
The Density Functional Theory Account of Interplaying Long-Range Exchange and Dispersion Effects in Supramolecular Assemblies of Aromatic Hydrocarbons with Spin

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
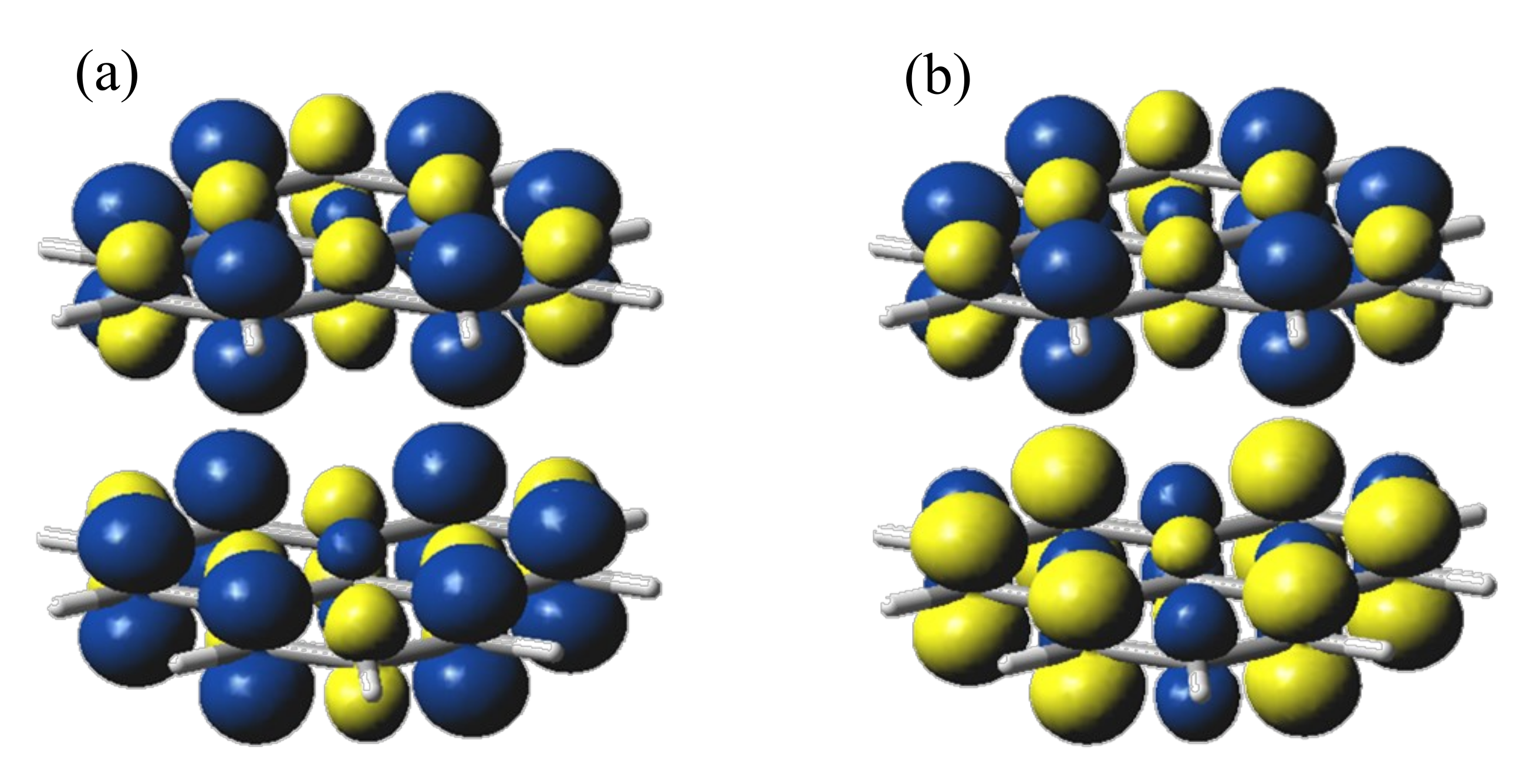
2.1. The Phenalenyl Unit as Spin Carrier
2.2. Broken-Symmetry DFT Calculations of the Phenalenyl Dimer
2.3. The Fit of Potential Energy Profiles and Spin-Coupling Curves
2.4. Genuine vs. Corrected Functionals. The B3LYP Tests
2.5. Testing Selected Functionals and Long-Range Correction Recipes
3. Methods
Computational Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hicks, R. Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron. Compounds; Wiley& Sons: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Ratera, I.; Veciana, J. Playing with organic radicals as building blocks for functional molecular materials. Chem. Soc. Rev. 2011, 41, 303–349. [Google Scholar] [CrossRef]
- Iwamura, H. High-Spin Organic Molecules and Spin Alignment in Organic Molecular Assemblies. Adv. Phys. Org. Chem. 1990, 26, 179–253. [Google Scholar] [CrossRef]
- Han, W.; Kawakami, R.K.; Gmitra, M.; Fabian, J. Graphene spintronics. Nat. Nanotechnol. 2014, 9, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Sanvito, S. Organic Electronics: Memoirs of a Spin. Nat. Nanotechnol. 2007, 2, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Dediu, V.A.; Hueso, L.E.; Bergenti, I.; Taliani, C. Spin Routes in Organic Semiconductors. Nat. Mater. 2009, 8, 707–716. [Google Scholar] [CrossRef]
- Suzuki, S.; Morita, Y.; Fukui, K.; Sato, K.; Shiomi, D.; Takui, T.; Nakasuji, K. Aromaticity on the Pancake-Bonded Dimer of Neutral Phenalenyl Radical as Studied by MS and NMR Spectroscopies and NICS Analysis. J. Am. Chem. Soc. 2006, 128, 2530–2531. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.E.; Brown, R.L. π-Electron Properties of Large Condensed Polyaromatic Hydrocarbon. J. Am. Chem. Soc. 1987, 109, 3721–3729. [Google Scholar] [CrossRef]
- Fernandez-Rossier, J.; Palacios, J.J. Magnetism in Graphene Nanoislands. Phys. Rev. Lett. 2007, 99, 177204. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Novoselov, K.S. The Rise of Graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Tung, V.C.; Kaner, R.B. Honeycomb Carbon: A Review of Graphene. Chem. Rev. 2010, 110, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Shurki, A.; Danovich, D.; Hiberty, P.C. A Different Story of π-Delocalization: The distortivityof π-Electrons and its Chemical Manifestations. Chem. Rev. 2001, 101, 1501–1540. [Google Scholar] [CrossRef]
- Putz, M.V.; Cimpoesu, F.; Ferbinteanu, M. Structural Chemistry, Principles, Methods, and Case Studies; Springer: Cham, Switzerland, 2018; pp. 408–423. [Google Scholar]
- Kivelson, S.; Chapman, O.L. Polyacene and a New Class of Quasi-One-Dimensional Conductors. Phys. Rev. B 1983, 28, 7236–7243. [Google Scholar] [CrossRef]
- Hegmann, F.A.; Tykwinski, R.R.; Lui, K.P.H.; Bullock, J.E.; Anthony, J.E. Picosecond Transient Photoconductivity in Functionalized Pentacene Molecular Crystals Probed by Terahertz Pulse Spectroscopy. Phys. Rev. Lett. 2002, 89, 227403. [Google Scholar] [CrossRef] [PubMed]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, Oligoacenenes, and Their Buckminsterfullerene Derivatives: The Brick and Mortar of Organic Electronics. Chem. Rev. 2004, 104, 4891–4945. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, H. What Role Has Organic Chemistry Played in the Development of Molecule-Based Magnets? Polyhedron 2013, 66, 3–14. [Google Scholar] [CrossRef]
- Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; et al. A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-tert-butyl-phenalenyl. J. Am. Chem. Soc. 1999, 121, 1619–1620. [Google Scholar] [CrossRef]
- Beer, L.; Mandal, S.K.; Reed, R.W.; Oakley, R.T.; Tham, F.S.; Donnadieu, B.; Haddon, R.C. The First Electronically Stabilized Phenalenyl Radical: Effect of Substituents on Solution Chemistry and Solid-State Structure. Crys. Growth Des. 2007, 7, 802–809. [Google Scholar] [CrossRef]
- Uchida, K.; Hirao, Y.; Kurata, H.; Kubo, T.; Hatano, S.; Inoue, K. Dual Association Modes of the 2,5,8-Tris(pentafluorophenyl)phenalenyl Radical. Chem. Asian J. 2014, 9, 1823–1829. [Google Scholar] [CrossRef]
- Pavliček, N.; Mistry, A.; Majzik, Z.; Moll, N.; Meyer, G.; Fox, D.J.; Gross, L. Synthesis and characterization of triangulene. Nat. Nanotechnol. 2017, 12, 308–311. [Google Scholar] [CrossRef]
- Mishra, S.; Beyer, D.; Eimre, K.; Liu, J.; Berger, R.; Gröning, O.; Pignedoli, C.A.; Müllen, K.; Fasel, R.; Feng, X.; et al. Synthesis and Characterization of π-Extended Triangulene. J. Am. Chem. Soc. 2019, 141, 10621–10625. [Google Scholar] [CrossRef]
- Su, J.; Telychko, M.; Hu, P.; Macam, G.; Mutombo, P.; Zhang, H.; Bao, Y.; Cheng, F.; Huang, Z.-Q.; Qiu, Z.; et al. Atomically precise bottom-up synthesis of π-extended [5] triangulene. Sci. Adv. 2019, 5, eaav7717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toader, A.M.; Buta, C.M.; Frecus, B.; Mischie, A.; Cimpoesu, F. Valence Bond Account of Triangular Polyaromatic Hydrocarbons with Spin: Combining Ab Initio and Phenomenological Approaches. J. Phys. Chem. C 2019, 123, 6869–6880. [Google Scholar] [CrossRef]
- Buta, M.C.; Frecus, B.; Enache, M.; Humelnicu, I.; Toader, A.M.; Cimpoesu, F. Intra- and Inter-Molecular Spin Coupling in Phenalenyl Dimeric Systems. J. Phys. Chem. A 2021, 125, 6893–6901. [Google Scholar] [CrossRef] [PubMed]
- Frecus, B.; Buta, M.C.; Oprea, C.I.; Stroppa, A.; Putz, M.V.; Cimpoesu, F. Noble Gas Endohedral Fullerenes, Ng@C60 (Ng= Ar, Kr): A Particular Benchmark for Assessing the Account of Non-Covalent Interactions by Density Functional Theory Calculations. Theor. Chem. Acc. 2016, 135, 133. [Google Scholar] [CrossRef]
- Szczepanik, D.W.; Solà, M.; Andrzejak, M.; Pawelek, B.; Dominikowska, J.; Kukulka, M.; Dyduch, K.; Krygowski, T.M.; Szatylowicz, H. The role of the long-range exchange corrections in the description of electron delocalization in aromatic species. J. Comput. Chem. 2017, 38, 1640–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R.; Gavezzotti, A. Crystal structures of polynuclear aromatic hydrocarbons. Classification, rationalization and prediction from molecular structure. Acta Crystallogr. Sect. B Struct. Sci. 1989, 45, 473–482. [Google Scholar] [CrossRef]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The Nature and Applications of π–π Interactions: A Perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Ferbinteanu, M.; Buta, C.; Toader, A.M.; Cimpoesu, F. The Spin Coupling in the Polyaromatic Hydrocarbons and Carbon-based Materials. In Carbon-Related Materials-in Recognition of Nobel Lectures by Prof. Akira Suzuki in ICCE; Kaneko, S., Ed.; Springer: Cham, Switzerland, 2017; pp. 327–371. [Google Scholar] [CrossRef]
- Mou, Z.; Uchida, K.; Kubo, T.; Kertesz, M. Evidence of σ- and π-Dimerization in a Series of Phenalenyls. J. Am. Chem. Soc. 2014, 136, 18009–18022. [Google Scholar] [CrossRef]
- Uchida, K.; Mou, Z.; Kertesz, M.; Kubo, T. Fluxional σ-Bonds of the 2,5,8-Trimethylphenalenyl Dimer: Direct Observation of the Sixfold σ-Bond Shift via a π-Dimer. J. Am. Chem. Soc. 2016, 138, 4665–4672. [Google Scholar] [CrossRef]
- Mou, Z.; Kertesz, M. Pancake Bond Orders: A Study of a Series of Triangulenes. Angew. Chem. Int. Ed. 2017, 56, 10188–10191. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An Unusual π-Stacking Interaction. Chem. Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Uchida, K.; Ito, S.; Nakano, M.; Abe, M.; Kubo, T. Biphenalenylidene: Isolation and Characterization of the Reactive Intermediate on the Decomposition Pathway of Phenalenyl Radical. J. Am. Chem. Soc. 2016, 138, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Noodleman, L. Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimmers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Noodleman, L.; Peng, C.Y.; Case, D.A.; Mouesca, J.M. Orbital Interactions, Electron Delocalization and Spin Coupling in Iron-Sulfur Clusters. Coord. Chem. Rev. 1995, 144, 199–244. [Google Scholar] [CrossRef]
- Heisenberg, W. ZurTheorie des Ferromagnetismus. Z. Für Phys. 1985, 49, 580–597. [Google Scholar] [CrossRef]
- Goll, E.; Werner, H.-J.; Stoll, H.; Leininger, T.; Gori-Giorgi, P.; Savin, A. A short-range gradient-corrected spin density functional in combination with long-range coupled-cluster methods: Application to alkali-metal rare-gas dimers. Chem. Phys. 2006, 329, 276–282. [Google Scholar] [CrossRef]
- Kamiya, M.; Tsuneda, T.; Hirao, K. A density functional study of van der Waals interactions. J. Chem. Phys. 2002, 117, 6010–6015. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T. Van der Waals interactions studied by density functional theory. Mol. Phys. 2005, 103, 1151–1164. [Google Scholar] [CrossRef]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Gori-Giorgi, P.; Savin, A. Properties of short-range and long-range correlation energy density functionals from electron-electron coalescence. Phys. Rev. A 2006, 73, 032506. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C. Valency and Bonding: A Natural Bond. Orbital Donor–Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Onishi, T.; Takano, Y.; Kitagawa, Y.; Kawakami, T.; Yoshioka, Y.; Yamaguchi, K. Theoretical study of the magnetic interaction for M–O–M type metal oxides. Comparison of broken-symmetry approaches. Polyhedron 2001, 20, 1177–1184. [Google Scholar] [CrossRef]
- Nagao, H.; Nishino, M.; Shigeta, Y.; Soda, T.; Kitagawa, Y.; Onishi, T.; Yoshioka, Y.; Yamaguchi, K. Theoretical studies on effective spin interactions, spin alignments and macroscopic spin tunneling in polynuclear manganese and related complexes and their mesoscopic clusters. Coord. Chem. Rev. 2000, 198, 265–295. [Google Scholar] [CrossRef]
- Morse, P.M. Diatomic Molecules According to the Wave Mechanics. II. Vibrational Levels. Phys. Rev. 1929, 34, 57–64. [Google Scholar] [CrossRef]
- Dong, S.-H.; Lemus, R.; Frank, A. Ladder operators for the Morse potential. Int. J. Quantum Chem. 2002, 86, 433–439. [Google Scholar] [CrossRef]
- Keyes, R.W. An Antibonding Morse Potential. Nature 1958, 182, 1071–1072. [Google Scholar] [CrossRef]
- Buta, M.C.; Toader, A.M.; Frecus, B.; Oprea, C.I.; Cimpoesu, F.; Ionita, G. Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals. Int. J. Mol. Sci. 2019, 20, 4733. [Google Scholar] [CrossRef] [Green Version]
- Slater, J.C. The Self-Consistent Field for Molecular and Solids, Quantum Theory of Molecular and Solids; McGraw-Hill: New York, NY, USA, 1974. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Efficient hybrid density functional calculations in solids: Assessment of the Heyd–Scuseria–Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, V.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Steric Modulations in the Reversible Dimerizations of Phenalenyl Radicals via Unusually Weak Carbon-Centered π- and σ-Bonds. J. Org. Chem. 2005, 71, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.-H.; Lischka, H.; Beneberu, H.Z.; Kertesz, M. Rotational Barrier in Phenalenyl Neutral Radical Dimer: Separating Pancake and van der Waals Interactions. J. Am. Chem. Soc. 2014, 136, 5539–5542. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian,Version 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Eaton, J.W.; Bateman, D.; Hauberg, S.; Wehbring, R. GNU Octave Version 3.8.1 Manual: A High-Level Interactive Language for Numerical Computations; CreateSpace Independent Publishing Platform: Scotts Valley, CA, USA, 2014. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Basis Set | 〈S02〉 | u | GOF | D kcal/mol | R0 (Å) | a (Å)−1 | J0 kcal/mol | p (Å)−1 | ρ (Å) | σ - | s (Å)−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HFS | 6-31+G* | 0.764 | 1 | 0.9986 | 17.946 | 3.082 | 1.63895 | 11.152 | 1.79592 | 3.821 | 1.02399 | 3.41507 |
| def2tzvp | 0.764 | 1 | 0.9968 | 17.759 | 3.051 | 1.73299 | 11.608 | 1.84319 | 3.929 | 1.02502 | 3.57648 | |
| SVWN | 6-31+G* | 0.756 | 1 | 0.9970 | 28.203 | 2.921 | 1.67267 | 15.594 | 1.7085 | 4.152 | 1.02147 | 3.32925 |
| def2tzvp | 0.756 | 1 | 0.9952 | 28.063 | 2.906 | 1.70599 | 15.779 | 1.6113 | 4.142 | 1.02151 | 3.44984 | |
| BP86 | 6-31+G* | 0.767 | 1 | 0.9871 | 2.510 | 3.329 | 2.21563 | 6.807 | 1.98562 | 3.712 | 1.01870 | 3.34216 |
| def2tzvp | 0.767 | 1 | 0.9636 | 1.439 | 3.362 | 2.44780 | 6.657 | 2.08065 | 3.706 | 1.02009 | 3.61832 | |
| BLYP | 6-31+G* | 0.765 | 0 | 0.9873 | 0.773 | 3.250 | 3.59274 | 7.549 | 2.10370 | 3.447 | 1.01908 | 3.61409 |
| def2tzvp | 0.765 | 0 | 0.9689 | 1.042 | 3.250 | 3.16227 | 7.502 | 2.15263 | 3.446 | 1.01832 | 3.63673 | |
| B3LYP | 6-31+G* | 0.799 | 1 | 0.9933 | 0.318 | 3.837 | 2.41695 | 2.598 | 2.24661 | 3.447 | 1.01907 | 3.61409 |
| def2tzvp | 0.798 | 0 | 0.9587 | 1.115 | 3.250 | 3.35913 | 5.729 | 2.13559 | 3.446 | 1.01832 | 3.63673 |
| Method | Basis Set | 〈S02〉 | u | GOF | D kcal/mol | R0 (Å) | a (Å)−1 | J0 kcal/mol | p (Å)−1 | ρ (Å) | σ - | s (Å)−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LC-HFS | 6-31+G* | 1.144 | 0 | 0.9983 | 3.480 | 3.250 | 2.64500 | 4.481 | 2.90744 | 3.821 | 1.02399 | 3.41507 |
| def2tzvp | 1.141 | 0 | 0.9906 | 2.527 | 3.250 | 1.89001 | 4.543 | 2.93621 | 3.929 | 1.02502 | 3.57648 | |
| LC-SVWN | 6-31+G* | 0,979 | 1 | 0.9981 | 0.658 | 3.906 | 1.72638 | 0.863 | 2.52575 | 2.876 | 1.00203 | 1.97545 |
| def2tzvp | 0,977 | 1 | 0.9849 | 0.265 | 3.848 | 2.33551 | 0.919 | 2.68017 | 2.841 | 1.00091 | 1.83531 | |
| LC-BP86 | 6-31+G* | 1.030 | 1 | 0.9973 | 12.108 | 3.105 | 2.13143 | 6.931 | 2.16669 | 3.026 | 1.01035 | 3.21305 |
| def2tzvp | 1.028 | 1 | 0.9927 | 10.304 | 3.116 | 2.10700 | 6.484 | 2.24239 | 2.937 | 1.00807 | 2.66578 | |
| LC-BLYP | 6-31+G* | 1.010 | 1 | 0.9920 | 5.423 | 3.355 | 1.72612 | 3.679 | 2.5073 | 2.784 | 0.99893 | 1.57951 |
| def2tzvp | 1.006 | 1 | 0.9928 | 5.253 | 3.324 | 2.00463 | 3.489 | 2.69383 | 2.897 | 1.00361 | 2.10122 | |
| CAM-B3LYP | 6-31+G* | 0.868 | 1 | 0.9780 | 4.450 | 3.391 | 2.24565 | 7.174 | 2.24928 | 3.144 | 1.00777 | 2.58372 |
| def2tzvp | 0.867 | 1 | 0.9796 | 1.201 | 3.499 | 2.61995 | 3.073 | 2.4510 | 3.106 | 1.00352 | 2.14649 | |
| HSEH1PBE | 6-31+G* | 0.823 | 1 | 0.9893 | 5.114 | 3.329 | 1.98489 | 5.122 | 2.03229 | 3.260 | 1.01873 | 3.73800 |
| def2tzvp | 0.823 | 1 | 0.9949 | 4.181 | 3.301 | 2.46109 | 5.402 | 2.21272 | 3.258 | 1.01849 | 3.80583 | |
| M11 | 6-31+G* | 0.879 | 1 | 0.9931 | 16.903 | 3.145 | 1.82830 | 6.020 | 2.05099 | 3.080 | 1.01722 | 4.35074 |
| def2tzvp | 0.872 | 1 | 0.9956 | 17.319 | 3.075 | 1.93448 | 7.728 | 2.03871 | 2.971 | 1.01641 | 4.18583 |
| Method | Basis Set | 〈S02〉 | u | GOF | D kcal/mol | R0 (Å) | a (Å)−1 | J0 kcal/mol | p (Å)−1 | ρ (Å) | σ - | s (Å)−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP GD3 | 6-31+G* | 0.799 | 1 | 0.9988 | 15.982 | 3.256 | 1.41408 | 5.926 | 2.14210 | 3.447 | 1.01908 | 3.61409 |
| def2tzvp | 0.798 | 1 | 0.9977 | 15.496 | 3.247 | 1.48886 | 5.798 | 2.19535 | 3.446 | 1.01832 | 3.63673 | |
| B97D | 6-31+G* | 0.773 | 1 | 0.9938 | 21.785 | 3.094 | 1.56362 | 8.979 | 2.00851 | 3.729 | 1.02388 | 3.57989 |
| def2tzvp | 0.774 | 1 | 0.9984 | 21.118 | 3.111 | 1.54651 | 9.167 | 1.93595 | 3.593 | 1.02162 | 3.67610 | |
| B97D3 | 6-31+G* | 0.773 | 1 | 0.9990 | 23.942 | 3.101 | 1.43868 | 9.323 | 1.86482 | 3.249 | 1.01954 | 3.30823 |
| def2tzvp | 0.774 | 1 | 0.9995 | 22.961 | 3.105 | 1.46734 | 9.167 | 1.93595 | 3.594 | 1.01819 | 3.34281 | |
| wB97XD | 6-31+G* | 0.850 | 1 | 0.9990 | 19.668 | 3.197 | 1.59813 | 6.366 | 2.0404 | 3.043 | 1.01459 | 3.56559 |
| def2tzvp | 0.851 | 1 | 0.9993 | 18.308 | 3.199 | 1.64489 | 6.243 | 2.07563 | 3.041 | 1.01441 | 3.60865 | |
| APFD | 6-31+G* | 0.818 | 1 | 0.9971 | 23.665 | 3.135 | 1.50090 | 7.676 | 2.03858 | 3.249 | 1.02002 | 4.04944 |
| def2tzvp | 0.817 | 1 | 0.9976 | 22.455 | 3.135 | 1.55166 | 7.592 | 2.06477 | 3.249 | 1.01938 | 4.06371 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toader, A.M.; Buta, M.C.; Mischie, A.; Putz, M.V.; Cimpoesu, F. The Density Functional Theory Account of Interplaying Long-Range Exchange and Dispersion Effects in Supramolecular Assemblies of Aromatic Hydrocarbons with Spin. Molecules 2022, 27, 45. https://doi.org/10.3390/molecules27010045
Toader AM, Buta MC, Mischie A, Putz MV, Cimpoesu F. The Density Functional Theory Account of Interplaying Long-Range Exchange and Dispersion Effects in Supramolecular Assemblies of Aromatic Hydrocarbons with Spin. Molecules. 2022; 27(1):45. https://doi.org/10.3390/molecules27010045
Chicago/Turabian StyleToader, Ana Maria, Maria Cristina Buta, Alice Mischie, Mihai V. Putz, and Fanica Cimpoesu. 2022. "The Density Functional Theory Account of Interplaying Long-Range Exchange and Dispersion Effects in Supramolecular Assemblies of Aromatic Hydrocarbons with Spin" Molecules 27, no. 1: 45. https://doi.org/10.3390/molecules27010045