
HPLC Determination of Colistin in Human Urine Using Alkaline Mobile Phase Combined with Post-Column Derivatization: Validation Using Accuracy Profiles



 and
and 
Abstract
:1. Introduction
2. Results and Discussion
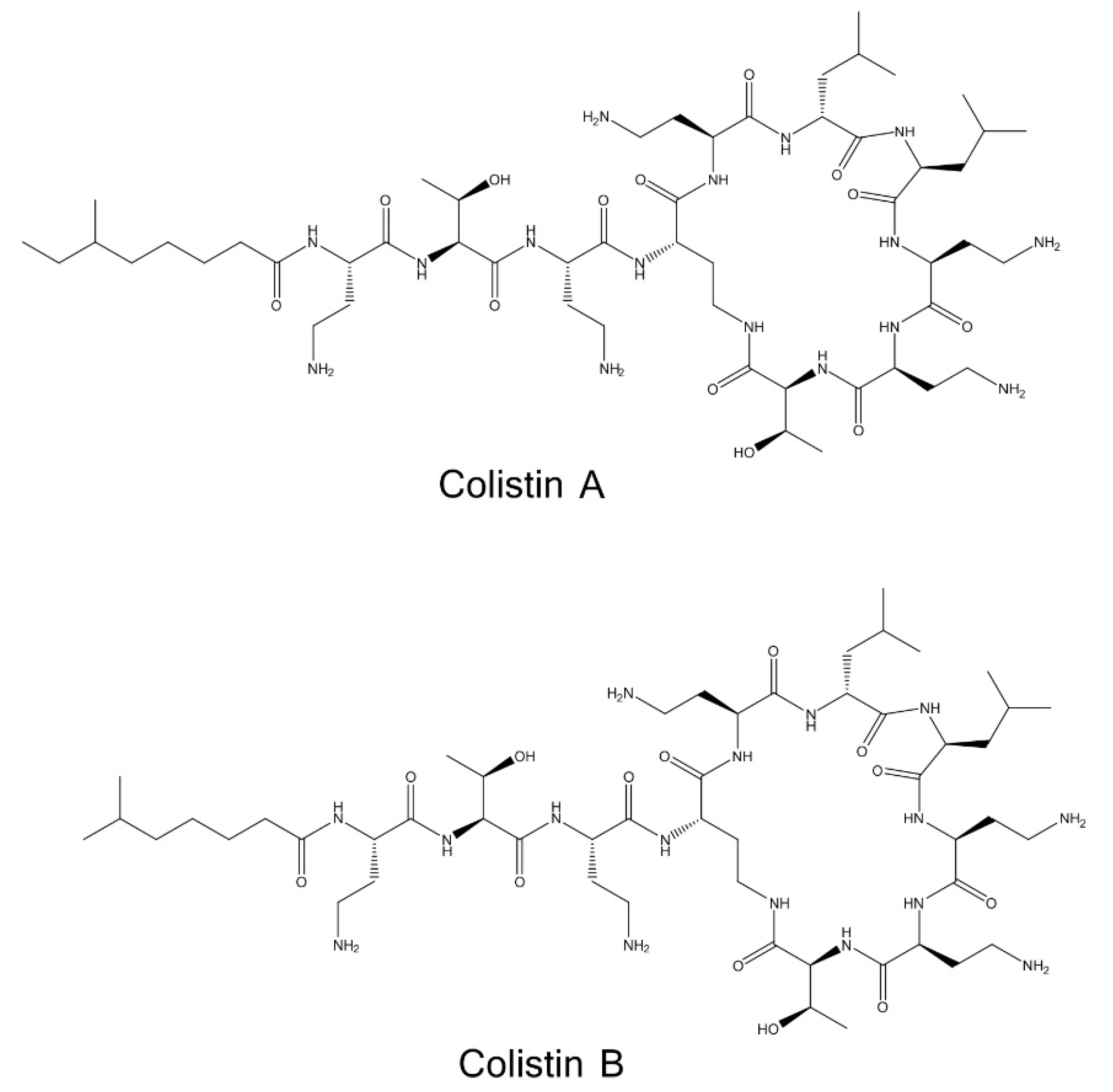
2.1. Determination of Colistin Sulfate Purity and Stoichiometry of Colistin A and B
2.2. Optimization of the HPLC Parameters
2.3. Optimization of the PCD Parameters Using Response Surface Methodology
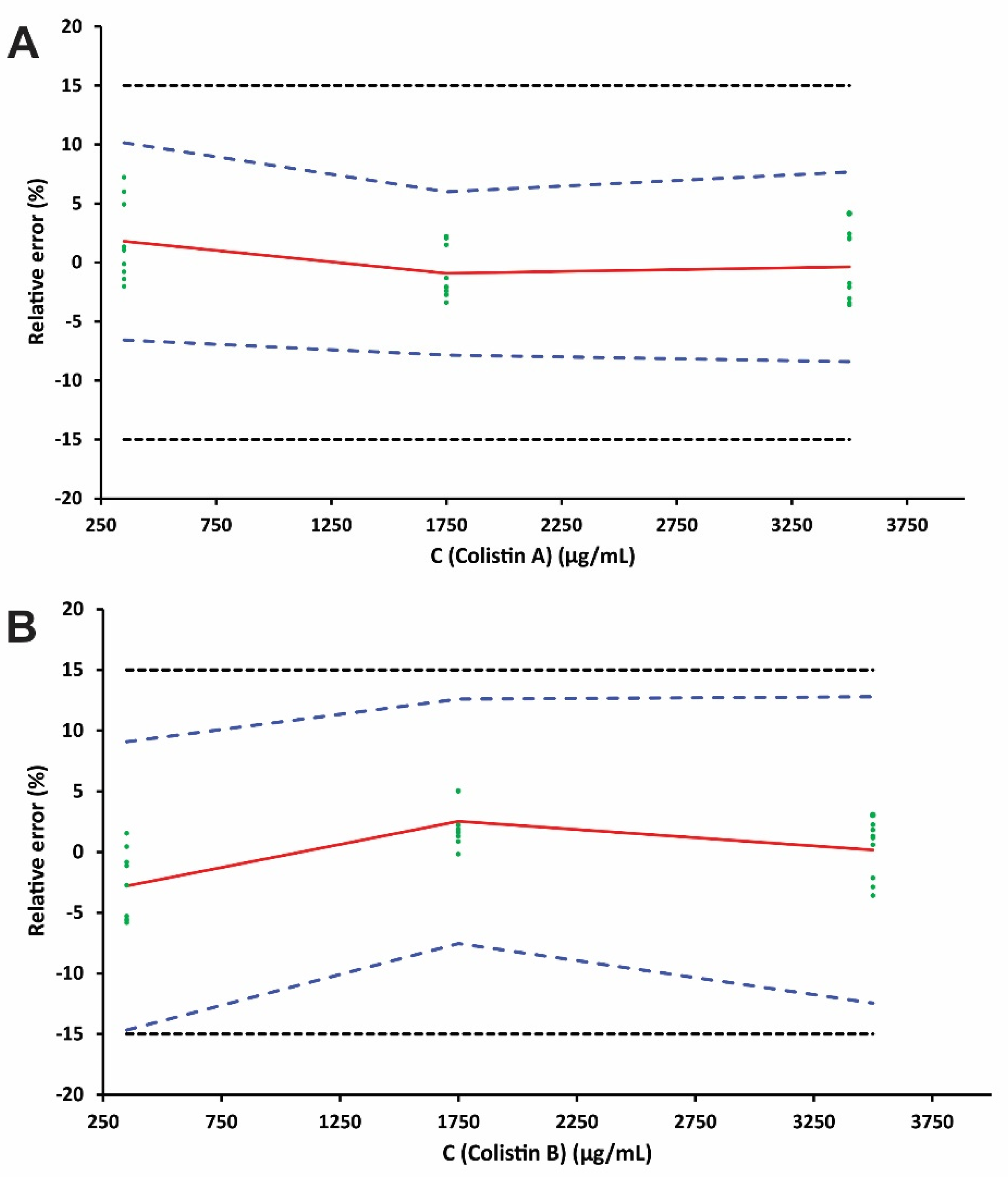
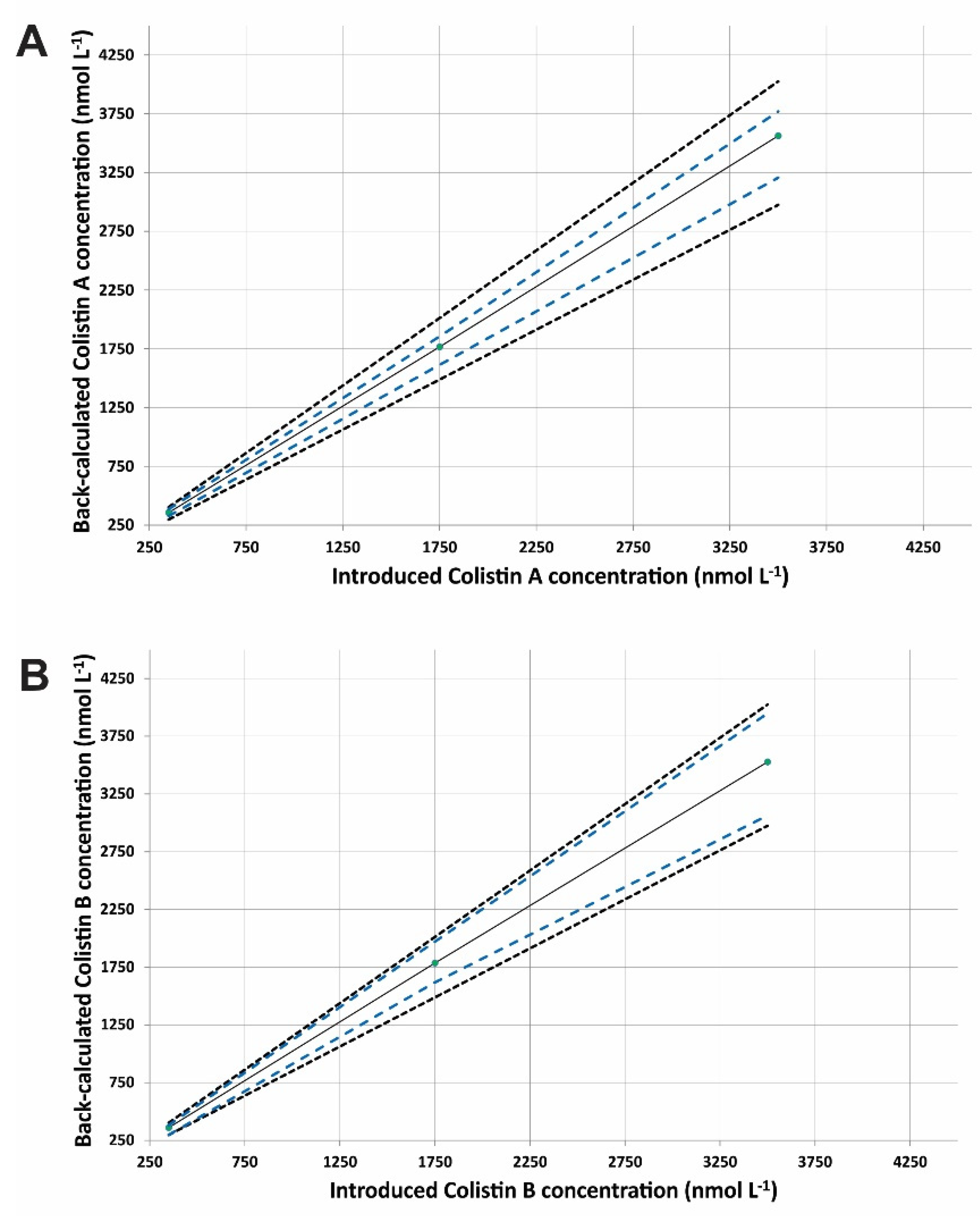
2.4. Method Validation
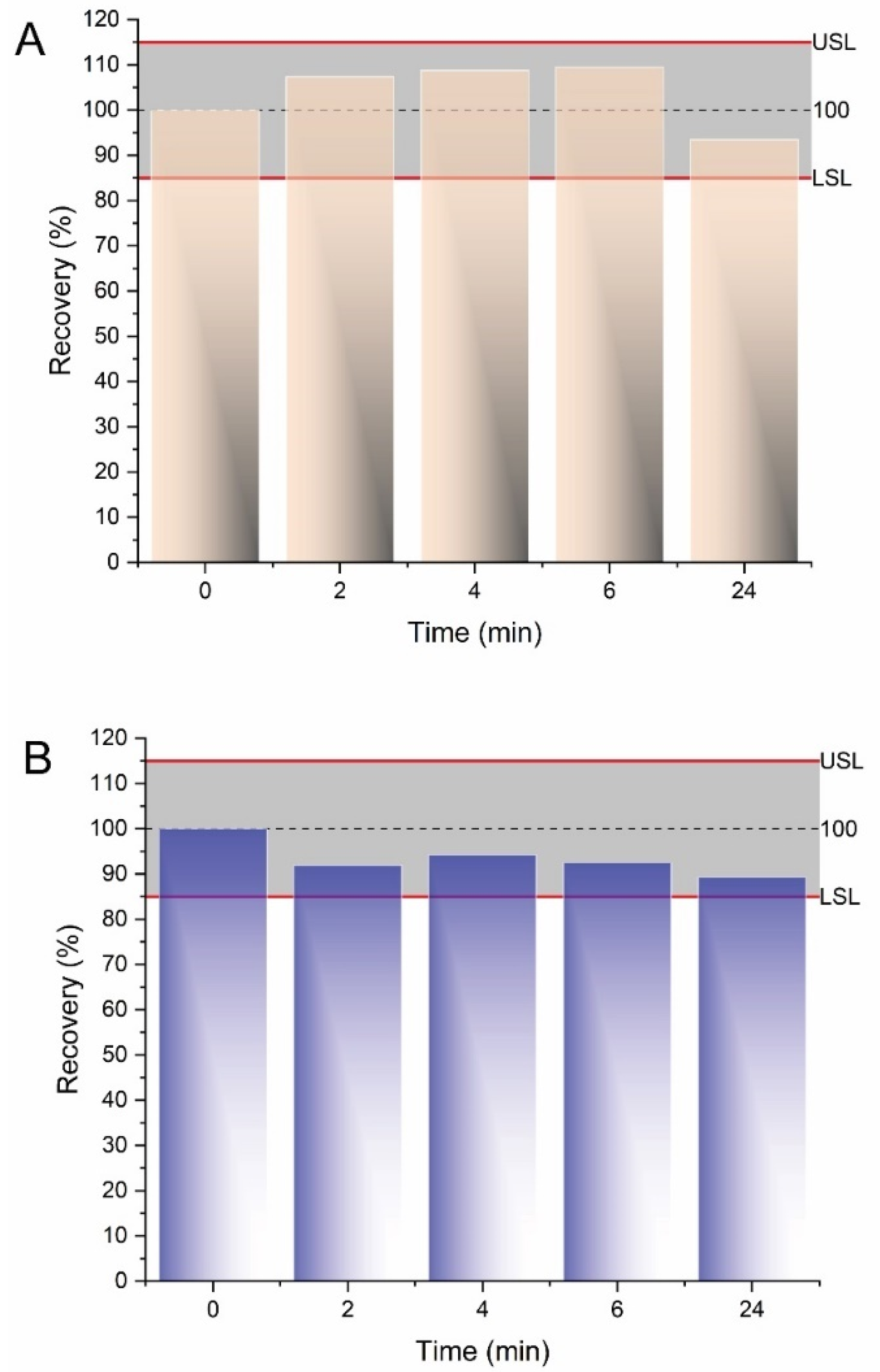
2.5. Sample Stability
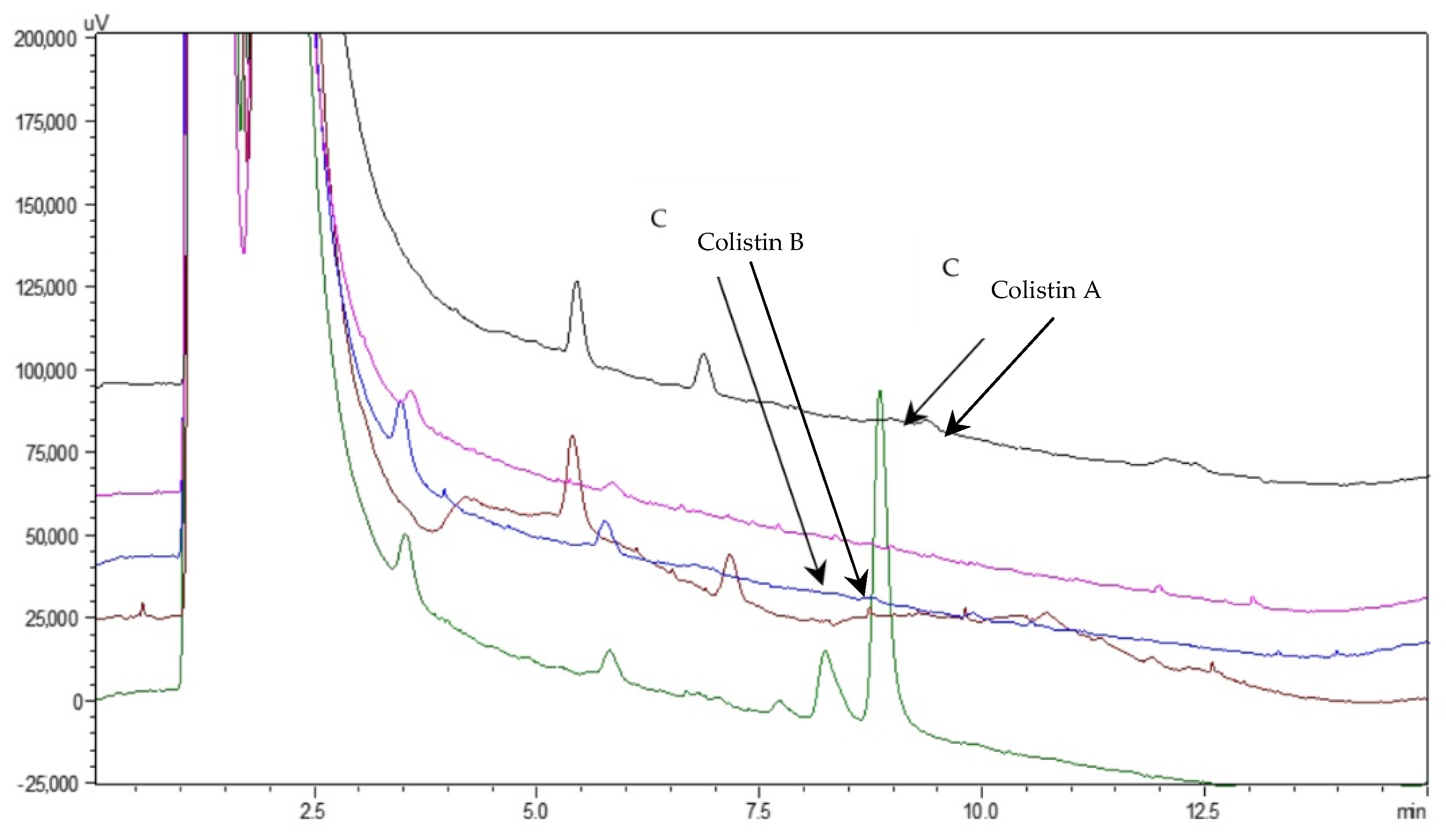
2.6. Sample Analysis
3. Materials and Methods
3.1. Reagents and Solutions
3.2. HPLC Instrumentation
3.3. HPLC–PCD Conditions
3.4. Sample Preparation
3.5. Method Validation Using Accuracy Profiles
- –
- is the estimate of the mean results of the j-th concentration level,
- –
- is the unknown “true value”,
- –
- p is the number of series,
- –
- n is the number of the independent replicate per series,
- –
- Qt (ν; (1 + β)/2) is the β quantile of the t-Student distribution with ν degrees of freedom,
- –
- is the within-series variance,
- –
- is the between-series variance.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kumar, H.; Chen, B.-H.; Kuca, K.; Nepovimova, E.; Kaushal, A.; Nagraik, R.; Bhatia, S.K.; Dhanjal, D.S.; Kumar, V.; Kumar, A.; et al. Understanding of Colistin Usage in Food Animals and Available Detection Techniques: A Review. Animals 2020, 10, 1892. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagla, I.; Karkoula, E.; Baira, E.; Tsarbopoulos, A.; Gikas, E. Analytical Methodologies Used for the Determination of Colistin in Biological Fluids. Is It Still a Challenge? J. Pharm. Biomed. Anal. 2019, 164, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Dagla, I.; Tsarbopoulos, A.; Gikas, E. A Novel Validated Injectable Colistimethate Sodium Analysis Combining Advanced Chemometrics and Design of Experiments. Molecules 2021, 26, 1546. [Google Scholar] [CrossRef] [PubMed]
- Dagla, I.; Tsarbopoulos, A.; Gikas, E. Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS. Molecules 2021, 26, 447. [Google Scholar] [CrossRef] [PubMed]
- Pristovšek, P.; Kidrič, J. Solution Structure of Polymyxins B and E and Effect of Binding to Lipopolysaccharide: An NMR and Molecular Modeling Study. J. Med. Chem. 1999, 42, 4604–4613. [Google Scholar] [CrossRef]
- Schindler, M.; Osborn, M.J. Interaction of Divalent Cations and Polymyxin B with Lipopolysaccharide. Biochemistry 1979, 18, 4425–4430. [Google Scholar] [CrossRef]
- Bergen, P.J.; Li, J.; Rayner, C.R.; Nation, R.L. Colistin Methanesulfonate Is an Inactive Prodrug of Colistin against Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Yu, S.; Chai, G.; Liu, J.; Zhou, Q.T. An LC-MS/MS Method for Simultaneous Analysis of the Cystic Fibrosis Therapeutic Drugs Colistin, Ivacaftor and Ciprofloxacin. J. Pharm. Anal. 2021, 11, 732–738. [Google Scholar] [CrossRef]
- Matar, K.M.; Al-Refai, B. Quantification of Colistin in Plasma by Liquid Chromatography-Tandem Mass Spectrometry: Application to a Pharmacokinetic Study. Sci. Rep. 2020, 10, 8198. [Google Scholar] [CrossRef]
- Qi, B.; Gijsen, M.; Van Brantegem, P.; De Vocht, T.; Deferm, N.; Abza, G.B.; Nauwelaerts, N.; Wauters, J.; Spriet, I.; Annaert, P. Quantitative Determination of Colistin A/B and Colistin Methanesulfonate in Biological Samples Using Hydrophilic Interaction Chromatography Tandem Mass Spectrometry. Drug Test. Anal. 2020, 12, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.; Gillard, N.; Robert, C.; Pierret, G.; Henrottin, J.; Richez, P.; Pelligand, L. Determination of Colistin in Luminal and Parietal Intestinal Matrices of Chicken by Ultra-high-performance Liquid Chromatography-tandem Mass Spectrometry. J. Vet. Pharmacol. Ther. 2021, 44, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.; Richez, P.; Azzariti, S.; Pelligand, L. Pharmacokinetics of Colistin in the Gastrointestinal Tract of Poultry Following Dosing via Drinking Water and Its Bactericidal Impact on Enteric Escherichia Coli. Front. Vet. Sci. 2021, 8, 634. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zuo, S.; Qiu, Y.; Fu, S.; Peng, L. Determination of Colistin in Contents Derived from Gastrointestinal Tract of Feeding Treated Piglet and Broiler. Antibiotics 2021, 10, 422. [Google Scholar] [CrossRef]
- Kumar, H.; Kumar, D.; Nepovimova, E.; Oulkar, D.; Kumar, A.; Azad, R.M.R.; Budakoti, S.K.; Upadhyay, N.K.; Verma, R.; Kuča, K. Determination of Colistin B in Chicken Muscle and Egg Using Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Int. J. Environ. Res. Public Health 2021, 18, 2651. [Google Scholar] [CrossRef]
- Decolin, D.; Leroy, P.; Nicolas, A.; Archimbault, P. Hyphenated Liquid Chromatographic Method for the Determination of Colistin Residues in Bovine Tissues. J. Chromatogr. Sci. 1997, 35, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Le Brun, P.P.H.; de Graaf, A.I.; Vinks, A.A.T.M.M. High-Performance Liquid Chromatographic Method for the Determination of Colistin in Serum. Ther. Drug Monit. 2000, 22, 589–593. [Google Scholar] [CrossRef]
- Cancho-Grande, B.; Rodríguez-Comesaña, M.; Simal-Gándara, J. Simple HPLC Determination of Colistin in Medicated Feeds by Pre-Column Derivatization and Fluorescence Detection. Chromatographia 2001, 54, 481–484. [Google Scholar] [CrossRef]
- Morales-Muñoz, S.; de Castro, M.D.L. Dynamic Ultrasound-Assisted Extraction of Colistin from Feeds with on-Line Pre-Column Derivatization and Liquid Chromatography-Fluorimetric Detection. J. Chromatogr. A 2005, 1066, 1–7. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Coulthard, K.; Johnson, D.W. A Simple Method for the Assay of Colistin in Human Plasma, Using Pre-Column Derivatization with 9-Fluorenylmethyl Chloroformate in Solid-Phase Extraction Cartridges and Reversed-Phase High-Performance Liquid Chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2001, 761, 167–175. [Google Scholar] [CrossRef]
- Chepyala, D.; Tsai, I.-L.; Sun, H.-Y.; Lin, S.-W.; Kuo, C.-H. Development and Validation of a High-Performance Liquid Chromatography-Fluorescence Detection Method for the Accurate Quantification of Colistin in Human Plasma. J. Chromatogr. B 2015, 980, 48–54. [Google Scholar] [CrossRef]
- Zacharis, C.K.; Tzanavaras, P.D. Liquid Chromatography Coupled to On-Line Post Column Derivatization for the Determination of Organic Compounds: A Review on Instrumentation and Chemistries. Anal. Chim. Acta 2013, 798, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Morovján, G.; Csokán, P.P.; Németh-Konda, L. HPLC Determination of Colistin and Aminoglycoside Antibiotics in Feeds by Post-Column Derivatization and Fluorescence Detection. Chromatographia 1998, 48, 32–36. [Google Scholar] [CrossRef]
- Gikas, E.; Bazoti, F.N.; Katsimardou, M.; Anagnostopoulos, D.; Papanikolaou, K.; Inglezos, I.; Skoutelis, A.; Daikos, G.L.; Tsarbopoulos, A. Determination of Colistin A and Colistin B in Human Plasma by UPLC–ESI High Resolution Tandem MS: Application to a Pharmacokinetic Study. J. Pharm. Biomed. Anal. 2013, 83, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Dotsikas, Y.; Markopoulou, C.K.; Koundourellis, J.E.; Loukas, Y.L. Validation of a Novel LC-MS/MS Method for the Quantitation of Colistin A and B in Human Plasma. J. Sep. Sci. 2011, 34, 37–45. [Google Scholar] [CrossRef]
- Cangemi, G.; Barco, S.; Castagnola, E.; Tripodi, G.; Favata, F.; D’Avolio, A. Development and Validation of UHPLC–MS/MS Methods for the Quantification of Colistin in Plasma and Dried Plasma Spots. J. Pharm. Biomed. Anal. 2016, 129, 551–557. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, X.J.; Fan, Y.X.; Guo, B.N.; Zhang, J. Development and Validation of a UHPLC–MS/MS Assay for Colistin Methanesulphonate (CMS) and Colistin in Human Plasma and Urine Using Weak-Cation Exchange Solid-Phase Extraction. J. Pharm. Biomed. Anal. 2016, 124, 303–308. [Google Scholar] [CrossRef]
- Tzanavaras, P.D.; Chatzitaki, A.T.; Karavasili, C.; Sarigiannis, Y.; Fatouros, D.G.; Zacharis, C.K. Analytical Quality-by-Design Optimization of UHPLC Method for the Analysis of Octreotide Release from a Peptide-Based Hydrogel in-Vitro. J. Pharm. Biomed. Anal. 2022, 214, 114699. [Google Scholar] [CrossRef]
- Sajid, M.; Płotka-Wasylka, J. Green Analytical Chemistry Metrics: A Review. Talanta 2022, 238, 123046. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Coulthard, K.; Valentine, J. Simple Method for Assaying Colistin Methanesulfonate in Plasma and Urine Using High-Performance Liquid Chromatography. Antimicrob. Agents Chemother. 2002, 46, 3304. [Google Scholar] [CrossRef] [Green Version]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of Strategies for the Validation of Quantitative Analytical Procedures: A SFSTP Proposal—Part I. J. Pharm. Biomed. Anal. 2004, 36, 579–586. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Sum of Squares | Df 1 | Mean Square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| Model | 1.578 × 105 | 5 | 31,567.80 | 34.45 | <0.0001 | significant |
| B-C(NAC) | 65,280.25 | 1 | 65,280.25 | 71.24 | <0.0001 | |
| C-Temperature | 15,006.25 | 1 | 15,006.25 | 16.38 | 0.0002 | |
| E-Qv (PCD) (Total) | 60,639.06 | 1 | 60,639.06 | 66.17 | <0.0001 | |
| B2 | 9409.42 | 1 | 9409.42 | 10.27 | 0.0027 | |
| C2 | 10,039.22 | 1 | 10,039.22 | 10.96 | 0.0020 | |
| Residual | 36,655.39 | 40 | 916.38 | |||
| LoF | 29,272.06 | 35 | 836.34 | 0.5664 | 0.8544 | not significant |
| Pure Error | 7383.33 | 5 | 1476.67 | |||
| Cor Total | 1.945 × 105 | 45 | ||||
| Validation Criteria | |||
|---|---|---|---|
| Response Function (Linear Unweighted) | Slope | Intercept | r |
| (k a = 3; m = 3; n = 3) (350–3500 nmol·L−1) | |||
| Day 1 | 307.96 | −55,640 | 0.9985 |
| Day 2 | 353.28 | −86,169 | 0.9997 |
| Day 3 | 345.59 | −80,842 | 0.9967 |
| Precision (k = 3; n = 3) | |||
| C (nmol·L−1) | sr (%) b | sR (%) c | |
| 350 | 3.4 | 3.4 | |
| 1750 | 1.7 | 2.3 | |
| 3500 | 2.7 | 3.1 | |
| Trueness (k = 3; n = 3) | |||
| C (nmol·L−1) | Relative bias (%) | ||
| 350 | 1.8 | ||
| 1750 | −0.9 | ||
| 3500 | −0.4 | ||
| Accuracy (k = 3; n = 3) | |||
| C (nmol·L−1) | Relative β-ΕΤΙ (%) | ||
| 350 | [−6.57, 10.15] | ||
| 1750 | [−7.84, 6.0] | ||
| 3500 | [−8.39, 7.66] | ||
| Linearity (k = 3; n = 3; m = 3) (350–3500 nmol·L−1) | |||
| Slope | 1.018 | ||
| Intercept | −5.74 | ||
| r2 | 0.9999 | ||
| LOD (nmol·L−1) | 100 | ||
| LLOQ (nmol·L−1) | 350 | ||
| Sample | Colistin A | Colistin B | ||||
|---|---|---|---|---|---|---|
| Fortification Level (nmol·L−1) | Recovery (%) | RSD (%) | Fortification Level (nmol·L−1) | Recovery (%) | RSD (%) | |
| Urine S1 | 865 | 108.1 | 1.3 | 865 | 111.4 | 1.6 |
| 2580 | 103.9 | 1.7 | 2580 | 114.1 | 1.1 | |
| Urine S2 | 865 | 87.0 | 2.7 | 865 | 105.7 | 2.1 |
| 2580 | 106.4 | 1.3 | 2580 | 100.9 | 1.7 | |
| Urine S3 | 865 | 86.7 | 1.2 | 865 | 108.3 | 2.0 |
| 2580 | 107.2 | 1.6 | 2580 | 100.6 | 3.2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papavasileiou, K.; Tsiasioti, A.; Tzanavaras, P.D.; Zacharis, C.K. HPLC Determination of Colistin in Human Urine Using Alkaline Mobile Phase Combined with Post-Column Derivatization: Validation Using Accuracy Profiles. Molecules 2022, 27, 3489. https://doi.org/10.3390/molecules27113489
Papavasileiou K, Tsiasioti A, Tzanavaras PD, Zacharis CK. HPLC Determination of Colistin in Human Urine Using Alkaline Mobile Phase Combined with Post-Column Derivatization: Validation Using Accuracy Profiles. Molecules. 2022; 27(11):3489. https://doi.org/10.3390/molecules27113489
Chicago/Turabian StylePapavasileiou, Kalliopi, Apostolia Tsiasioti, Paraskevas D. Tzanavaras, and Constantinos K. Zacharis. 2022. "HPLC Determination of Colistin in Human Urine Using Alkaline Mobile Phase Combined with Post-Column Derivatization: Validation Using Accuracy Profiles" Molecules 27, no. 11: 3489. https://doi.org/10.3390/molecules27113489