1.45 | 122.89 | 0 | 453.85 | 11 | 0 | 66.60 | 363.34 |
| 5l | 0.01 | −0.28 | −0.35 | −0.22 | −0.24 | −0.03 | 1.26 | 132.12 | 1 | 483.87 | 12 | 0 | 63.41 | 388.89 |
| 5m | −0.20 | −0.60 | −0.36 | −0.19 | −0.34 | 0.02 | 2.66 | 114.17 | 0 | 471.90 | 10 | 0 | 69.63 | 396.81 |
| 5n | 0.03 | −0.29 | −0.14 | −0.32 | −0.16 | 0.11 | 2.02 | 113.80 | 0 | 478.90 | 10 | 0 | 69.73 | 393.82 |
| 5o | 0.12 | −0.28 | −0.29 | −0.36 | −0.18 | 0.16 | 2.46 | 74.73 | 0 | 325.80 | 7 | 0 | 83.21 | 288.68 |
| 5p | 0.13 | −0.26 | −0.22 | −0.26 | −0.12 | 0.14 | 3.47 | 74.73 | 0 | 353.85 | 7 | 0 | 83.21 | 322.28 |
| 5q | 0.07 | −0.20 | −0.12 | −0.20 | −0.09 | 0.08 | 0.24 | 149.46 | 2 | 551.39 | 14 | 0 | 57.43 | 430.79 |
| 5r | 0.07 | −0.23 | −0.12 | −0.19 | −0.08 | 0.08 | 0.75 | 149.46 | 2 | 551.39 | 14 | 0 | 57.43 | 447.59 |
| 5-FU | −2.60 | −1.95 | −2.62 | −3.04 | −3.15 | −1.56 | −0.59 | 65.72 | 0 | 130.08 | 4 | 2 | 86.32 | 96.91 |
| Compound | HeLa | HUH-7 | NIH/3T3 |
|---|---|---|---|
| 5a | 11.2 ± 0.1 | ≫10 | ND 1 |
| 5b | ≫10 | ≫10 | ND |
| 5c | ≫10 | ≫10 | ND |
| 5d | ≫10 | ≫10 | ND |
| 5e | ≫10 | ≫10 | ND |
| 5f | ≫10 | 10.8 ± 0.5 | ND |
| 5g | ≫10 | ≫10 | ND |
| 5h | 4.5 ± 0.5 | 7.7 ± 0.3 | ND |
| 5i | ≫10 | 9.0 ± 0.8 | ND |
| 5j | 10 ± 1.5 | ≫10 | ND |
| 5k | ≫10 | ≫10 | ND |
| 5l | ≫10 | ≫10 | ND |
| 5m | ≫10 | ≫10 | ND |
| 5n | 9.6 ± 1.2 | ≫10 | ND |
| 5o | ≫10 | ≫10 | ND |
| 5p | ≫10 | ≫10 | ND |
| 5q | ≫10 | ≫10 | ND |
| 5r | 9.6 ± 0.9 | ≫10 | ND |
| 5-FU | 4.6 ± 0.2 | 30 ± 2.5 | NA 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, N.N.; Hung, S.-J.; Swamy, M.K.; Sanjeev, A.; Rao, V.S.; Rohini, R.; Raju, A.K.; Bhaskar, K.; Hu, A.; Reddy, P.M. Synthesis and Rational Design of New Appended 1,2,3-Triazole-uracil Ensembles as Promising Anti-Tumor Agents via In Silico VEGFR-2 Transferase Inhibition. Molecules 2021, 26, 1952. https://doi.org/10.3390/molecules26071952
Reddy NN, Hung S-J, Swamy MK, Sanjeev A, Rao VS, Rohini R, Raju AK, Bhaskar K, Hu A, Reddy PM. Synthesis and Rational Design of New Appended 1,2,3-Triazole-uracil Ensembles as Promising Anti-Tumor Agents via In Silico VEGFR-2 Transferase Inhibition. Molecules. 2021; 26(7):1952. https://doi.org/10.3390/molecules26071952
Chicago/Turabian StyleReddy, Nadipolla Naresh, Sung-Jen Hung, Merugu Kumara Swamy, Ananthula Sanjeev, Vankadari Srinivasa Rao, Rondla Rohini, Atcha Krishnam Raju, Kuthati Bhaskar, Anren Hu, and Puchakayala Muralidhar Reddy. 2021. "Synthesis and Rational Design of New Appended 1,2,3-Triazole-uracil Ensembles as Promising Anti-Tumor Agents via In Silico VEGFR-2 Transferase Inhibition" Molecules 26, no. 7: 1952. https://doi.org/10.3390/molecules26071952