
Highlights on Steroidal Arylidene Derivatives as a Source of Pharmacologically Active Compounds: A Review




1
CICS-UBI—Health Sciences Research Centre, University of Beira Interior, Av. Infante D. Henrique, 6200-506 Covilhã, Portugal
2
CNC—Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(7), 2032; https://doi.org/10.3390/molecules26072032
Submission received: 15 March 2021
/
Revised: 29 March 2021
/
Accepted: 30 March 2021
/
Published: 2 April 2021
(This article belongs to the Special Issue Molecules Medicinal Chemistry Reviews)
Abstract
:Steroids constitute a unique class of chemical compounds, playing an important role in physiopathological processes, and have high pharmacological interest. Additionally, steroids have been associated with a relatively low toxicity and high bioavailability. Nowadays, multiple steroidal derivatives are clinically available for the treatment of numerous diseases. Moreover, different structural modifications on their skeleton have been explored, aiming to develop compounds with new and improved pharmacological properties. Thus, steroidal arylidene derivatives emerged as a relevant example of these modifications. This family of compounds has been mainly described as 17β-hydroxysteroid dehydrogenase type 1 and aromatase inhibitors, as well as neuroprotective and anticancer agents. Besides, due to their straightforward preparation and intrinsic chemical reactivity, steroidal arylidene derivatives are important synthetic intermediates for the preparation of other compounds, particularly bearing heterocyclic systems. In fact, starting from arylidenesteroids, it was possible to develop bioactive steroidal pyrazolines, pyrazoles, pyrimidines, pyridines, spiro-pyrrolidines, amongst others. Most of these products have also been studied as anti-inflammatory and anticancer agents, as well as 5α-reductase and aromatase inhibitors. This work aims to provide a comprehensive overview of steroidal arylidene derivatives described in the literature, highlighting their bioactivities and importance as synthetic intermediates for other pharmacologically active compounds.
1. Introduction
Steroids are natural products that share a 17-carbon-atom skeleton and are composed of four fused rings: three cyclohexanes (A, B, and C rings) and one cyclopentane (D ring). These compounds vary on the attached functional groups, their position, and configuration [1]. In addition, steroids represent a unique class of chemical products, playing an important role in several biological processes, being the most important group of regulatory and signaling molecules [2,3]. In fact, this class of compounds is widely found in animal and plant kingdoms. In humans, the different steroids are biosynthesized from cholesterol via several enzyme-mediated transformations. Usually, steroids are lipophilic and readily enter cells, being able to interact with nuclear receptors as well as with membrane proteins. Therefore, they are associated with most physiological functions and pathological conditions. In addition, due to their low toxicity, less vulnerability to multidrug resistance, and high bioavailability [4,5,6,7,8], steroid-based therapeutic drugs have called attention to the scientific academia and industry for a long time. Consequently, steroidal derivatives constitute an important class of pharmaceutically active compounds for a large number of diseases, including brain tumors, breast and prostate cancers, osteoarthritis, adrenal insufficiencies, fungal and microbial infections, in addition to cardiovascular and autoimmune diseases [3,9,10,11,12,13].
Due to their relevance, several modified steroids have been synthesized and biologically evaluated, being verified that their relevant pharmacological properties depend on the structural features of the steroidal four-ring skeleton and side-chain [5,14]. In fact, even a minor structural variation on the steroidal nucleus can lead to marked changes in their physiological activity [15]. Therefore, aiming to improve their pharmacological properties and/or develop compounds with different bioactivities, structural modifications of steroids have been an important focus of research over the last decades [16,17,18,19,20].
One of the diverse groups of modified steroids is arylidenesteroids, which are the focus of the present review. Generally, arylidene derivatives have been associated with a wide range of pharmacological activities, including antifungal, antimicrobial, antioxidant, anti-arrhythmic, anticancer, antimalarial, and anti-inflammatory [21,22,23,24,25,26,27,28,29,30]. The general structure of these compounds comprises a natural or synthetic core bearing an aryl group linked through an exocyclic double bound, which is part of a α,β-unsaturated ketone system. An advantage of this system is the inherent chemical reactivity due to the simultaneous presence of electrophilic and nucleophilic points. Therefore, these derivatives are especially useful in the preparation of several bioactive heterocyclic systems [4,31,32]. Thus, considering the pharmacological relevance of steroids and the biological and synthetic value of arylidene derivatives, it is not surprising that several studies involved the synthesis and biological evaluation of steroidal arylidenes, despite some potential problems of these compounds. In fact, in addition to the known possible hormonal effects, usually, these steroids have higher lipophilicity and the consequent risk of bioavailability problems. Moreover, the presence and inherent reactivity of their α,β-unsaturated ketone system can also be associated with potential toxicity problems, including mutagenesis and carcinogenesis [31,32]. Despite this, over the years, various arylidenesteroids have been studied, namely as hydroxysteroid dehydrogenases, aromatase, and 5α-reductases inhibitors, as well as skeletal muscle relaxants, antimicrobial, neuroprotective, antiparasitic, and antiproliferative agents [17,33,34,35,36,37,38,39,40]. Additionally, other research works have been demonstrating their use as synthetic intermediates, mainly in the preparation of aza-heterocycles with relevant potential applications [41,42].
In the past few years, several reviews have been published reporting the importance and synthetic approaches for chemical modifications on the steroidal skeleton [43,44,45]. However, arylidene steroidal derivatives and their application were only superficially presented, being inexistent an extensive and comprehensive review on this important topic. Therefore, this review aims for the first time, to our knowledge, to highlight steroidal arylidene derivatives described in the literature, particularly 2- and 16E-arylideneandrostanes, 21E-arylidenepregnanes and 16E-arylidenoestranes, with relevant pharmacological activity, as well as their main chemical synthesis and application as synthetic intermediates of relevant bioactive molecules, particularly heterocyclic compounds.
2. Synthetic Approaches to Prepare Arylidene Steroidal Derivatives
Arylidenesteroids are usually obtained through an aldol condensation between a steroid and an aldehyde. In an aldol condensation, an enol or enolate reacts with a carbonyl in the presence of an acid or base catalyst to form a β-hydroxyaldehyde or a β-hydroxyketone, followed by dehydration to afford a conjugated enone [46]. More particularly, the reaction between an aldehyde/ketone capable of forming an enol/enolate and an aromatic carbonyl without an α-hydrogen (usually an aromatic aldehyde) is named Claisen–Schmidt condensation. In steroids, this reaction was mainly performed in C-16 or C-21 positions and less often in C-2.
In general, the reactions to prepare arylidenesteroids occur at room temperature (RT), under basic catalysis, and the solvent is, in most cases, methanol (MeOH) or ethanol (EtOH) (Figure 1). The basicity of the reaction medium is conferred by sodium hydroxide (NaOH) or potassium hydroxide (KOH) [38,47,48,49,50,51] However, depending on reaction substrate and reagents, the duration of the reaction and observed yields are extremely variable. In fact, according to the literature, these reactions can take between 1 to 48 h to be concluded and yields ranging from 23 to 98% were described [32,42,52,53]. Androstane, pregnane, and estrane derivatives, namely dehydroepiandrosterone, androstenedione, estrone, pregnenolone, and progesterone have been described as the main starting materials.
Less often, the preparation of 2-arylidenesteroids from dehydroepiandrosterone (DHEA) derivatives, dihydrotestosterone (DHT), and cholestanone was described [31,54,55]. The synthesis of these derivatives was based on the classical approach for aldol condensation, occurring at RT and under basic catalysis, using MeOH or EtOH as solvent [31,54,55].
Interestingly, over the last years, other approaches different from the classical reaction conditions have been explored. As a representative example, Guo et al. performed the same reaction using MeOH as solvent and NaOH as a base, but at higher temperatures, to afford excellent yields (91–95%) [37]. In addition, Huang et al. and Yu et al. applied a different strategy to prepare the corresponding aldol condensation products from 4-azaandrost-3,17-dione (1) and dehydroepiandrosterone (DHEA) (Figure 2). As alternative for the use of NaOH or KOH, these authors used potassium fluoride on aluminum oxide (KF/Al2O3) as a recyclable basic catalyst and performed the reaction at EtOH under reflux temperature. Remarkably, shorter reaction times (1 h) were observed, and excellent yields of the products (90–94%) were achieved [41,56]. This transformation was also performed using molecular iodine-loaded aluminum oxide (I2-Al2O3) as heterogeneous catalyst and under microwave (MW) irradiation (Figure 3). These conditions allowed extremely short reaction times (5–7 min) and good yields of the products (79–85%) [52]. More recently, Mótyán et al. reported the synthesis of 16E-benzylidene-estrone-3-methyl ether derivatives by a MW-assisted (100 °C, 20 min) Claisen–Schmidt condensation, using EtOH as a solvent and KOH [53].
The most different approach to prepare steroidal arylidene derivatives was employed by Riebe et al., in the synthesis of 16E-arylidene-3-methoxy-estrone derivatives from commercially available 3-methoxyestrone (Figure 4). Firstly, an enol intermediate using sodium methoxide and ethyl formate in benzene and under reflux was prepared, followed by a reaction with formaldehyde in pyridine at room temperature. Then, a palladium-catalyzed Mizoroki–Heck reaction allowed the preparation of 16E-arylidenoestrane derivatives in fair to excellent yields (14–99%), strongly dependent on the electronic character of the aryl halide [54].
3. Bioactivity of 2- and 16E-arylideneandrostane Derivatives
The 16E-arylideneandrostane steroidal skeleton has emerged as a relevant template to develop potential anticancer agents. Among all different types of arylidene steroids this is the most common. On the other hand, A-ring arylideneandrostane derivatives were also described but much less frequently [31,54,55]. In addition to cytotoxic effects, several studies reported other biological activities, such as aromatase inhibition, anti-inflammatory, neuroprotective and skeletal muscle relaxing activities and tissue-selective androgen receptor modulator effects [38,57,58]. A selection of the most relevant 16E-arylideneandrostane derivatives is presented in Table 1. The compounds selected presented better results than the respective positive controls in biological assays. When data for positive control is not present, the steroid with the best results is presented. Concerning relevant antiproliferative compounds, several arylideneandrostane derivatives, namely the steroids 2–7 (Table 1), have shown promising results [16,55,57,59,60,61]. In this context, Huang et al. reported the cytotoxic effects of a series of novel DHEA derivatives bearing a modified v-triazole ring at the C-16 position on different human cancer cells. Through the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay, these authors observed that compound 2 (Table 1), which contains a 4-iodophenyl system attached to the triazole ring, displayed the highest antiproliferative activity in HepG2 and MCF-7 cells [half maximal inhibitory concentration (IC50) of 9.10 and 9.18 μM, respectively]. Interestingly, these IC50 values are even lower than the estimated for the positive control 5-fluorouracil (5-FU), an antineoplastic drug widely used in clinical practice to treat multiple solid tumors [58]. Furthermore, flow cytometry experiments in the HepG2 cell line demonstrated that steroid 2 exerted antiproliferative effects by arresting cells in the G2 phase, inducing apoptosis [16]. The androstane derivative 3 (Table 1), bearing a 3-chlorobenzylidene at C-16, and analogues were synthesized by Vosooghi et al. and their cytotoxicity was evaluated through the MTT assay. The results evidenced that this steroid 3 was the most potent, especially against KB and T47-D cells (IC50 values of 0.6 and 1.7 μM, respectively) [60]. In another study, the amide androstane 4 with a chlorine atom attached to C-3 (Table 1) revealed potent antiproliferative effects against a panel of leukemia cell lines. In fact, the determined IC50 values for this steroid 4 against CCRF-CEM, K-562, RPMI-8226 and SR cells were 3.94, 2.61, 6.90, and 1.79 μM, respectively [61]. More recently, the synthesis and biological evaluation of ferrocenyl androstane conjugates was reported. The antiproliferative activity of these compounds was assessed and the steroid 5 (Table 1) was the most potent, being determined an IC50 of 1.2 μM in colon cancer HT-29 cells. This IC50 value is very relevant when compared with the positive control, cisplatin (Cis) (IC50 = 66 μM) [59]. Besides in vitro studies using cell lines, Chattopadhaya et al. also performed an in vivo cytotoxicity evaluation using a mouse model, the hollow fiber assay. In this study, interesting results were observed for the 3,4,5-trimetoxibenzylidene 6 (Table 1). In fact, for this steroid, a good score for inhibition of tumoral cells in the subcutaneous (S.C.) fiber implants (8/20) was determined, while for intraperitoneal (I.P.) fiber implants, the value significantly decreased (2/20) [57]. In a very similar study, Bansal et al. synthesized and tested a series of 16E-arylideneandrostene derivatives bearing tertiary amine functionalities. Of these, the aminosteroid 7 (Table 1), possessing a diethylaminoalkoxy group, was the most promising molecule with a score of 12 for I.P. and 8 for S.C. in the in vivo hollow fiber assay [62].
Several arylidenesteroids have been reported as aromatase inhibitors, particularly the 16E-arylideneandrostane derivatives 8–11 (Table 1). Aromatase is a cytochrome P450 enzyme (CYP19) responsible for the conversion of androgens into estrogens through an aromatization reaction. This enzyme plays a crucial role in endocrine physiology and in estrogen-dependent tumors such as breast and endometrial cancers [63]. Therefore, potent aromatase inhibitors (AIs) have been clinically relevant in the treatment of the referred diseases. However, the side effects of AIs used in clinical practice, such as muscle-skeletal pain, osteoporosis, and cardiovascular disease, are limiting and justify further research in this field [64,65,66,67,68]. In this context, the steroids 8–11 (Table 1) revealed potent aromatase inhibitory activity in in vitro assays. Noteworthy, the determined IC50 values for the enzyme inhibition are much lower than the observed for aminoglutethimide, used as a positive control [54,69,70,71].
16E-Arylidene steroidal derivatives were also studied as a new class of neuroprotective agents for the treatment of Alzheimer’s and Parkinson´s diseases. In this scope, Singh et al. reported some interesting results, being the 4-aza-16E-pyridylene derivative 12, prepared from DHEA (Table 1), the most promising. This compound was found to be a relevant neuroprotective agent, producing effects on TNF-α levels (a pro-inflammatory cytokine) in brain serum of rats comparable to the standard drug celecoxib (CEL) and better results than dexamethasone (DEX) (Table 1) [72].
More recently, Li et al. prepared 2-arylideneandrostene derivatives and evaluated their potential anti-inflammatory properties, including their effect on NO release by microglia BV-2 cells activated by lipopolysaccharide (LPS), as well as their cytotoxicity. Among all synthesized derivatives, steroids 13 and 14 (Table 1), presenting, respectively, 3-chloro and 3,4,5-trimethoxy groups at C-2 position, displayed the most relevant results. In fact, the calculated IC50 values for anti-inflammatory effect were 2.69 and 3.28 μM, respectively, which were lower than the positive control, minocycline (5.97 μM) [73].
Despite these promising results, other studies carried out with 2- and 16E-arylideneandrostrane derivatives were described, but less relevant biological effects were observed [17,38,50,55,64,67]. In addition, Semenenko and coworkers prepared 16E-arylidenedehydroepiandrosterones, as well as 2-arylidenesteroidal derivatives of cholestanone, to be used as chiral dopants for cholesteric liquid crystal composition [74]. Another study reported the synthesis of a new series of quaternary ammonium salts of 16E-[4-(2-alkylaminoethoxy)-3-methoxybenzylidene]androstane derivatives as skeletal muscle relaxants. The biological results showed that these compounds produced varied degrees of muscle relaxant activity. However, they also inhibited acetylcholinesterase activity in low concentrations and therefore are not suitable for use as muscle relaxants [36]. Additionally, Acharya and coworkers prepared new 16E-arylidene steroids linked to nitrogen mustards as hybrid compounds and evaluated their cytotoxic effects aiming to develop antineoplastic agents against leukemia. However, the specificity of these compounds towards leukemia cells still remains unaffected and reduced potency was observed when compared to earlier reports for 16E-arylidene steroids [61,62]. Further studies suggested that these derivatives present different mechanisms of action in leukemia cells [17]. More recently, Brito et al. reported the synthesis, antiproliferative effects, and in silico studies of novel 16E-arylidene-4-azaandrost-5-enes as prostate cell growth inhibitors. Despite their less potent antiproliferative effects in comparison with the positive control (5-FU), an interesting selectivity toward cancer cell lines was found for all azaandrostenes, presenting low cytotoxicity in non-tumoral human fibroblasts. Furthermore, molecular docking studies predicted that these 4-azaandrostene derivatives can interact with 5β-reductase, a surrogate of 5AR, and with other common targets of steroidal drugs, which could be important for further studies [48].
4. Bioactivity of 21E-arylidenepregnene Derivatives
Arylidenesteroidal derivatives of progesterone and pregnenolone and other similar pregnanes constitute a smaller group than arylideneandrostanes, and they have mainly been studied as potential antitumoral agents. Of these, the most potent arylidenepregnene derivatives reported until now are presented in Table 2. The steroids 15 (with a 4-fluorophenyl group attached to C-21) and 16 (bearing a 4-nitrophenyl group attached to C-21 and an imine group at C-20) were prepared and tested by Banday and coworkers. Their effects on tumoral cell proliferation were assessed through National Cancer Institute-60 (NCI-60) Human Tumor Cell Lines Screen. This NCI-60 method involves 60 different human tumor cell lines, representing leukemia, melanoma, and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney cancers, to identify and characterize novel compounds with growth inhibition or killing of tumor cells. This method was designed to screen up to 3000 small molecules for potential anticancer activity in a short period of time [75]. The best results for compounds 15 and 16 were observed in HCT-15 and MCF-7 cell lines, respectively, being determined an IC50 value of 0.81 μM for compound 15 in HCT-15 cells, and an IC50 value of 0.60 μM for the 21E-arylidenepregnene derivative 16 in MCF-7 cells [69]. A more recent study described the synthesis of novel 4′-acylamino modified 21E-benzylidene steroidal derivatives and their cytotoxic activity. The toxicity of these steroids was assessed by a brine shrimp microtiter-plate method and by the MTT assay against HeLa and MCF-7 cells [18,38]. Within these arylidenesteroids, the 21E-4-(2-fluorobenzamido)benzylidene derivative 17 (Table 2) seemed to be the most active. In fact, at a concentration of 30 μg·mL−1, this steroid originated a 58 and 64% growth inhibition of HeLa and MCF-7 cells, respectively, and their toxicity against brine shrimp was considered weak (median lethal concentration (LC50) > 50 μg·mL−1) [38]. Interestingly, Abood and coworkers reported the synthesis and antimicrobial study of 2,21E-arylidene progesterone derivatives. These compounds were tested using Streptococcus pneumoniae and Staphylococcus aureus as gram positive and Pseudomonas aeruginosa and Escherichia Coli as gram negative bacteria and their antifungal activity were studied against Candida albicans and Aspergillus niger, and ampicillin (AMP) (at 100 ng·mL−1) was used as a standard drug (positive control). The obtained results indicated that compound 18, at 100 ng·mL−1 (Table 2) has an inhibition zone (in mm) of S. pneumoniae and S. aureus proliferation (30 and 24 mm, respectively) larger than the standard drug (20 and 22 mm) [76].
5. Bioactivity of 16E-arylidenoestrone and 16E-arylidenoestradiol Derivatives
It is well known that estrogenic hormones have an important contribution to estrogen-dependent diseases, being breast cancers primarily initiated and stimulated by estrogens the majority of these conditions [77]. Consequently, the structural modification of estrone and estradiol in different positions to prepare bioactive compounds in this context has been the focus of intensive research. In this context, 16E-arylidenoestrone derivatives have also been prepared, and their bioactivity has been evaluated. For example, these arylidenes were studied as 17β-HSD type 1 inhibitors. This enzyme is responsible for the reduction of the 17-ketone group of estrone to afford 17β-estradiol, the most potent estrogen. Therefore, 17β-HSD type 1 is considered a relevant therapeutic target in the treatment of estrogen-dependent cancers and endometriosis [78,79,80,81]. Thus, over the years, potent 17β-HSD type 1 inhibitors have been designed and synthesized. In addition, selectivity for 17β-HSD type 1 over other isoforms and absence of estrogenic effects are of high relevance [82]. In this context, Allan et al. and Poirier et al. designed and prepared 16E-arylidenoestrone derivatives as 17β-HSD type 1 inhibitors, and promising results were achieved (Table 3). For example, steroid 19 (Table 3), prepared by Poirier and coworkers, presented a high inhibitory effect with an IC50 of 3.4 μM, which is a value lower than the observed for the positive control, estradiol (7.3 μM) [35]. In addition, Allan et al. modified estrone in different positions, namely C-6, C-16, and C-17, and the prepared structures were tested against 17β-HSD type 1 and type 2. Among all these derivatives, steroid 20 (Table 3), bearing a 4-dimethylaminobenzylidene group at C-16, presented very relevant results. In fact, at 10 μM, this derivative led to a 72% inhibition of 17β-HSD type 1 activity, while the 17β-HSD type 2 inhibition was only 13%. This research group also verified that further modifications of steroid 20 led to a loss of selectivity [83]. More recently, Wang et al. developed a new series of 16E-benzylidene modified 2-methoxyestradiol analogues. 2-Methoxyestradiol (2ME) is an endogenous estradiol metabolite that has been studied as an antitumor agent and several mechanisms of action for this compound have been explored. Interestingly, this steroid seems to mainly lead to neoangiogenesis inhibition, microtubule disruption, and upregulation of the extrinsic and intrinsic apoptotic pathways [84]. In this scope, Wang and coworkers used 2ME as starting material for the synthesis of 16E-benzylidenestradiol derivatives and their potential antitumoral interest was evaluated. Of these prepared compounds, steroid 21 (Table 3) showed a potent antiangiogenic activity. Moreover, further studies suggested that this compound suppresses the tumor growth in about 50% in human breast cancer (MCF-7) xenograft models without relevant side effects. The action mechanism studies suggested that steroid 21 targeted the epithelial to mesenchymal transition process in MCF-7 cells and inhibited human umbilical vein endothelial cells (HUVEC) migration, contributing to angiogenesis interruption [85].
Minu and coworkers also synthesized novel 16E-arylidene derivatives from estrone, and their biological activity was assessed. These compounds have been studied at NCI for their antineoplastic activity against a 60-cell lines panel and have also been tested for their in vitro estrogenic and anti-estrogenic activities, but the results obtained were not as promising as observed in other previously reported studies. Moreover, the measured ER binding affinity was also assessed, and the values determined were low [86]. In addition, novel 21E-benzylidene steroidal derivatives were synthesized from progesterone by Fan et al. These novel compounds were tested through MTT assay for their cytotoxicity against brine shrimp and murine Lewis lung carcinoma (LLC) cells. In general, this series of new steroids showed a weak cytotoxicity toward these cells [87]. Shan and coworkers also reported the preparation of 3β,7α,11α-trihydroxy-pregn-21-benzyliden-5-en-20-one derivatives and their cytotoxic activity. However, all these compounds showed a lower cytotoxic activity against EC109 cells than the positive control, oridonin [88]. More recently, Canário et al. prepared Δ9,11-estrones as potential antiproliferative agents, including a 16E-benzylidene derivative from Δ9,11-estrone. The effect of this compound on cell proliferation was assessed against several cell lines and it was verified that the introduction of a ∆9,11 double bond and a 16E-benzylidene group in estrone lead to an increase of the cytotoxic activity against the hormone-dependent breast cancer cells MCF-7 and T47-D, with IC50 values of 25.14 and 25.06 μM, respectively. However, when compared to 5-FU, as the positive control, this compound showed to be less potent (IC50 = 1.71 and 0.54 μM, respectively) [89].
6. Importance of Steroidal Arylidene Derivatives as Synthetic Intermediates of Bioactive Molecules
In addition to their biological activity, steroidal arylidenes are also versatile synthetic intermediates in the preparation of other bioactive structures. In fact, these steroids have been used in the introduction of diverse chemical groups present in bioactive compounds, such as oximes, hydroxyl and hydrazones [69,90,91,92,93], and are particularly useful in several heterocyclization reactions. In this context, over the years, a large number of bioactive heterocyclic steroidal derivatives have been synthesized, and some of them are already being clinically used [94,95]. Interestingly, diverse heterocyclic compounds, including arylpyrazolines and pyrazoles, arylpyrimidines, oxindoles, pyridones and pyridines as well as spiro-pyrrolidines were prepared from arylidenesteroids [4,18,42,49,53,56,76,93,96,97,98,99,100,101,102,103,104,105,106].
Pyrazolines seem to be the most frequently prepared heterocycles from steroidal arylidenes (Figure 5). A common approach to synthesize C-17 pyrazolinyl derivatives is the heterocyclization of 21E-arylpregnenolones in the presence of hydrazine hydrate (NH2·NH2·H2O) in glacial acetic acid (AcOH) under reflux during approximately 2 h [4,96,107]. Diverse approaches to obtain 16,17-pyrazolinyl steroids from 16E-arylidenedehydroepiandrosterone derivatives have also been reported (Figure 5). Thus, different androstane-N-acylated pyrazolines were prepared in the presence of NH2NH2.H2O in AcOH or propionic acid at reflux temperature after 2–9 h [97,100,108]. In addition, Singh et al. reported the synthesis of pyrazolinyl androstanes by using only NH2NH2.H2O in 1,4-dioxane under reflux for 5 h [97]. Moreover, the reaction of these 16E-arylideneandrostanes with phenylhydrazine hydrate in anhydrous MeOH under reflux during 12 h afforded the corresponding phenylpyrazolinyl substituted derivatives [97]. Furthermore, Naggar et al. synthesized estrone-N-substituted pyrazolines from 16E-arylidenoestrones by condensation with methylhydrazine or phenylhydrazine in refluxing dioxane for 5 h [99]. To stress that the information about the stereochemistry of 16,17-pyrazolinyl steroids is not always clear. In fact, some authors indicated that C-C bound between C-16 and C-5′ is β [97,100]. On the other hand, the study reported by El-Naggar et al. do not present any information about the stereochemistry of these compounds [99].
The synthesis of steroidal arylpyrimidines and pyridines from 16E- and 21E-arylidenesteroids was also described (Figure 6). In fact, Ke et al. described the preparation of steroidal[17,16-d]pyrimidines using 3 as starting material and guanidine nitrate under basic catalysis and reflux during 1 h [49]. In addition, Huang et al. reported the synthesis of steroidal[17,16-d]triazolopyrimidines using 3-aminotriazole, again using a basic catalyst at reflux during 30 h [41]. Furthermore, 2’-aminopyrimidines and 2’-cyanoiminopyrimidines were prepared from 16E-arylidenoestrone derivatives using guanidine hydrochloride or 2-cyanoguanidine and sodium ethoxide in EtOH, and the mixture was refluxed for 4–6 h [102]. Different steroidal pyridines were also synthesized from 21E-arylidenesteroids in the presence of malononitrile and sodium ethoxide, at reflux temperature for 8 h, in a single reaction. Interestingly, sodium ethoxide acted as a bifunctional species, being simultaneously the ethoxy source and the promoter of reactions between α,β-unsaturated ketones and malononitrile [101].
The preparation of steroidal spiro-oxindoles and spiro-pyrrolidines from arylidenesteroids was reported by Yu et al. and Gavaskar et al. (Figure 7). In this context, a series of novel steroidal spiro-pyrrolidinyl oxindoles was synthesized through a 1,3-dipolar cycloaddition of azomethine ylides generated from the decarboxylative condensation of isatin and sarcosine. For this, the arylidenesteroids 23 were mixed with isatin and sarcosine in 1,4-dioxane/MeOH (1:1) and this reactional mixture was left under reflux for 5 h to afford the referred products in excellent yields [56]. More recently, Gavaskar et al. reported a 1,3-dipolar cycloaddition to prepare novel steroidal dispiropyrrolidine heterocycles. This reaction occurred through an azomethine ylide generated from 1,2-phenylenediamine, ninhydrin, sarcosine, and a 16E-arylidenoestrone derivative in the ionic liquid N-(1-acryloyl)-N-(4-cyclopentyl)piperazinium phosphate, previously synthesized by this group, 120 °C [104,109]. Most of these final heterocyclic products have been developed and evaluated as antiproliferative and anti-inflammatory agents, as well as aromatase and 5α-reductase (5AR) inhibitors. 5AR is the enzyme responsible for the NADPH-dependent conversion of testosterone (T) to the more potent androgen dihydrotestosterone (DHT). There are two distinct isozymes, differentially expressed in human tissues, the type 1 5AR (5AR-1) and type 2 5AR (5AR-2) [107,108]. Additionally, DHT have an important role in benign prostatic hyperplasia (BHP), a common disease in aging men [110]. These facts have contributed to the increase of the interest in finding potent 5AR inhibitors. Finasteride and dutasteride are the most effective steroidal analogs clinically used. However, both molecules cause several side effects such as erectile dysfunction, abnormal ejaculation, impotence, abnormal sexual function, decreased sexual desire, and gynecomastia [111,112]. Hence, it becomes important to develop more potent and safer 5AR inhibitors.
The most promising steroidal heterocyclic derivatives prepared from arylidenesteroids that have been reported until the moment are presented in Table 4. Of these, steroid 24 has relevant antiproliferative effects, as a very low IC50 values (0.24 and 0.25 μM, respectively) against HT-29 and HCT-15 cells were determined [4]. Furthermore, pyrimidine 25 has also exhibited interesting antiproliferative effects against HepG2, Huh-7 and SGC-790 cells, presenting IC50 values lower than positive control (5.41, 5.65 and 10.64 μM, respectively). In addition, the steroids 26–30 were evaluated as potential 5AR inhibitors, and some interesting results were obtained (Table 4). Compound 26 presented relevant 5AR-1 inhibitory effects in comparison with finasteride (IC50 values are 14.5 μM and 21.6 μM, respectively), whereas derivatives 27–30 revealed potent 5AR-2 inhibitory effects [96,98]. Of these, pyrazolines 29 and 30 showed the most promising results (the IC50 values for 5AR-2 inhibition were 7.3 and 8.2 nM, respectively), while 27 and 28 presented apparently lower inhibitory activity for 5AR-2 (the determined IC50 were 13.90 and 14.20 μM, respectively) [96,98]. Steroid 31 (Table 4) also showed to be a potential antiproliferative agent against NCI-H460 and HeLa cells, presenting good IC50 values (10.30 and 12.50 μM, respectively). Particularly, in HeLa cell line, this compound led to cell cycle arrest at S phase [18]. Yu and coworkers also reported some molecules with interesting results for antiproliferative activity, namely the steroidal spiropyrrolidinyl oxindoles 32–34, synthesized from DHEA (Table 4). Particularly, pyrazolidine 32 showed good antiproliferative activity against SMMC-7721 (IC50 = 4.30 μM) and MCF-7 (IC50 = 2.06 μM) cells, being more potent than positive control (5-FU) [56]. Steroid 33 also presented relevant results in SMMC-7721 and MGC-803 cell lines (the determined IC50 were 6.05 and 5.79 μM, respectively). In addition, flow cytometry studies demonstrated that this compound caused cellular early apoptosis and cell cycle arrest at G2/M phase in a concentration and time-independent mode [56]. Additionally, compound 34 showed to be the most potent derivative in SMMC-7721 cells (IC50 = 0.71 μM) [56]. Steroid 35 (Table 4) also stood out from novel pregnenolone derivatives synthesized by Choudhary et al., presenting an interesting effect against MDA-MB 231 cells (the determined IC50 value for antiproliferative activity was 0.91 μM). In addition, this result showed that steroid 35 is more potent than positive control, DOX (IC50 = 1.23 μM) [93].
Mitchell and coworkers synthesized 16-substituted 4-azasteroids as tissue-selective androgen receptor modulators, using 16E-arylideneandrostane derivatives as synthetic intermediates. In general, this class of steroids displayed potent AR binding and agonist activity. Of these novel molecules, 36 (Table 4) showed the most promising results, exhibiting an osteoanabolic and tissue-selective profile in ovariectomized (OVX) and orchiectomized (ORX) rat models. In OVX rats (models with bone loss that simulate estrogen deficiency), the main evaluated parameter was bone formation rate (BFR) and it was reported as a percentage of the DHT effect at a standard dose. On the other hand, the selectivity was assessed by measuring the effects on ventral prostate (VP) and seminal vesicles (SV) through ORX male rat models. Interestingly, compound 36 exhibited a BFR of 120%, relative to DHT, an increase in the VP of 3% and a 21% of increase in SV weight, confirming its tissue selectivity (Table 4) [113]. Furthermore, the preparation and biological evaluation of a new series of 16E-arylidene substituted steroidal oximes was also reported from 16E-arylideneandrostanes [57,114]. In this context, Chattopadhaya et al. used the hollow fiber assay to assess the effects of novel 16E-arylideneandrostane oxime derivatives [57]. Interestingly, the oxime 37 presented the best result, exhibiting a good score for inhibition of tumoral cells in the S.C. fiber implants (4/20) and for I.P. fiber implants (6/20) (Table 4). Moreover, Dubey and coworkers also synthesized 16E-benzylidene substituted steroidal oximes from 16E-arylideneepiandrosterone derivatives [114]. These compounds were evaluated for in vitro antineoplastic activity at NCI against three cell lines. Within these oximes, the 3β-hydroxy derivative 38 (Table 4) was found as the most potent. The results indicated that in the 3 cell lines tested, NCI-H460 (lung), MCF-7 (breast) and SF-268 (central nervous system), this oxime 38 showed negative values for the percentage of cell growth (−44, −44 and −79, respectively) (Table 4) [114]. In addition, the same authors also reported the preparation of 16E-benzylidene substituted 3,17-dioximinoandrostene derivatives as anticancer agents and interesting results were obtained [92]. These steroids were also biologically tested at NCI against the same 3 cell lines. Of these, compound 39 showed to be the most active dioxime against the tumoral cells, presenting the lowest percentage values for cell growth in NCI-H460, MCF-7 and SF-268 cells (−11, 5, and −8, respectively) (Table 4).
7. Conclusions
Steroids constitute an important group of structurally related natural, semi-synthetic, and synthetic compounds with remarkable functions, including regulatory and signaling activities. In the last three decades, steroidal arylidene derivatives have been prepared and screened for a range of biological activities and used as synthetic intermediates, with special attention to bioactive heterocyclic steroids.
Arylidenesteroids are usually prepared by an aldol condensation under mild reactional conditions and mainly comprise three distinct groups based on starting materials: 2- and 16E-arylideneandrostane, 21E-arylidenepregnane, and 16E-arylidenoestrone derivatives. A substantial number of these steroids have shown significant antiproliferative activity when compared to reference compounds. In addition to this cytotoxic activity, a considerable number of 16E-arylideneandrostane derivatives were also studied as aromatase inhibitors with promising results, and a few 16E-arylidenoestrone derivatives were studied as 17β-HSD type 1 inhibitors. In general, the most active arylidenesteroids usually comprise heteroatoms or halogens on their structures.
In addition, steroidal arylidene derivatives have also emerged as very important synthetic intermediates to obtain steroidal heterocycles as arylpyrazolines and pyrazoles, arylpyrimidines and pyridines, spiro-oxindoles, and spiro-pyrrolidines, among other bioactive systems. Within these, arylpyrazoline derivatives stand out as 5AR inhibitors and antiproliferative agents.
In conclusion, due to the straightforward synthesis of arylidenesteroids and their bioactivity, as well as the inherent chemical reactivity of α,β-unsaturated ketones, useful in the preparation of other derivatives, this class of compounds has been of high interest in the last years. This review highlighted the most relevant points in this context and evidenced, for the first time, the great relevance of this particular group of steroids, which should be of high interest in further studies looking to new, more active, and safer bioactive compounds with potential clinical interest.
Author Contributions
Conceptualization, S.S. and V.B.; methodology, S.S. and V.B.; software, ChemDraw version 20.0, Mendeley Desktop, version 1.19.8.; investigation, S.S. and V.B.; writing—original draft preparation, V.B.; writing—review and editing, S.S., G.A. and P.A.; supervision, S.S.; funding acquisition, S.S., G.A., P.A. and V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by FEDER funds through the POCI-COMPETE 2020-Operational Programme Competitiveness and Internationalization in Axis I-Strengthening Research, Technological Development and Innovation (Project No. 007491), National Funds by Foundation for Science and Technology (FCT) (Project UID/ Multi/00709 and grant SFRH/BD/131059/2017) and C4-Cloud Computing Competences Center project (CENTRO-01-0145-FEDER-000019).
Acknowledgments
All authors acknowledge the previously referred funding support, including the PhD grant SFRH/BD/131059/2017 (VB). Thanks are also due to Sílvia Socorro (CICS-UBI) and University of Beira Interior.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lednicer, D. Steroid Chemistry at a Glance; Wiley: Hoboken, NJ, USA, 2011; ISBN 9780470660850. [Google Scholar]
- Burger, A.; Abraham, D.J.; Rotella, D.P. Burger’s Medicinal Chemistry, Drug Discovery and Development; Wiley: Hoboken, NJ, USA, 2010; Volume 7, ISBN 9780470770085. [Google Scholar]
- Latham, K.A.; Zamora, A.; Drought, H.; Matejuk, A.; Offner, H.; Edward, F. Estradiol Treatment Redirects the Isotype of the Autoantibody Response and Prevents the Development of Autoimmune Arthritis. J. Immunol. 2003, 171, 5820–5827. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.H.; Mir, B.P.; Lone, I.H.; Suri, K.A.; Kumar, H.M.S. Studies on Novel D-Ring Substituted Steroidal Pyrazolines as Potential Anticancer Agents. Steroids 2010, 75, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, M.A.; Nafie, M.S.; Elmegeed, G.A.; Ali, I.A.I. Auspicious Role of the Steroidal Heterocyclic Derivatives as a Platform for Anti-Cancer Drugs. Bioorg. Chem. 2017, 73, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Vil, V.A.; Terent, A.O.; Savidov, N.; Gloriozova, T.A. Hydroperoxy Steroids and Triterpenoids Derived from Plant and Fungi: Origin, Structures and Biological Activities. J. Steroid Biochem. Mol. Biol. 2019, 190, 76–87. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Progress in Lipid Research Antitumor and Hepatoprotective Activity of Natural and Synthetic Neo Steroids. Prog. Lipid Res. 2020, 79, 101048. [Google Scholar] [CrossRef]
- Xiao, J.; Gao, M.; Fei, B.; Huang, G.; Diao, Q. Fitoterapia Nature-Derived Anticancer Steroids Outside Cardica Glycosides. Fitoterapia 2020, 147, 104757. [Google Scholar] [CrossRef]
- Dubey, R.K.; Oparil, S.; Imthurn, B.; Jackson, E.K. Sex Hormones and Hypertension. Cardiovasc. Res. 2002, 53, 688–708. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, P.J.; Blum, K.; Trachtenberg, M.C. Steroid Receptors and Disease: Cancer, Autoimmune, Bone, and Circulatory Disorders. Trends Pharmacol. Sci. 1988, 10, 122. [Google Scholar] [CrossRef]
- Holst, J.P.; Soldin, S.J.; Tractenberg, R.E.; Guo, T.; Kundra, P.; Verbalis, J.G.; Jonklaas, J.; Clinical, G. Use of Steroid Profiles in Determining the Cause of Adrenal Insufficiency. Steroids 2006, 72, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Jursic, B.S.; Kumar, S.; Creech, C.C.; Neumann, D.M. Novel and Efficient Synthesis and Antifungal Evaluation of 2,3-Functionalized Cholestane and Androstane Derivatives. Bioorg. Med. Chem. Lett. 2020, 20, 7372–7375. [Google Scholar] [CrossRef]
- Banday, A.H.; Zargar, M.I.; Ganaie, B.A. Synthesis and Antimicrobial Studies of Chalconyl Pregnenolones. Steroids 2011, 76, 1358–1362. [Google Scholar] [CrossRef]
- Na, M.S.; Tantawy, M.A.; Elmgeed, G.A. Screening of Different Drug Design Tools to Predict the Mode of Action of Steroidal Derivatives as Anti-Cancer Agents. Steroids 2019, 152. [Google Scholar] [CrossRef]
- Lemke, T.L.; Williams, D.A.; Roche, V.F.; Zito, S.W. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Huang, X.; Shen, Q.; Zhang, H.; Li, J.; Tian, Y.; Quan, Z. Design and Synthesis of Novel Dehydroepiandrosterone Analogues as Potent Antiproliferative Agents. Molecules 2018, 23, 2243. [Google Scholar] [CrossRef] [Green Version]
- Acharya, P.C.; Bansal, R.; Kharkar, P.S.; Res, D.; Bansal, R. Hybrids of Steroid and Nitrogen Mustard as Antiproliferative Agents: Synthesis, in Vitro Evaluation and in Silico Inverse Screening Authors. Drug Res. (Stuttg). 2017. [Google Scholar] [CrossRef] [Green Version]
- Fan, N.; Tang, J.; Li, H.; Li, X.; Luo, B.; Gao, J. Synthesis and Cytotoxic Activity of Some Novel Steroidal C-17 Pyrazolinyl Derivatives. Eur. J. Med. Chem. 2013, 69, 182–190. [Google Scholar] [CrossRef]
- Gogoi, J.; Bezbaruah, P.; Saikia, P.; Goswami, J.; Gogoi, P.; Boruah, R.C. Synthesis of a Novel Class of Steroidal Tetrazolo[1,5-a]Pyridines Via Intramolecular 1,3-Dipolar Cycloadditions. Tetrahedron Lett. 2012, 53, 1497–1500. [Google Scholar] [CrossRef]
- Chowdhury, P.; Borah, J.M.; Goswami, P.; Das, A.M. A Convenient Synthesis of the Side Chain of Loteprednol Etabonate - an Ocular Soft Corticosteroid from 20-Oxopregnanes Using Metal-Mediated Halogenation as a Key Reaction. Steroids 2011, 76, 497–501. [Google Scholar] [CrossRef]
- Delong, W.; Yongling, W.; Lanying, W.; Juntao, F.; Xing, Z. Design, Synthesis and Evaluation of 3-Arylidene Azetidin-2-Ones as Potential Antifungal Agents Against Alternaria Solani Sorauer. Bioorg. Med. Chem. 2017, 25, 6661–6673. [Google Scholar] [CrossRef]
- Szymanska, E.; Kiec-Kononowicz, K. Antimycobacterial Activity of 5-Arylidene Aromatic Derivatives of Hydantoin. Farm. 2002, 57, 355–362. [Google Scholar] [CrossRef]
- Ottanà, R.; Maccari, R.; Giglio, M.; Del, A.; Cappiello, M.; Mura, U.; Cosconati, S.; Marinelli, L.; Novellino, E.; Sartini, S.; et al. Identification of 5-Arylidene-4-Thiazolidinone Derivatives Endowed with Dual Activity as Aldose Reductase Inhibitors and Antioxidant Agents for the Treatment of Diabetic Complications. Eur. J. Med. Chem. 2011, 46, 2797–2806. [Google Scholar] [CrossRef]
- Stadnicka, K.; Broda, A.; Filipek, B.; Kiec, K. Synthesis, Structure–Activity Relationship of Some New Anti-Arrhythmic 5-Arylidene Imidazolidine-2,4-Dione Derivatives. Eur. J. Med. Chem. 2005, 40, 259–269. [Google Scholar] [CrossRef]
- Li, N.; Xin, W.; Yao, B.; Wang, C.; Cong, W.; Zhao, F.; Li, H.; Hou, Y.; Meng, Q.; Hou, G. Antitumor Agents with Biological Evaluation in Vitro and in Vivo. Eur. J. Med. Chem. 2018, 147, 21–33. [Google Scholar] [CrossRef]
- Amr, A.E.; Mohamed, A.M.; Mohamed, S.F.; Abdel-hafez, N.A.; Hammam, A.E.G. Anticancer Activities of Some Newly Synthesized Pyridine, Pyrane and Pyrimidine Derivatives. Bioorg. Med. Chem. 2006, 14, 5481–5488. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumar, A.; Saini, D. Experimental Parasitology Novel Arylidene Derivatives of Quinoline Based Thiazolidinones: Synthesis, in Vitro, in Vivo and in Silico Study as Antimalarials. Exp. Parasitol. 2018, 185, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Valla, A.; Valla, B.; Cartier, D.; Le, R.; Labia, R.; Florent, L.; Charneau, S.; Schrevel, J.; Potier, P. New Syntheses and Potential Antimalarial Activities of New ‘Retinoid-like Chalcones. ’ Eur. J. Med. Chem. 2006, 41, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Maria, R.; Ottanà, R.; Rocchiccioli, M.; Marrazzo, A.; Cardile, V.; Carol, A.; Graziano, E.; Amodeo, P.; Mura, U.; et al. Structure-Activity Relationships and Molecular Modelling of New 5-Arylidene-4-Thiazolidinone Derivatives as Aldose Reductase Inhibitors and Potential Anti-Inflammatory Agents. Eur. J. Med. Chem. 2014, 81, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hee, S.; Seog, G.; Yeong, J.; Yu, X.; Kim, H.; Hwan, D. Heme Oxygenase 1 Mediates Anti-Inflammatory Effects of 2′,4′,6′-Tris(Methoxymethoxy) Chalcone. Eur. J. Pharmacol. 2006, 532, 178–186. [Google Scholar] [CrossRef]
- Taylor, P.; Schultz, T.W.; Yarbrough, J.W. Trends in Structure-Toxicity Relationships for Carbonyl-Containing α,β-Unsaturated Compounds. SAR QSAR Environ. Res. 2007, 2, 37–41. [Google Scholar] [CrossRef]
- Eder, E.; Scheckenbach, S.; Deininger, C.; Hoffman, C. The Possible Role of α,β-Unsaturated Carbonyl Compounds in Mutagenesis and Carcinogenesis. Toxicol. Lett. 1993, 67, 87–103. [Google Scholar] [CrossRef]
- Aguilera, E.; Perdomo, C.; Espindola, A.; Corvo, I.; Faral-tello, P.; Robello, C.; Serna, E.; Torres, S.; De Bilbao, N.I.V.; Yalu, G. A Nature-Inspired Design Yields a New Class of Steroids Against Trypanosomatids. Molecules 2019, 8, 3800. [Google Scholar] [CrossRef] [Green Version]
- Al-masoudi, N.A.; Abdul-rida, N.A.; Kadhim, R.A.; Krug, S.J.; Engel, M.; Saeed, B.A. Synthesis and CYP17 a Hydroxylase Inhibition Activity of New 3α- and 3β-Ester Derivatives of Pregnenolone and Related Ether Analogues. Med. Chem. Res. 2016, 25, 310–321. [Google Scholar] [CrossRef]
- Poirier, D.; Chang, H.; Azzi, A.; Boivin, R.P.; Lin, S. Estrone and Estradiol C-16 Derivatives as Inhibitors of Type 1 17b-Hydroxysteroid Dehydrogenase. Mol. Cell. Endocrinol. 2006, 248, 236–238. [Google Scholar] [CrossRef]
- Bansal, R.; Guleria, S.; Young, L.C.; Harvey, A.L. Synthesis of Quaternary Ammonium Salts of 16E-[4-(2-Alkylaminoethoxy)-3-Methoxybenzylidene]Androstene Derivatives as Skeletal Muscle Relaxants. Steroids 2011, 76, 254–260. [Google Scholar] [CrossRef]
- Guo, H.; Wu, H.; Yang, J.; Xiao, Y.; Altenbach, H.; Qiu, G. Synthesis, Characterization and Biological Evaluation of Some 16E-Arylidene Androstane Derivatives as Potential Anticancer Agents. Steroids 2011, 76, 709–723. [Google Scholar] [CrossRef]
- Fan, N.; Han, Y.; Li, Y.; Gao, J.; Tang, J. Synthesis of Novel 4′-Acylamino Modified 21E-Benzylidene Steroidal Derivatives and Their Cytotoxic Activities. Steroids 2017, 123, 20–26. [Google Scholar] [CrossRef]
- Dubey, S.; Kaur, P.; Jindal, D.P.; Satyanarayan, Y.D.; Piplani, P. Synthesis, Evaluation and QSAR Studies of 16-(4 & 3,4-Substituted) Benzylidene Androstene Derivatives as Anticancer Agents. Med Chem 2008, 4, 229–236. [Google Scholar] [CrossRef]
- Jiang, C.; Guo, X.; Gong, J.; Zhu, T.; Zhang, H.; Guo, Y. Synthesis and Biological Evaluation of 21-Arylidenepregnenolone Derivatives as Neuroprotective Agents. Bioorg. Med. Chem. Lett. 2012, 22, 2226–2229. [Google Scholar] [CrossRef]
- Huang, L.H.; Zheng, Y.F.; Song, C.J.; Wang, Y.G.; Xie, Z.Y.; Lai, Y.W.; Lu, Y.Z.; Liu, H.M. Synthesis of Novel D-Ring Fused 7′-Aryl-Androstano[17,16-d][1,2,4]Triazolo[1,5-a]Pyrimidines. Steroids 2012, 77, 367–374. [Google Scholar] [CrossRef]
- Huang, L.-H.; Zheng, Y.-F.; Lu, Y.-Z.; Song, C.-J.; Wang, Y.-G.; Yu, B.; Liu, H.-M. Synthesis and Biological Evaluation of Novel Steroidal[17,16-d][1,2,4]Triazolo[1,5-a]Pyrimidines. Steroids 2012, 77, 710–715. [Google Scholar] [CrossRef]
- Ke, S. Recent Progress of Novel Steroid Derivatives and Their Potential Biological Properties. Mini-Reviews Med. Chem. 2018, 18, 745–775. [Google Scholar] [CrossRef]
- Hanson, J.R. Steroids: Partial Synthesis in Medicinal Chemistry. Nat. Prod. Rep. 2010, 27, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Frank, É.; Schneider, G. Synthesis of Sex Hormone-Derived Modified Steroids Possessing Antiproliferative Activity. J. Steroid Biochem. Mol. Biol. 2013, 137, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. Vogel’s Textboom of Practical Organic Chemistry, 5th ed.; Pearson: Delhi, India, 1989. [Google Scholar]
- Karatas, S.; Çapan, İ.; Servi, S. Synthesis of Indole and Benzimidazole Substituted Novel 16-Arylidene Steroid Derivatives. Lett. Org. Chem. 2019, 16, 884–890. [Google Scholar] [CrossRef]
- Brito, V.; Santos, A.O.; Almeida, P.; Silvestre, S. Novel 4-Azaandrostenes as Prostate Cancer Cell Growth Inhibitors: Synthesis, Antiproliferative Effects and Molecular Docking Studies. Comptes Rendus Chim. 2018, 22, 73–83. [Google Scholar] [CrossRef]
- Ke, S.; Shi, L.; Zhang, Z.; Yang, Z. Pyrimidines Derived from Dehydroepiandrosterone: A Convenient Synthesis, Antiproliferation Activity, Structure-Activity Relationships, and Role of Heterocyclic Moiety. Nat. Publ. Gr. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, S.; Kaur, P.; Paul, D. Synthesis and QSAR Studies of 16-(3-Methoxy-4-Substitued Benzylidene) Androstene Derivatives as Anticancer Agents. Indian J. Chem. 2010, 49B, 948–955. [Google Scholar] [CrossRef]
- Dubey, S.; Piplani, P.; Jindal, D.P. Synthesis and in Vitro Antineoplastic Evaluation of Certain 16-(4-Substituted Benzylidene) Derivatives of Androst-5-Ene. Chem. Biodivers. 2004, 1, 1529–1536. [Google Scholar] [CrossRef]
- Kakati, D.; Sarma, R.K.; Saikia, R.; Barua, N.C.; Sarma, J.C. Rapid Microwave Assisted Synthesis and Antimicrobial Bioevaluation of Novel Steroidal Chalcones. Steroids 2013, 78, 321–326. [Google Scholar] [CrossRef]
- Mótyána, G.; Molnár, B.; Wölfling, J.; Frank, É. Microwave-Assisted Stereoselective Heterocyclization to Novel Ring D-Fused Arylpyrazolines in the Estrone Series. Molecules 2019, 24, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Riebe, S.; Jopp, S.; Ehlers, P.; Frank, E.; Schneider, G.; Wölfling, J.; Villinger, A.; Langer, P. Synthesis of 16-E-([Aryl]Idene)-3-Methoxy-Estrones by a Palladium Catalysed Mizoroki-Heck Reaction. Tetrahedron Lett. 2017, 58, 2801–2803. [Google Scholar] [CrossRef]
- Peña-López, M.; Ayán-Varela, M.; Sarandeses, L.A.; Pérez Sestelo, J. Palladium-Catalyzed Cross-Coupling Reactions of Organogold(I) Reagents with Organic Electrophiles. Chemistry 2010, 16, 9905–9909. [Google Scholar] [CrossRef]
- Yu, B.; Shi, X.; Qi, P.; Yu, D.; Liu, H. Design, Synthesis and Biological Evaluation of Novel Steroidal Spiro-Oxindoles as Potent Antiproliferative Agents. J. Steroid Biochem. Mol. Biol. 2014, 141, 121–134. [Google Scholar] [CrossRef]
- Chattopadhaya, R.; Jindal, D.P.; Minu, M.; Gupta, R. Synthesis and Cytotoxic Studies of Hydroximino Derivatives of Some 16E-Arylidenosteroids. Arzneim. Forshung Drug Res. 2004, 556, 551–556. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Narváez-Pita, X.; Rheingold, A.L.; Meléndez, E. Ferrocene-Steroid Conjugates: Synthesis, Structure and Biological Activity. J. Organomet. Chem. 2017, 846, 113–120. [Google Scholar] [CrossRef]
- Vosooghi, M.; Yahyavi, H.; Divsalar, K.; Shamsa, H.; Kheirollahi, A.; Safavi, M.; Ardestani, S.K.; Sadeghi-Neshat, S.; Mohammadhosseini, N.; Edraki, N.; et al. Synthesis and in Vitro Cytotoxic Activity Evaluation of (E)-16-(Substituted Benzylidene) Derivatives of Dehydroepiandrosterone. Daru 2013, 21, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, R.; Acharya, P.C. Synthesis and Antileukemic Activity of 16E-[4-(2-Carboxy)Ethoxybenzylidene]-Androstene Amides. Steroids 2012, 77, 552–557. [Google Scholar] [CrossRef]
- Bansal, R.; Guleria, S. Synthesis of 16E-[3-Methoxy-4-(2-Aminoethoxy)Benzylidene]Androstene Derivatives as Potent Cytotoxic Agents. Steroids 2008, 73, 1391–1399. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, Aromatase Inhibitors, and Breast Cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Lonning, P.E.; Eikesdal, H.P. Aromatase Inhibition 2013: Clinical State of the Art and Questions That Remain to Be Solved. Endocr. Relat. Cancer 2013, 20, 183–201. [Google Scholar] [CrossRef] [Green Version]
- Coates, A.S.; Keshaviah, A.; Thu, B.; Del Mastro, L.; Smith, I.; Chirgwin, J.; Nogaret, J.; Pienkowski, T.; Wardley, A. Five Years of Letrozole Compared With Tamoxifen As Initial Adjuvant Therapy for Postmenopausal Women With Endocrine-Responsive Early Breast Cancer: Update of Study BIG 1-98. J. Clin. Oncol. 2007, 25, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Seruga, B.; Niraula, S.; Carlsson, L.; Ocaña, A. Toxicity of Adjuvant Endocrine Therapy in Postmenopausal Breast Cancer Patients: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2011, 103, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobral, A.F.; Amaral, C.; Correia-da-silva, G.; Teixeira, N. Unravelling Exemestane: From Biology to Clinical Prospects. J. Steroid Biochem. Mol. Biol. 2016, 163, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gasi, K.M.P.; Stankovic, S.M.; Sakac, M.N.; Stojanovic, S.Z.; Andric, S.; Molnar, D.; Kovac, R. New D-Modified Androstane Derivatives as Aromatase Inhibitors. Steroids 2001, 66, 645–653. [Google Scholar] [CrossRef]
- Banday, A.H.; Akram, S.M.M.; Shameem, S.A. Benzylidine Pregnenolones and Their Oximes as Potential Anticancer Agents: Synthesis and Biological Evaluation. Steroids 2014, 84, 64–69. [Google Scholar] [CrossRef]
- Bansal, R.; Thota, S.; Karkra, N.; Minu, M.; Zimmer, C.; Hartmann, R.W. Synthesis and Aromatase Inhibitory Activity of Some New 16E-Arylidenosteroids. Bioorg. Chem. 2012, 45, 36–40. [Google Scholar] [CrossRef]
- Bansal, R.; Guleria, S.; Thota, S.; Hartmann, R.W.; Zimmer, C. Synthesis of Imidazole-Derived Steroidal Hybrids as Potent Aromatase Inhibitors. Med. Chem. Res. 2013, 22, 692–698. [Google Scholar] [CrossRef]
- Singh, R.; Bansal, R. Investigations on 16-Arylideno Steroids as a New Class of Neuroprotective Agents for the Treatment of Alzheimer’s and Parkinson’s Diseases. ACS Chem. Neurosci. 2016. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, Y.; Gao, P.; An, X.; Sun, Y.; Sun, X.; Hou, Y.; Shan, L. Synthesis and Anti-Inflammatory Activity Evaluation of 2-Dehydroepiandrosterone Benzene Methyl Derivatives. Chinese J. Org. Chem. 2019, 39, 2625–2631. [Google Scholar] [CrossRef]
- Semenenko, A.N.; Babak, N.L.; Popov, S.S.; Svitlana, V.; Mazepa, A.V.; Lipson, V.V.; Semenenko, A.N.; Babak, N.L.; Popov, S.S.; Svitlana, V. New Ylidene and Spirocyclopropyl Derivatives of Cholestanone and Dehydroepiandrosterone Series and Their Ability to Induce Cholesteric Mesophase in Nematic Solvent. Synth. Commun. 2018, 48, 1008–1015. [Google Scholar] [CrossRef]
- Boyd, M.R. The NCI Human Tumor Cell Line (60-Cell) Screen. In Cancer Drug Discovery and Development: Anticancer Drugs Development Guide: Preclinical Screening, Clinical Trials, and Approval—Part I; Teicher, B.A., Andrews, P.A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2004; pp. 41–61. [Google Scholar]
- Abood, N.K.; Ibraheem, H.H. Synthesis, Characterize and Antimicrobial Study of New Chalcones and Pyrazole Derivatives from Progesterone. Int. J. Sci. Res. 2016, 5, 2319–7064. [Google Scholar] [CrossRef]
- Bernstein, L.; Ross, R.K. Endogenous Hormones and Breast Cancer Risk. Epidemiol. Rev. 1993, 15, 48–65. [Google Scholar] [CrossRef]
- Smuc, T.; Hevir, N.; Ribiˇ, M.; Husen, B.; Thole, H.; Laniˇ, T. Disturbed Estrogen and Progesterone Action in Ovarian Endometriosis. Mol. Cell. Endocrinol. 2009, 301, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Sawetawan, C.; Milewich, L.; Word, R.A.; Carr, B.R.; Rainey, W.E. Compartmentalization of Type I 17β-Hydroxysteroid Oxidoreductase in the Human Ovary. Mol. Cell. Endocrinol. 1994, 99, 161–168. [Google Scholar] [CrossRef]
- Fournet-Dulguerov, N.; Maclusky, N.J.; Leranth, C.Z.; Todd, R.; Mendelson, C.R.; Simpson, E.R.; Naftolin, F. Immunohistochemical Localization of Aromatase Cytochrome P-450 and Estradiol Dehydrogenase in the Syncytiotrophoblast of the Human Placenta*. J. Clin. Endocrinol. Metab. 1987, 65, 757–764. [Google Scholar] [CrossRef]
- Vihko, P.; Herrala, A. Control of Cell Proliferation by Steroids: The Role of 17HSDs. Mol. Cell. Endocrinol. 2006, 248, 141–148. [Google Scholar] [CrossRef]
- Kruchten, P.; Werth, R.; Marchais-oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Development of a Biological Screening System for the Evaluation of Highly Active and Selective 17β-HSD1-Inhibitors as Potential Therapeutic Agents. Mol. Cell Endocrinol. 2009, 301, 154–157. [Google Scholar] [CrossRef]
- Allan, G.M.; Lawrence, H.R.; Cornet, J.; Bubert, C.; Fischer, D.S.; Vicker, N.; Smith, A.; Tutill, H.J.; Purohit, A.; Day, J.M.; et al. Modification of Estrone at the 6, 16, and 17 Positions: Novel Potent Inhibitors of 17 -Hydroxysteroid Dehydrogenase Type 1. J. Med. Chem. 2006, 49, 1325–1345. [Google Scholar] [CrossRef]
- Mueck, A.O.; Seeger, H. 2-Methoxyestradiol- Biology and Mechanism of Action. Steroids 2010, 75, 625–631. [Google Scholar] [CrossRef]
- Wang, C.; Li, L.; Fu, D.; Qin, T.; Ran, Y.; Xu, F.; Du, X. Discovery of Chalcone-Modified Estradiol Analogs as Antitumour Agents That Inhibit Tumour Angiogenesis and Epithelial to Mesenchymal Transition. Eur. J. Med. Chem. 2019, 176, 135–148. [Google Scholar] [CrossRef]
- Minu, M.; Jindal, D.P. Synthesis and Biological Activity of 16-Arylidene Derivatives of Estrone and Estrone Methyl Ether. Indian J. Chem. 2003, 42, 166–172. [Google Scholar] [CrossRef]
- Fan, N.-J.; Bai, Y.-B.; Zhang, F.-Y.; Luo, B.; Tang, J.-J.; Zhang, Q.-Z.; Gao, J.-M. Synthesis and Cytotoxicity of Some Novel 21E-Benzylidene Steroidal Derivatives. Steroids 2013, 78, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Liu, H.; Huang, K.; Dai, G.; Cao, C.; Dong, R. Synthesis of 3b, 7a, 11a-Trihydroxy-Pregn-21-Benzylidene-5-En-20-One Derivatives and Their Cytotoxic Activities. Bioorg. Med. Chem. Lett. 2009, 19, 6637–6639. [Google Scholar] [CrossRef] [PubMed]
- Canário, C.; Matias, M.; De Brito, V.; Adriana, O.; Falcão, A.; Silvestre, S.; Alves, G. ∆9,11-Estrone Derivatives as Potential Antiproliferative Agents: Synthesis, in Vitro Biological Evaluation and Docking Studies. Comptes Rendus Chim. 2020, 23, 201–217. [Google Scholar] [CrossRef]
- Kolo, A.M.; İpek, E.; Çapan, İ.; Servi, S. Synthesis of Heterocyclic-Substituted Novel Hydroxysteroids with Regioselective and Stereoselective Reactions. J. Heterocycl. Chem. 2018, 55, 492–497. [Google Scholar] [CrossRef]
- Thamotharan, S.; Parthasarathi, V.; Gupta, R. Two Androst-5-Ene Derivatives: 16-[4-(3-Chloropropoxy)-3-Methoxybenzylidene]-17-Oxoandrost-5-En-3β-Ol and 16-[3-Methoxy-4-(2-Pyrrolidin-1-Ylethoxy)Benzylidene]-3β-Pyrrolidinoandrost-5-En-17β-Ol Monohydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2004, 60, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Dubey, S.; Piplani, P.; Jindal, D.P. Synthesis and Evaluation of Some 16-Benzylidene Substituted 3,17-Dioximino Androstene Derivatives as Anticancer Agents. Lett. Drug Des. Discov. 2005, 2, 537–545. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Alam, M.S.; Yousuf, S.; Wu, Y.; Lin, A.; Shaheen, F. Pregnenolone Derivatives as Potential Anticancer Agents. Steroids 2011, 76, 1554–1559. [Google Scholar] [CrossRef]
- Bryce, A.; Ryan, C.J. Development and Clinical Utility of Abiraterone Acetate as an Androgen Synthesis Inhibitor. Clin. Pharmacol. Ther. 2009, 91, 101–108. [Google Scholar] [CrossRef]
- Bastos, D.A.; Antonarakis, E.S. Galeterone for the Treatment of Advanced Prostate Cancer: The Evidence to Date. Drug Des. Devel. Ther. 2016, 10, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Banday, A.H.; Shameem, S.A.; Jeelani, S. Steroidal Pyrazolines and Pyrazoles as Potential 5α-Reductase Inhibitors: Synthesis and Biological Evaluation. Steroids 2014, 92, 13–19. [Google Scholar] [CrossRef]
- Singh, R.; Thota, S.; Bansal, R. Studies on 16,17-Pyrazoline Substituted Heterosteroids as Anti-Alzheimer and Anti-Parkinsonian Agents Using LPS Induced Neuroinflammation Models of Mice and Rats. ACS Chem. Neurosci. 2017. [Google Scholar] [CrossRef]
- Banday, A.H.; Shameem, S.A.; Banday, J.A.; Ganaie, B.A. Synthesis, 17α-Hydroxylase-C 17,20-Lyase Inhibitory and 5AR Reductase Activity of Novel Pregnenolone Derivatives. Anticancer. Agents Med. Chem. 2018, 18, 1919–1926. [Google Scholar] [CrossRef]
- El-Naggar, M.; Amr, A.E.E.; Fayed, A.A.; Elsayed, E.A. Potent Anti-Ovarian Cancer with Inhibitor Activities on Both Topoisomerase II and V600E BRAF of Synthesized Substituted Estrone Candidates. Molecules 2019, 24, 2054. [Google Scholar] [CrossRef] [Green Version]
- Amr, A.E.E.; Abdel-latif, N.A.; Abdalla, M.M. Synthesis and Antiandrogenic Activity of Some New 3-Substituted Androstano [17,16-c]-5′-Aryl-Pyrazoline and Their Derivatives. Bioorg. Med. Chem. 2006, 14, 373–384. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, B.; Shi, X.; Zhao, Y. Synthesis and Biological Evaluation of New Steroidal Pyridines as Potential Anti-Prostate Cancer Agents. Eur. J. Med. Chem. 2018, 145, 11–22. [Google Scholar] [CrossRef]
- Amr, A.E.E.; Elsayed, E.A.; Al-omar, M.A.; Eldin, H.O.B.; Nossier, E.S.; Abdallah, M.M. Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives. Molecules 2019, 24, 416. [Google Scholar] [CrossRef] [Green Version]
- Babu, A.R.S.; Raghunathan, R. An Easy Access to Novel Steroidal Dispiropyrrolidines through 1, 3-Dipolar Cycloaddition of Azomethine Ylides. Tetrahedron Lett. 2008, 49, 4618–4620. [Google Scholar] [CrossRef]
- Gavaskar, D.; Babu, A.R.S.; Raghunathan, R.; Dharani, M.; Balasubramanian, S. An Expedient Sequential One-Pot Four Component Synthesis of Novel Steroidal Spiro-Pyrrolidine Heterocycles in Ionic Liquid. Steroids 2016, 109, 1–6. [Google Scholar] [CrossRef]
- Mótyána, G.; Gopisetty, M.K.; Kiss-Faludy, R.E.; Kulmány, Á.; Zupkó, I.; Frank, É.; Kiricsi, M. Anti-Cancer Activity of Novel Dihydrotestosterone-Derived Ring A-Condensed Pyrazoles on Androgen Non-Responsive Prostate Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 2170. [Google Scholar] [CrossRef] [Green Version]
- Iványi, Z.; Szabó, N.; Wölfling, J.; Szécsi, M.; Julesz, J.; Schneider, G. Novel Series of 17β-Pyrazolylandrosta-5,16-Diene Derivatives and Their Inhibitory Effect on 17α-Hydroxylase/C17,20-Lyase. Steroids 2012, 77, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Wilson, J.D. Steroid 5α-Reductase: Two Genes/Two Enzymes. Annu. Rev. Biochem. 1994, 63, 25–61. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, A.E.; Silver, R.; Guileyardo, J.M.; Casey, I.M.L.; Mcconnell, J.D.; Russell, D.W. Tissue Distribution and Ontogeny of Steroid 5α-Reductase Isozyme. J. Clin. Invest. 1993, 92, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Lall, S.I.; Mancheno, D.; Castro, S.; Behaj, V.; Cohen, J.I.; Engel, R. Polycations. Part X. LIPs, a New Category of Room Temperature Ionic Liquid Based on Polyammonium Salts. R. Soc. Chem. 2000, 2413–2414. [Google Scholar] [CrossRef]
- Azzouni, F.; Godoy, A.; Li, Y.; Mohler, J. The 5α-Reductase Isozyme Family: A Review of Basic Biology and Their Role in Human Diseases. Adv. Urol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Sudduth, S.L.; Koronkowski, M.J. Finasteride: The First 5α-Reductase Inhibitor. Pharmacotherapy 1993, 13, 309. [Google Scholar] [CrossRef]
- Kurup, A.; Garg, R.; Hansch, C. Comparative QSAR Analysis of 5α-Reductase Inhibitors. Chem. Rev. 2000, 100, 909–924. [Google Scholar] [CrossRef]
- Mitchell, H.J.; Dankulich, W.P.; Hartman, G.D.; Prueksaritanont, T.; Schmidt, A.; Vogel, R.L.; Bai, C.; Mcelwee-witmer, S.; Zhang, H.Z.; Chen, F.; et al. Design, Synthesis, and Biological Evaluation of 16-Substituted 4-Azasteroids as Tissue-Selective Androgen Receptor Modulators (SARMs). J. Med. Chem. 2009, 52, 4578–4581. [Google Scholar] [CrossRef]
- Dubey, S.; Jindal, D.P.; Piplani, P. Synthesis and Antineoplastic Activity of Some 16-Benzylidene Substituted Steroidal Oximes. Indian J. Chem. 2005, 44, 2126–2137. [Google Scholar] [CrossRef]
Figure 1.
Claisen–Schmidt condensation to prepare 16E- and 21E-arylidenesteroids.
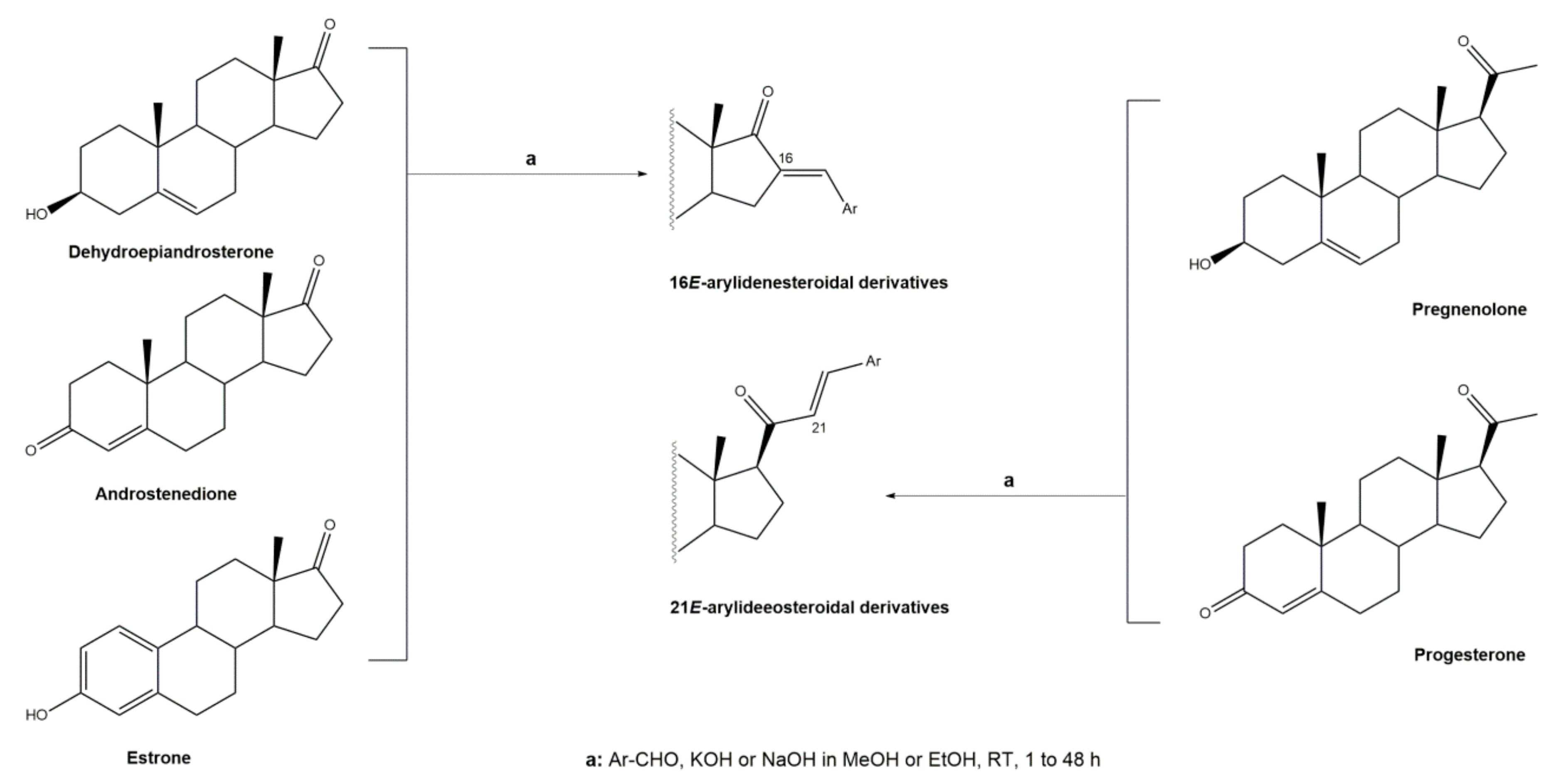
Figure 2.
Aldol condensation using KF/Al2SO3 or KF/Al2O3 as catalysts in EtOH, under reflux, to synthesize 16E-arylidenesteroids from 1 and dehydroepiandrosterone.
Figure 2.
Aldol condensation using KF/Al2SO3 or KF/Al2O3 as catalysts in EtOH, under reflux, to synthesize 16E-arylidenesteroids from 1 and dehydroepiandrosterone.
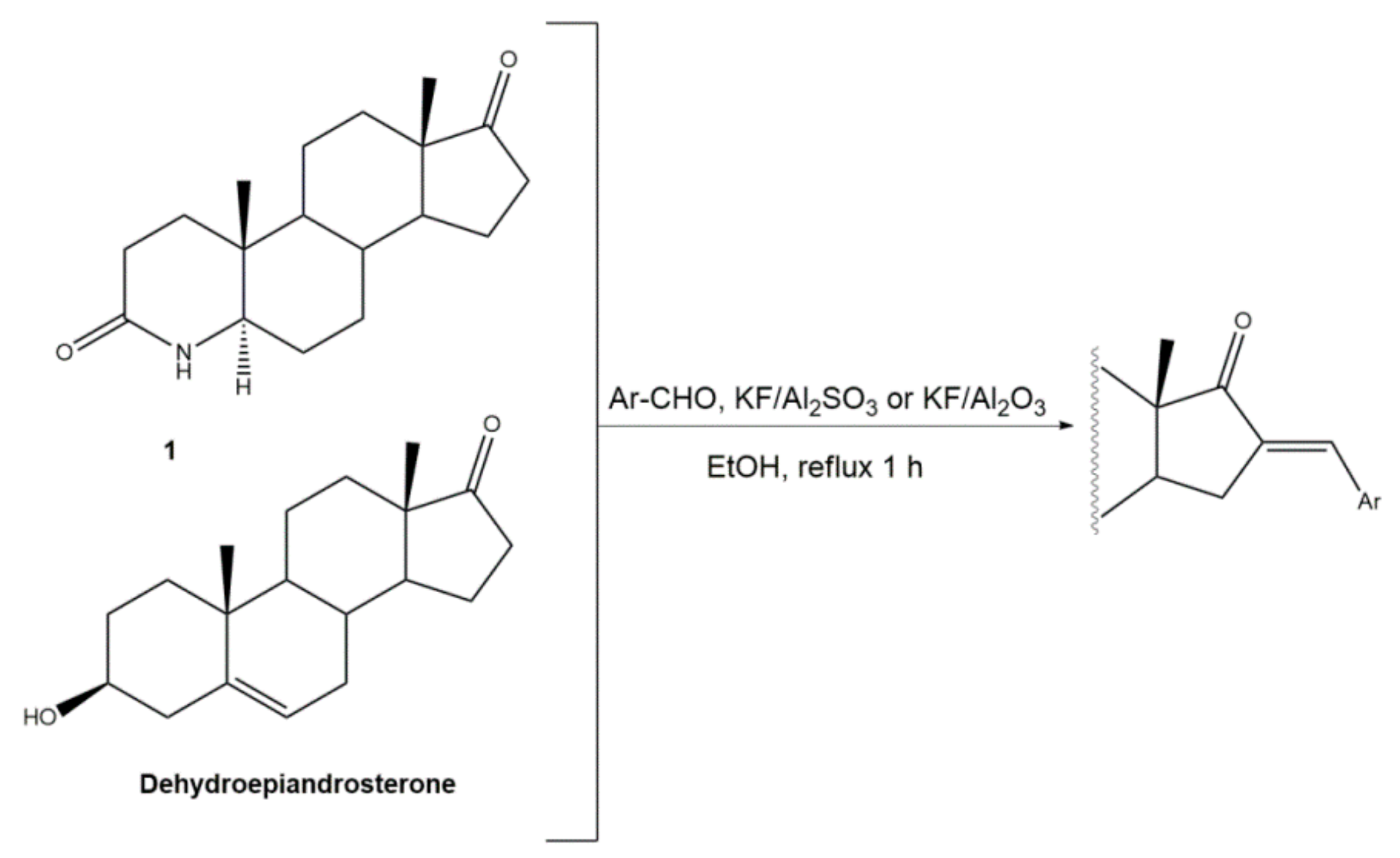
Figure 3.
Preparation of 21E-arylidenepregnenolone acetate derivatives, catalyzed by I2-Al2O3 under microwave irradiation [52].
Figure 3.
Preparation of 21E-arylidenepregnenolone acetate derivatives, catalyzed by I2-Al2O3 under microwave irradiation [52].
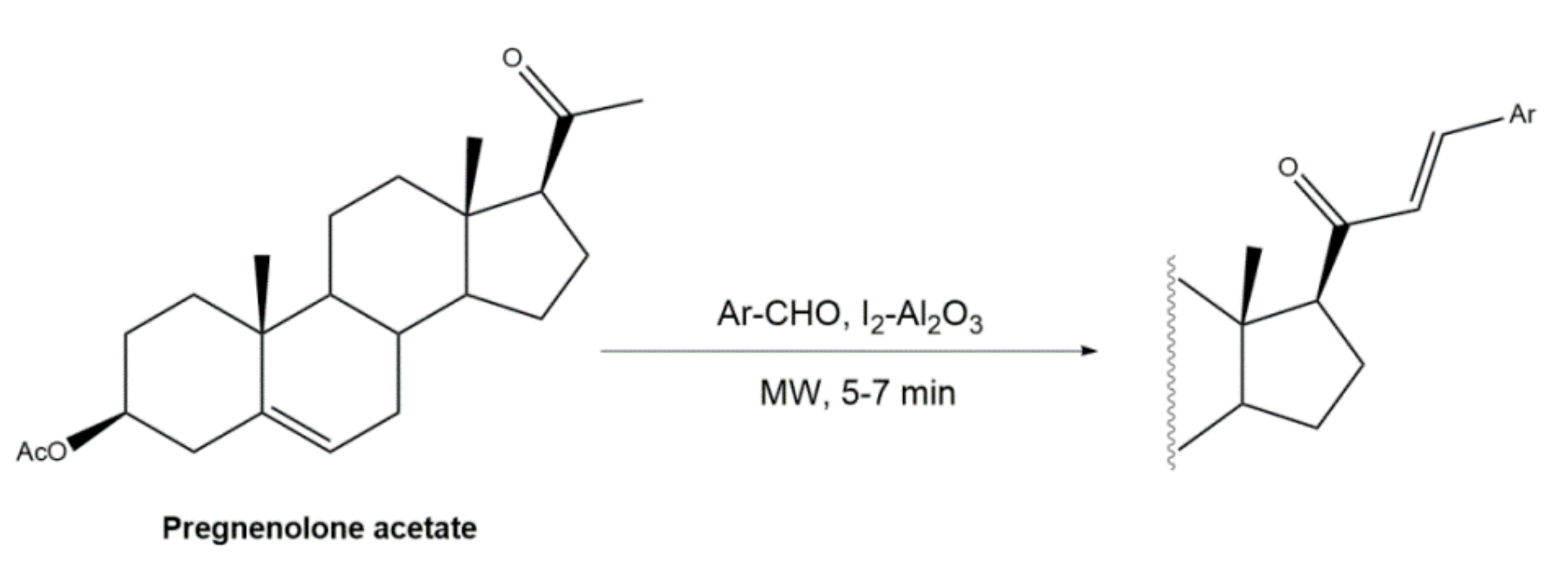
Figure 4.
Alternative approach to obtain 16E-arylidenoestrane derivatives by the employment of Mizoroki–Heck reaction [54].
Figure 4.
Alternative approach to obtain 16E-arylidenoestrane derivatives by the employment of Mizoroki–Heck reaction [54].

Figure 5.
Synthesis of steroidal arylpyrazolines from 16E- and 21E-arylidenesteroids.

Figure 6.
Synthesis of steroidal arylpyrimidines and pyridines from 16E- and 21E-arylidenesteroids.
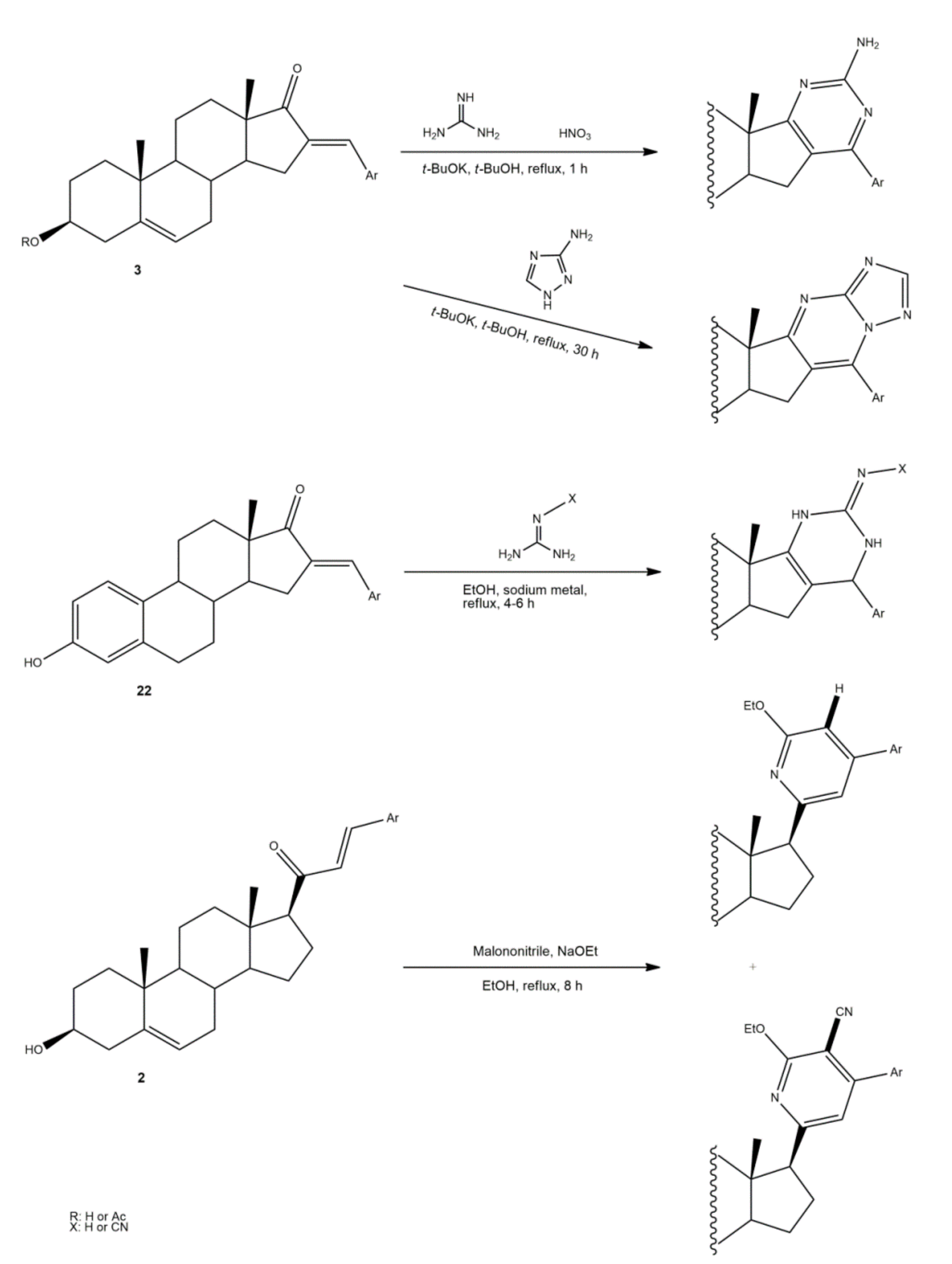
Figure 7.
Preparation of steroidal spiro-oxindoles and spiro-pyrrolidines from 16E-arylidenesteroids [56,104].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Examples of relevant bioactive 2- and 16E-arylideneandrostane derivatives. Positive controls, when shown, are identified by (+) symbol.
Table 1.
Examples of relevant bioactive 2- and 16E-arylideneandrostane derivatives. Positive controls, when shown, are identified by (+) symbol.
| Compound | Bioactivity Data | Ref. | |||||
|---|---|---|---|---|---|---|---|
 2 | Antiproliferative activity (IC50 μM) | [16] | |||||
| Cell line | 2 | 5-FU (+) | |||||
| HepG2 | 9.10 | 10.59 | |||||
| MCF-7 | 9.18 | 28.11 | |||||
| Cell cycle arrest at G2 phase in HepG2 | |||||||
 3 | Antiproliferative activity (IC50 ± SEM μM) | [60] | |||||
| Cell line | 3 | Etoposide (+) | |||||
| KB | 0.6 ± 2.0 | 2.8 ± 16.8 | |||||
| T47-D | 1.7 ± 14.8 | 1.2 ± 8 | |||||
 4 | Antiproliferative activity (IC50 μM) | [61] | |||||
| Cell line | 4 | ||||||
| CCRF-CEM | 3.94 | ||||||
| K-562 | 2.61 | ||||||
| RPMI-8226 | 6.90 | ||||||
| SR | 1.79 | ||||||
 5 | Antiproliferative activity (IC50 μM ± SD μM) | [59] | |||||
| Cell line | 5 | Cis (+) | |||||
| HT-29 | 1.2 ± 0.4 | 66 ± 2 | |||||
 6 | Cytotoxic activity–in vivo hollow fiber assay (score) | [57] | |||||
| 6 | Taxol (+) | ||||||
| I.P. | 2 | Data not shown | |||||
| S.C. | 8 | ||||||
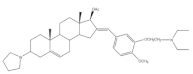 7 | Cytotoxic activity–in vivo hollow fiber assay (score) | [62] | |||||
| 7 | Taxol (+) | ||||||
| I.P. | 12 | Data not shown | |||||
| S.C. | 8 | ||||||
 8 | Aromatase inhibitory activity (IC50 μM) | [70] | |||||
| 8 | Aminoglutethimide (+) | ||||||
| 5.2 | 28.5 | ||||||
 9 | Aromatase inhibitory activity (IC50 μM) | [70] | |||||
| 9 | Aminoglutethimide (+) | ||||||
| 6.4 | 28.5 | ||||||
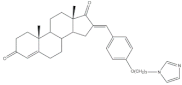 10 | Aromatase inhibitory activity (IC50 μM) | [71] | |||||
| 10 | Aminoglutethimide (+) | ||||||
| 4.4 | 28.5 | ||||||
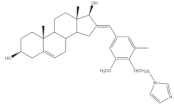 11 | Aromatase inhibitory activity (IC50 μM) | [71] | |||||
| 11 | Aminoglutethimide (+) | ||||||
| 2.4 | 28.5 | ||||||
 12 | Anti-inflammatory activity TNF-α levels (pg.mg−1protein ± SD) | [72] | |||||
| 12 | CEL (+) | DEX (+) | |||||
| 88.6 ± 1.8 | 68.2 ± 1.1 | 89.6 ± 2.0 | |||||
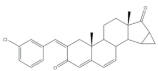 13 | Anti-inflammatory activity (IC50 μM) (NO release of LPS-activated mouse microglial cell line BV2) | [73] | |||||
| 13 | Minocycline (+) | ||||||
| 2.69 | 5.97 | ||||||
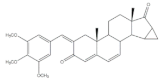 14 | Anti-inflammatory activity (IC50 μM) (NO release of LPS-activated mouse microglial cell line BV2) | [73] | |||||
| 14 | Minocycline (+) | ||||||
| 3.28 | 5.97 | ||||||
Table 2.
The most active 21E-arylidenepregnene derivatives. Positive controls, when shown, are identified by (+) symbol.
Table 2.
The most active 21E-arylidenepregnene derivatives. Positive controls, when shown, are identified by (+) symbol.
| Compound | Bioactivity Data | Ref. | ||
|---|---|---|---|---|
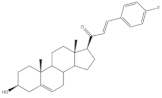 15 | Antiproliferative activity (IC50 μM) | [69] | ||
| Cell line | 15 | |||
| HCT-15 | 0.81 | |||
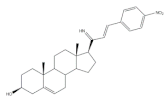 16 | Antiproliferative activity (IC50 μM) | [69] | ||
| Cell line | 16 | |||
| MCF-7 | 0.60 | |||
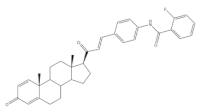 17 | Antiproliferative activity Growth inhibition (%) | [38] | ||
| Cell line | 17 | Cis (+) | ||
| HeLa | 58 | 99 | ||
| MCF-7 | 64 | 88 | ||
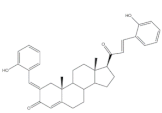 18 | Antimicrobial activity Zone of inhibition (mm) | [76] | ||
| Gram positive | 18 | AMP (+) | ||
| Streptococcus pneumoniae | 30 | 20 | ||
| Staphylococcus aureus | 24 | 22 | ||
Table 3.
The most active 16E-arylidenoestrone and -estradiol derivatives. Positive controls, when shown, are identified by (+) symbol.
Table 3.
The most active 16E-arylidenoestrone and -estradiol derivatives. Positive controls, when shown, are identified by (+) symbol.
| Compound | Bioactivity Data | Ref. | |
|---|---|---|---|
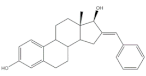 19 | 17β-HSD1 inhibitory activity (IC50 μM) | [35] | |
| 19 | Estradiol (+) | ||
| 3.4 | 7.3 | ||
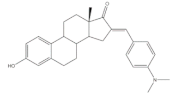 20 | 17β-HSD inhibitory activity (% at 10 μM) | [83] | |
| 20 | |||
| Type 1 | Type 2 | ||
| 72 | 13 | ||
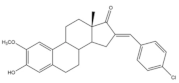 21 | Tumor weight reduction (%) Human breast cancer (MCF-7) xenografts models | [85] | |
| 21 (45 mg/kg/day) | 2ME (+) (45 mg/kg/day) | ||
| About 50% Significative reduction compared with negative control | About 50% Significative reduction compared with negative control | ||
Table 4.
The most active heterocyclic steroidal derivatives obtained from arylidenesteroids. Positive controls, when shown, are identified by (+) symbol.
Table 4.
The most active heterocyclic steroidal derivatives obtained from arylidenesteroids. Positive controls, when shown, are identified by (+) symbol.
| Compound | Bioactivity Data | Ref. | |||
|---|---|---|---|---|---|
 24 | Antiproliferative activity (IC50 μM) | [4] | |||
| Cell line | 24 | ||||
| HT-29 | 0.24 | ||||
| HCT-15 | 0.25 | ||||
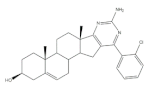 25 | Antiproliferative activity (IC50 μM) | [49] | |||
| Cell line | 25 | 5-FU (+) | |||
| HepG-2 | 5.41 | >100 | |||
| Huh-7 | 5.65 | >95 | |||
| SGC-790 | 10.64 | >100 | |||
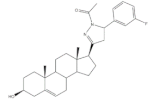 26 | 5AR-1 Inhibition (IC50 ± SEM μM) | [96] | |||
| 26 | Finasteride (+) | ||||
| 14.50 ± 0.48 | 21.6 ± 0.62 | ||||
 27 | 5AR-2 Inhibition (IC50 ± SEM μM) | [96] | |||
| 27 | Finasteride (+) | ||||
| 13.90 ± 0.75 | 15.4 ± 0.58 | ||||
 28 | 5AR-2 Inhibition (IC50 ± SEM μM) | [96] | |||
| 28 | Finasteride (+) | ||||
| 14.20 ± 0.75 | 15.4 ± 0.58 | ||||
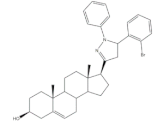 29 | 5AR-2 Inhibition (IC50 ± SEM nM) | [98] | |||
| 29 | Finasteride (+) | ||||
| 7.30 ± 0.62 | 2.4 ± 0.15 | ||||
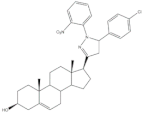 30 | 5AR-2 Inhibition (IC50 ± SEM nM) | [98] | |||
| 30 | Finasteride (+) | ||||
| 8.20 ± 0.55 | 2.4 ± 0.58 | ||||
 31 | Antiproliferative activity (IC50 μg.mL−1) | [18] | |||
| Cell line | 31 | 5-FU (+) | Cis (+) | ||
| NCI-H460 | 10.30 | 2.48 | 0.699 | ||
| HeLa | 12.50 | 0.887 | 2.03 | ||
| Cell cycle arrest at S phase in HeLa cells | |||||
 32 | Antiproliferative activity (IC50 μM) | [56] | |||
| Cell line | 32 | 5-FU (+) | |||
| SMMC-7721 | 4.30 | 9.78 | |||
| MCF-7 | 2.06 | 7.54 | |||
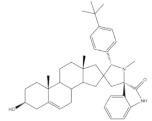 33 | Antiproliferative activity (IC50 ± SEM μM) | [56] | |||
| Cell line | 33 | 5-FU (+) | |||
| SMMC-7721 | 6.05 ± 0.48 | 9.78 ± 0.99 | |||
| MGC-803 | 5.79 ± 0.76 | 6.92 ± 0.35 | |||
| Cell cycle arrest at G2/M phase in MGC cells | |||||
 34 | Antiproliferative activity (IC50 ± SEM μM) | [56] | |||
| Cell line | 34 | 5-FU (+) | |||
| SMMC-7721 | 0.71 ± 0.11 | 9.78 ± 0.99 | |||
 35 | Antiproliferative activity (IC50 μM) | [93] | |||
| Cell line | 35 | DOX (+) | |||
| MDA-MB 231 | 0.91 | 1.23 | |||
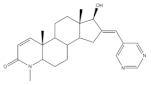 36 | Osteoanabolic activity and tissue-selectivity (% of the effect of DHT) | [113] | |||
| OVX | 36 | DHT | |||
| BRF | 120 | 100 | |||
| ORX | |||||
| VP | 3 | 100 | |||
| SV | 21 | 100 | |||
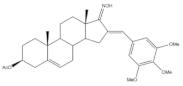 37 | Cytotoxic activity-in vivo hollow fiber assay (score) | [57] | |||
| 37 | Taxol (+) | ||||
| I.P. | 4 | Data not shown | |||
| S.C. | 6 | ||||
 38 | Antiproliferative activity Cell growth (%) | [114] | |||
| Cell line | 38 | ||||
| NCI-H460 | −44 | ||||
| MFC-7 | −44 | ||||
| SF-268 | −79 | ||||
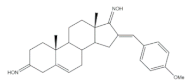 39 | Antiproliferative activity Cell growth (%) | [92] | |||
| Cell line | 39 | ||||
| NCI-H460 | −11 | ||||
| MCF-7 | 5 | ||||
| SF-268 | −8 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brito, V.; Alves, G.; Almeida, P.; Silvestre, S. Highlights on Steroidal Arylidene Derivatives as a Source of Pharmacologically Active Compounds: A Review. Molecules 2021, 26, 2032. https://doi.org/10.3390/molecules26072032
AMA Style
Brito V, Alves G, Almeida P, Silvestre S. Highlights on Steroidal Arylidene Derivatives as a Source of Pharmacologically Active Compounds: A Review. Molecules. 2021; 26(7):2032. https://doi.org/10.3390/molecules26072032
Chicago/Turabian StyleBrito, Vanessa, Gilberto Alves, Paulo Almeida, and Samuel Silvestre. 2021. "Highlights on Steroidal Arylidene Derivatives as a Source of Pharmacologically Active Compounds: A Review" Molecules 26, no. 7: 2032. https://doi.org/10.3390/molecules26072032