
A Computational Analysis of the Reaction of SO2 with Amino Acid Anions: Implications for Its Chemisorption in Biobased Ionic Liquids




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results
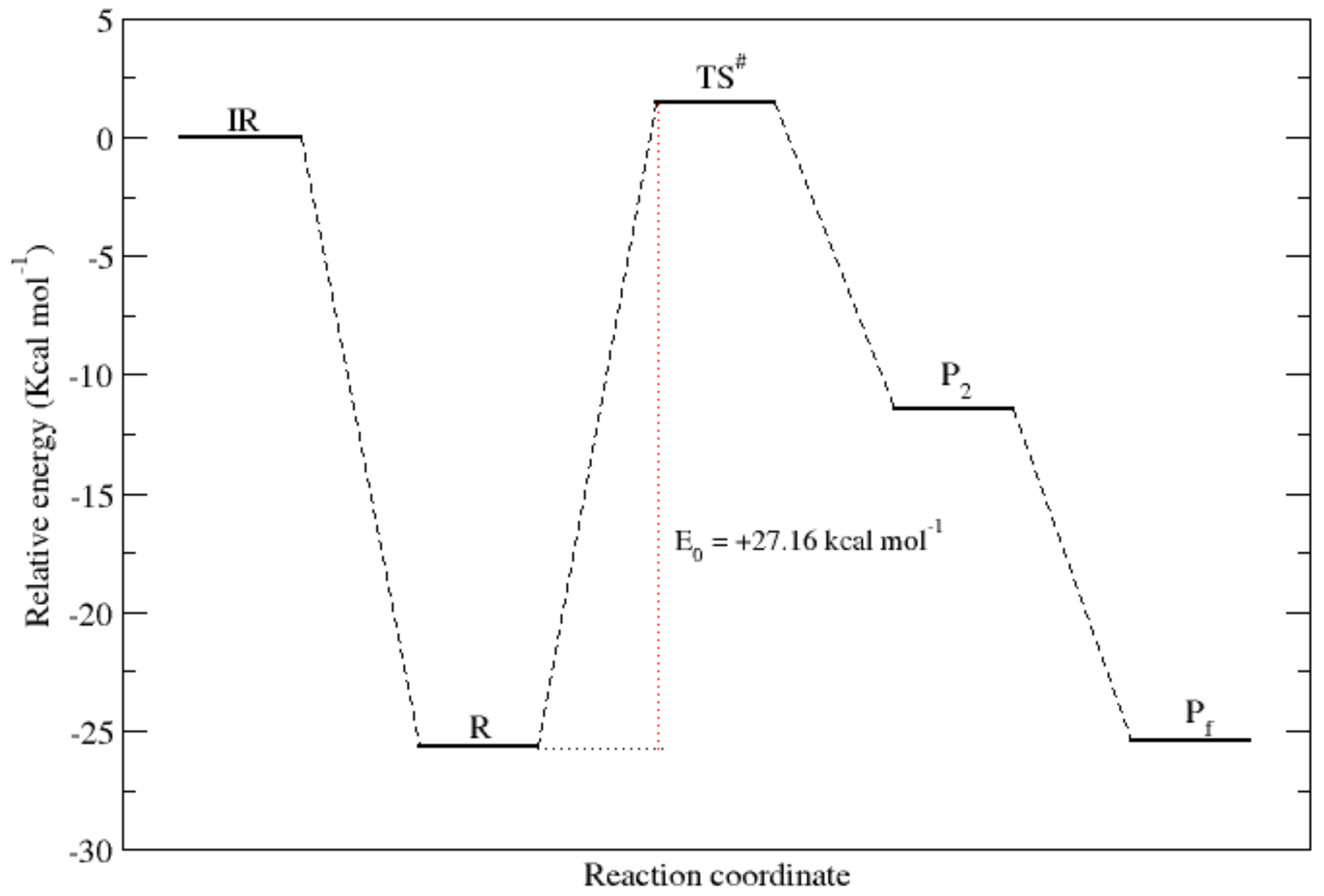
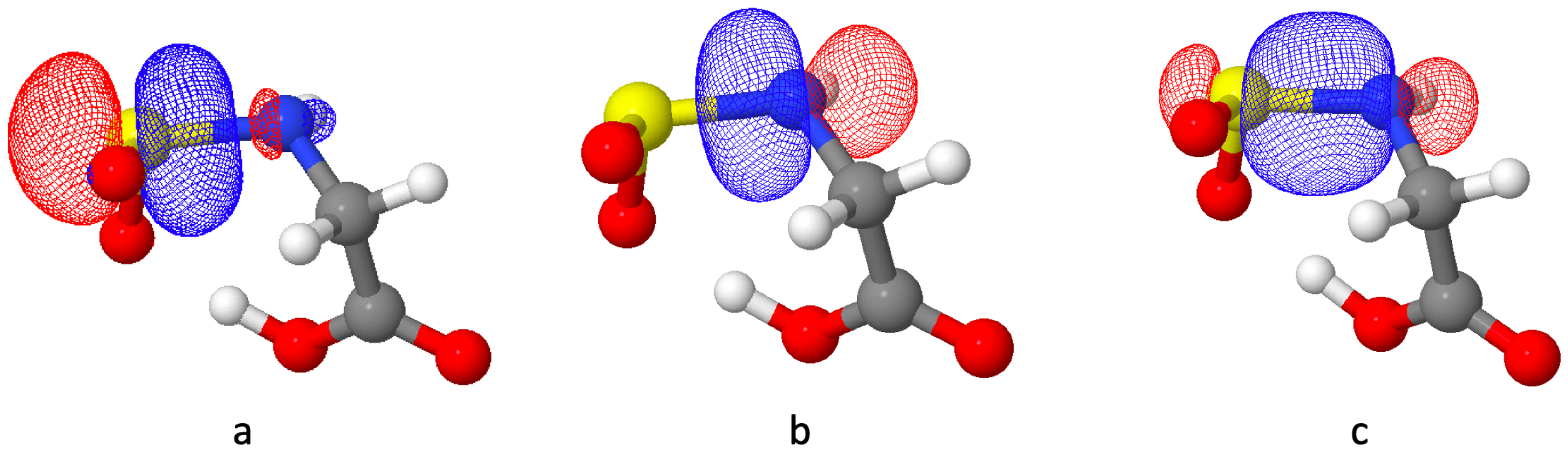
3.1. The Glycinate Reaction
3.2. The Cysteinate Reaction
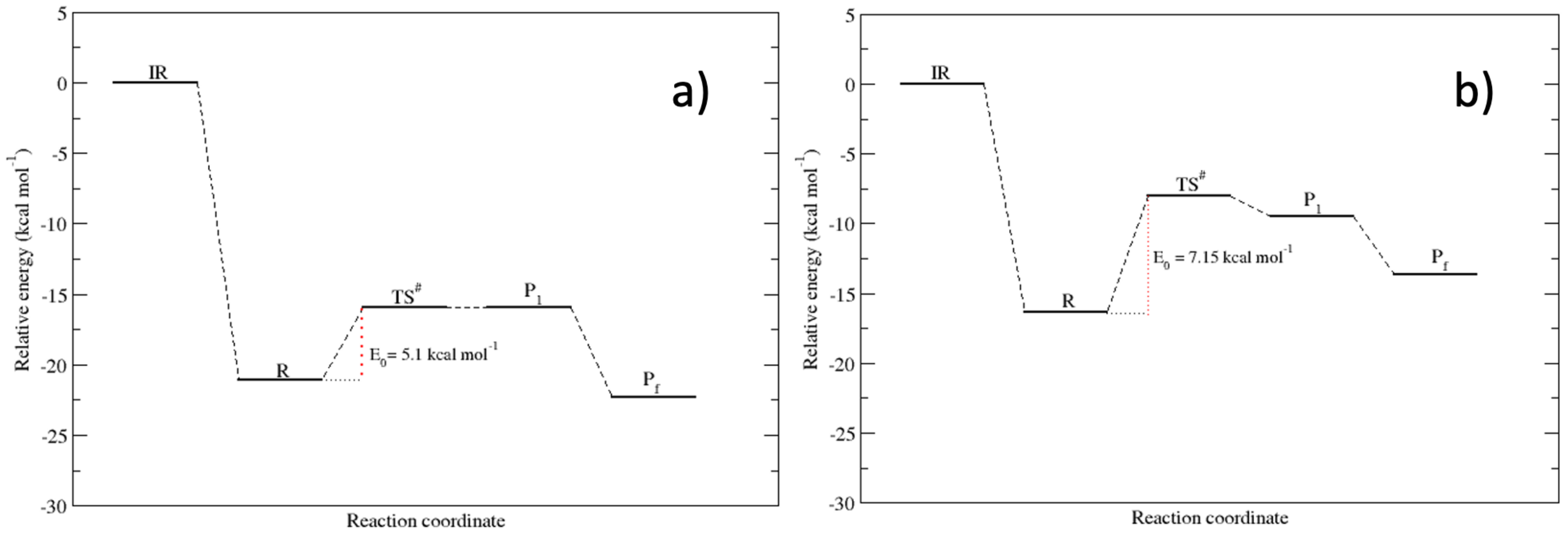
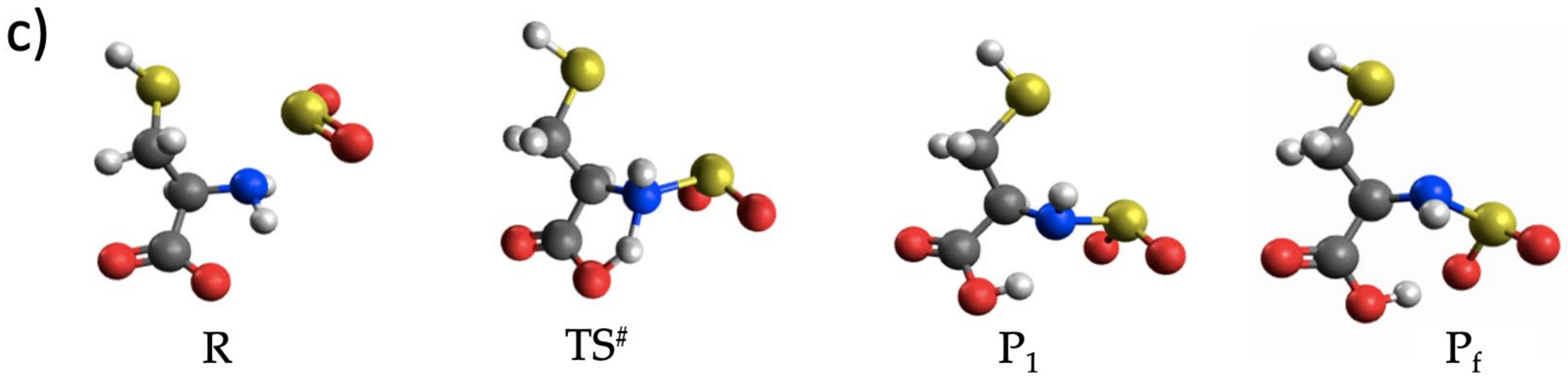
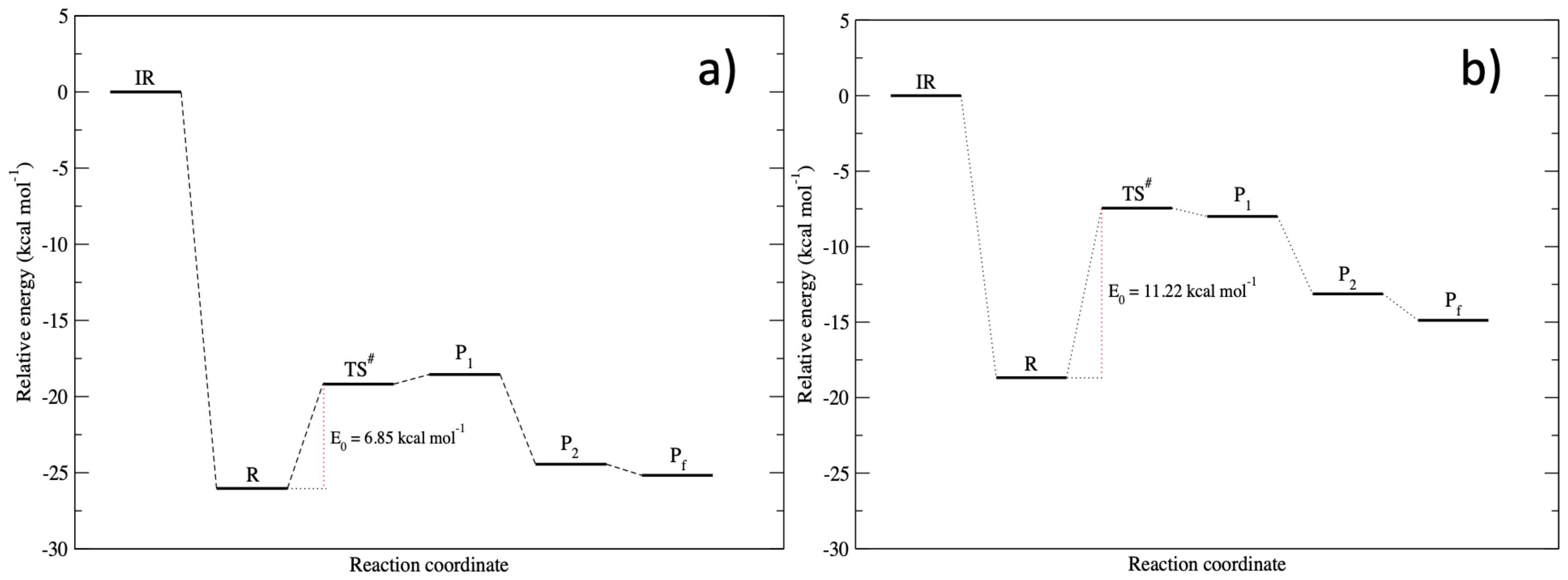
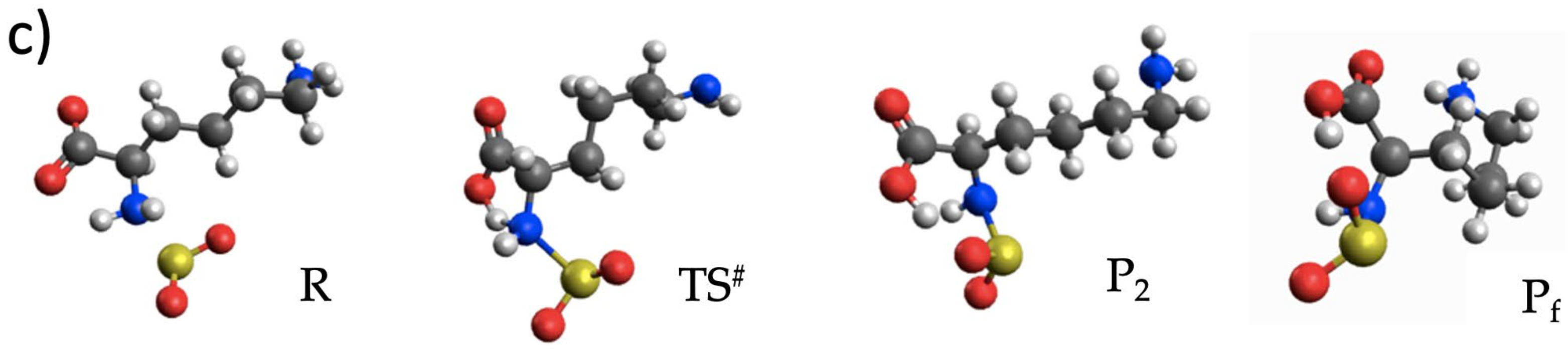
3.3. The Lysinate Reaction
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (Ed.) Air Quality Guidelines: Global Update 2005: Particulate Matter, Ozone, Nitrogen Dioxide, and Sulfur Dioxide; World Health Organization: Copenhagen, Denmark, 2006; ISBN 978-92-890-2192-0. [Google Scholar]
- Hoesly, R.M.; Smith, S.J.; Feng, L.; Klimont, Z.; Janssens-Maenhout, G.; Pitkanen, T.; Seibert, J.J.; Vu, L.; Andres, R.J.; Bolt, R.M.; et al. Historical (1750–2014) Anthropogenic Emissions of Reactive Gases and Aerosols from the Community Emissions Data System (CEDS). Geosci. Model Dev. 2018, 11, 369–408. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Li, Y.; Yao, Q. The Health Costs of the Industrial Leap Forward in China: Evidence from the Sulfur Dioxide Emissions of Coal-Fired Power Stations. China Econ. Rev. 2018, 49, 68–83. [Google Scholar] [CrossRef]
- Lawrence, G.B.; Hazlett, P.W.; Fernandez, I.J.; Ouimet, R.; Bailey, S.W.; Shortle, W.C.; Smith, K.T.; Antidormi, M.R. Declining Acidic Deposition Begins Reversal of Forest-Soil Acidification in the Northeastern U.S. and Eastern Canada. Environ. Sci. Technol. 2015, 49, 13103–13111. [Google Scholar] [CrossRef] [PubMed]
- Kahl, J.S.; Stoddard, J.L.; Haeuber, R.; Paulsen, S.G.; Birnbaum, R.; Deviney, F.A.; Webb, J.R.; DeWalle, D.R.; Sharpe, W.; Driscoll, C.T.; et al. Peer Reviewed: Have U.S. Surface Waters Responded to the 1990 Clean Air Act Amendments? Environ. Sci. Technol. 2004, 38, 484A–490A. [Google Scholar] [CrossRef] [Green Version]
- Hansen, B.B.; Kiil, S.; Johnsson, J.E.; Sønder, K.B. Foaming in Wet Flue Gas Desulfurization Plants: The Influence of Particles, Electrolytes, and Buffers. Ind. Eng. Chem. Res. 2008, 47, 3239–3246. [Google Scholar] [CrossRef]
- Mirdrikvand, M.; Moqadam, S.I.; Kharaghani, A.; Roozbehani, B.; Jadidi, N. Optimization of a Pilot-Scale Amine Scrubber to Remove SO2: Higher Selectivity and Lower Solvent Consumption. Chem. Eng. Technol. 2016, 39, 246–254. [Google Scholar] [CrossRef]
- Ren, S.; Hou, Y.; Zhang, K.; Wu, W. Ionic Liquids: Functionalization and Absorption of SO2. Green Energy Environ. 2018, 3, 179–190. [Google Scholar] [CrossRef]
- Bates, E.D.; Mayton, R.D.; Ntai, I.; Davis, J.H. CO2 Capture by a Task-Specific Ionic Liquid. J. Am. Chem. Soc. 2002, 124, 926–927. [Google Scholar] [CrossRef]
- Giernoth, R. Task-Specific Ionic Liquids. Angew. Chem. Int. Ed. 2010, 49, 2834–2839. [Google Scholar] [CrossRef]
- Yan, S.; Han, F.; Hou, Q.; Zhang, S.; Ai, S. Recent Advances in Ionic Liquid-Mediated SO2 Capture. Ind. Eng. Chem. Res. 2019, 58, 13804–13818. [Google Scholar] [CrossRef]
- Rashid, T.U. Ionic Liquids: Innovative Fluids for Sustainable Gas Separation from Industrial Waste Stream. J. Mol. Liq. 2021, 321, 114916. [Google Scholar] [CrossRef]
- Zeng, S.; Gao, H.; Zhang, X.; Dong, H.; Zhang, X.; Zhang, S. Efficient and Reversible Capture of SO2 by Pyridinium-Based Ionic Liquids. Chem. Eng. J. 2014, 251, 248–256. [Google Scholar] [CrossRef]
- Jiang, L.; Mei, K.; Chen, K.; Dao, R.; Li, H.; Wang, C. Design and Prediction for Highly Efficient SO2 Capture from Flue Gas by Imidazolium Ionic Liquids. Green Energy Environ. 2022, 7, 130–136. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Liu, Y.; Xie, H.; Xu, Y.; Wei, J. SO2 Absorption in Pure Ionic Liquids: Solubility and Functionalization. J. Hazard. Mater. 2020, 392, 122504. [Google Scholar] [CrossRef]
- Mao, F.-F.; Zhou, Y.; Zhu, W.; Sang, X.-Y.; Li, Z.-M.; Tao, D.-J. Synthesis of Guanidinium-Based Poly(Ionic Liquids) with Nonporosity for Highly Efficient SO2 Capture from Flue Gas. Ind. Eng. Chem. Res. 2021, 60, 5984–5991. [Google Scholar] [CrossRef]
- Geng, Z.; Xie, Q.; Fan, Z.; Sun, W.; Zhao, W.; Zhang, J.; Chen, J.; Xu, Y. Investigation of Tertiary Amine-Based PILs for Ideal Efficient SO2 Capture from CO2. J. Environ. Chem. Eng. 2021, 9, 105824. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, Q.; Gao, M.; Ren, S.; Wu, W. Absorption and Conversion of SO2 in Functional Ionic Liquids: Effect of Water on the Claus Reaction. ACS Omega 2022, 7, 10413–10419. [Google Scholar] [CrossRef]
- Onofri, S.; Adenusi, H.; Le Donne, A.; Bodo, E. CO2 Capture in Ionic Liquids Based on Amino Acid Anions With Protic Side Chains: A Computational Assessment of Kinetically Efficient Reaction Mechanisms. ChemistryOpen 2020, 9, 1153–1160. [Google Scholar] [CrossRef]
- Onofri, S.; Bodo, E. CO2 Capture in Biocompatible Amino Acid Ionic Liquids: Exploring the Reaction Mechanisms for Bimolecular Absorption Processes. J. Phys. Chem. B 2021, 125, 5611–5619. [Google Scholar] [CrossRef]
- Hou, X.-D.; Liu, Q.-P.; Smith, T.J.; Li, N.; Zong, M.-H. Evaluation of Toxicity and Biodegradability of Cholinium Amino Acids Ionic Liquids. PLoS ONE 2013, 8, e59145. [Google Scholar] [CrossRef]
- Gontrani, L.; Scarpellini, E.; Caminiti, R.; Campetella, M. Bio Ionic Liquids and Water Mixtures: A Structural Study. RSC Adv. 2017, 7, 19338–19344. [Google Scholar] [CrossRef] [Green Version]
- Gontrani, L. Choline-Amino Acid Ionic Liquids: Past and Recent Achievements about the Structure and Properties of These Really “Green” Chemicals. Biophys. Rev. 2018, 10, 873–880. [Google Scholar] [CrossRef]
- Caparica, R.; Júlio, A.; Baby, A.; Araújo, M.; Fernandes, A.; Costa, J.; Santos de Almeida, T. Choline-Amino Acid Ionic Liquids as Green Functional Excipients to Enhance Drug Solubility. Pharmaceutics 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Donne, A.; Adenusi, H.; Porcelli, F.; Bodo, E. Structural Features of Cholinium Based Protic Ionic Liquids through Molecular Dynamics. J. Phys. Chem. B 2019, 123, 5568–5576. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, X.; Li, J.; Qiu, R.; Lin, J. Cooperative CO2 Absorption by Amino Acid-Based Ionic Liquids with Balanced Dual Sites. RSC Adv. 2020, 10, 7751–7757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodo, E. Modelling Biocompatible Ionic Liquids Based on Organic Acids and Amino Acids: Challenges for Computational Models and Future Perspectives. Org. Biomol. Chem. 2021, 19, 4002–4013. [Google Scholar] [CrossRef] [PubMed]
- Dhattarwal, H.S.; Kashyap, H.K. Unique and Generic Structural Features of Cholinium Amino Acid-Based Biocompatible Ionic Liquids. Phys. Chem. Chem. Phys. 2021, 23, 10662–10669. [Google Scholar] [CrossRef]
- Bodo, E. Perspectives in the Computational Modeling of New Generation, Biocompatible Ionic Liquids. J. Phys. Chem. B 2022, 126, 3–13. [Google Scholar] [CrossRef]
- Hussain, M.A.; Soujanya, Y.; Sastry, G.N. Evaluating the Efficacy of Amino Acids as CO2 Capturing Agents: A First Principles Investigation. Environ. Sci. Technol. 2011, 45, 8582–8588. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Shah, F.U. Ether Functionalized Choline Tethered Amino Acid Ionic Liquids for Enhanced CO2 Capture. ACS Sustain. Chem. Eng. 2016, 4, 5441–5449. [Google Scholar] [CrossRef]
- Firaha, D.S.; Kirchner, B. Tuning the Carbon Dioxide Absorption in Amino Acid Ionic Liquids. ChemSusChem 2016, 9, 1591–1599. [Google Scholar] [CrossRef]
- Chen, F.-F.; Huang, K.; Zhou, Y.; Tian, Z.-Q.; Zhu, X.; Tao, D.-J.; Jiang, D.; Dai, S. Multi-Molar Absorption of CO2 by the Activation of Carboxylate Groups in Amino Acid Ionic Liquids. Angew. Chem. Int. Ed. 2016, 55, 7166–7170. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Yao, J.; Li, H. Equilibrium in Protic Ionic Liquids: The Degree of Proton Transfer and Thermodynamic Properties. J. Phys. Chem. B 2018, 122, 309–315. [Google Scholar] [CrossRef]
- Latini, G.; Signorile, M.; Crocellà, V.; Bocchini, S.; Pirri, C.F.; Bordiga, S. Unraveling the CO2 Reaction Mechanism in Bio-Based Amino-Acid Ionic Liquids by Operando ATR-IR Spectroscopy. Catal. Today 2019, 336, 148–160. [Google Scholar] [CrossRef]
- Davarpanah, E.; Hernández, S.; Latini, G.; Pirri, C.F.; Bocchini, S. Enhanced CO2 Absorption in Organic Solutions of Biobased Ionic Liquids. Adv. Sustain. Syst. 2020, 4, 1900067. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wang, J.; Jiang, H.; Shi, X.; Hu, Y. 2-Ethyl-4-Methylimidazolium Alaninate Ionic Liquid: Properties and Mechanism of SO2 Absorption. Energy Fuels 2017, 31, 2996–3001. [Google Scholar] [CrossRef]
- Wang, H.; Wu, P.; Li, C.; Zhang, J.; Deng, R. Reversible and Efficient Absorption of SO2 with Natural Amino Acid Aqueous Solutions: Performance and Mechanism. ACS Sustain. Chem. Eng. 2022, 10, 4451–4461. [Google Scholar] [CrossRef]
- Wu, C.; Lü, R.; Gates, I.D. Computational Study on the Absorption Mechanisms of SO2 by Ionic Liquids. ChemistrySelect 2018, 3, 4330–4338. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Ashraf, M.; AlMayef, T.; Chawla, M.; Poater, A.; Cavallo, L. Amino Acid Ionic Liquids as Potential Candidates for CO2 Capture: Combined Density Functional Theory and Molecular Dynamics Simulations. Chem. Phys. Lett. 2020, 745, 137239. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Nikolaienko, T.Y.; Bulavin, L.A. Localized Orbitals for Optimal Decomposition of Molecular Properties. Int. J. Quantum Chem. 2019, 119, e25798. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Kaur, R.P.; Singh, H. Effect of Cyclization on Bond Dissociation Enthalpies, Acidities and Proton Affinities of Carbamate Molecules: A Theoretical Study. Results Chem. 2019, 1, 100003. [Google Scholar] [CrossRef]
- Bennett, E.L.; Song, C.; Huang, Y.; Xiao, J. Measured Relative Complex Permittivities for Multiple Series of Ionic Liquids. J. Mol. Liq. 2019, 294, 111571. [Google Scholar] [CrossRef]
- Le Donne, A.; Bodo, E. Isomerization Patterns and Proton Transfer in Ionic Liquids Constituents as Probed by Ab-Initio Computation. J. Mol. Liq. 2018, 249, 1075–1082. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piacentini, V.; Le Donne, A.; Russo, S.; Bodo, E. A Computational Analysis of the Reaction of SO2 with Amino Acid Anions: Implications for Its Chemisorption in Biobased Ionic Liquids. Molecules 2022, 27, 3604. https://doi.org/10.3390/molecules27113604
Piacentini V, Le Donne A, Russo S, Bodo E. A Computational Analysis of the Reaction of SO2 with Amino Acid Anions: Implications for Its Chemisorption in Biobased Ionic Liquids. Molecules. 2022; 27(11):3604. https://doi.org/10.3390/molecules27113604
Chicago/Turabian StylePiacentini, Vanessa, Andrea Le Donne, Stefano Russo, and Enrico Bodo. 2022. "A Computational Analysis of the Reaction of SO2 with Amino Acid Anions: Implications for Its Chemisorption in Biobased Ionic Liquids" Molecules 27, no. 11: 3604. https://doi.org/10.3390/molecules27113604