
Structural Elucidation of Alkali Degradation Impurities of Favipiravir from the Oral Suspension: UPLC-TQ-ESI-MS/MS and NMR

 , ,
, ,  ,
,  , and
, and
Abstract
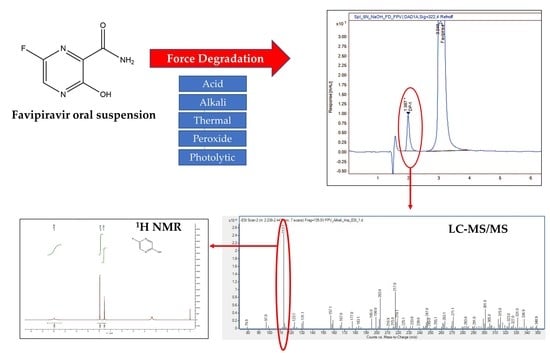
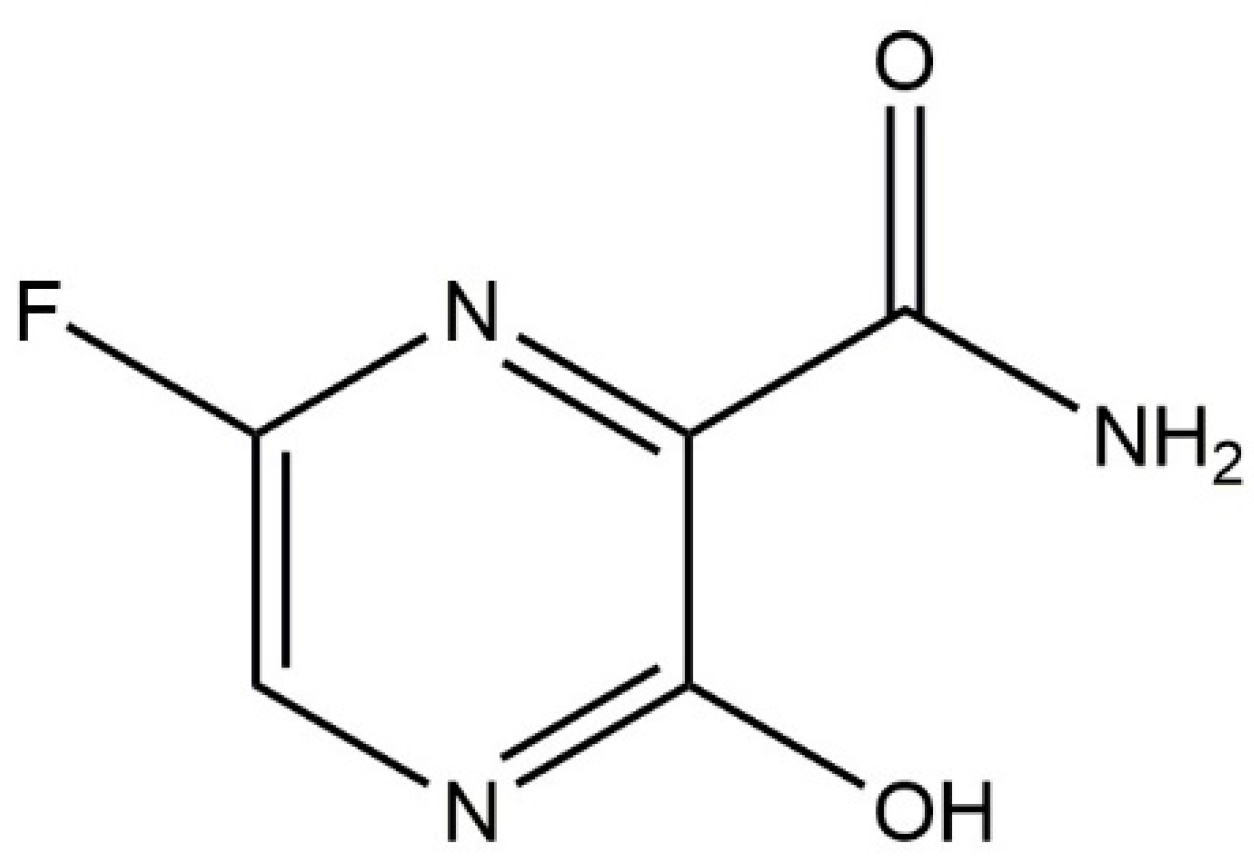
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instruments
2.3. Chromatographic Conditions
2.3.1. RP-HPLC Conditions
2.3.2. UPLC-ESI-TQ-MS/MS Conditions
2.4. Preparation of Analytical Solution
2.4.1. Standard Preparations
2.4.2. Sample Preparation
2.5. Method Validation
2.5.1. System Suitability
2.5.2. Specificity
Acid Degradation
Alkali Degradation
Oxidative Degradation
Thermal Degradation
Photolytic Degradation
2.5.3. Linearity
2.5.4. Accuracy
2.5.5. Precision
2.5.6. Limit of Detection and Quantification
2.5.7. Robustness
2.5.8. Solution Stability
3. Results and Discussion
3.1. Method Development and Optimization
3.2. Method Validation
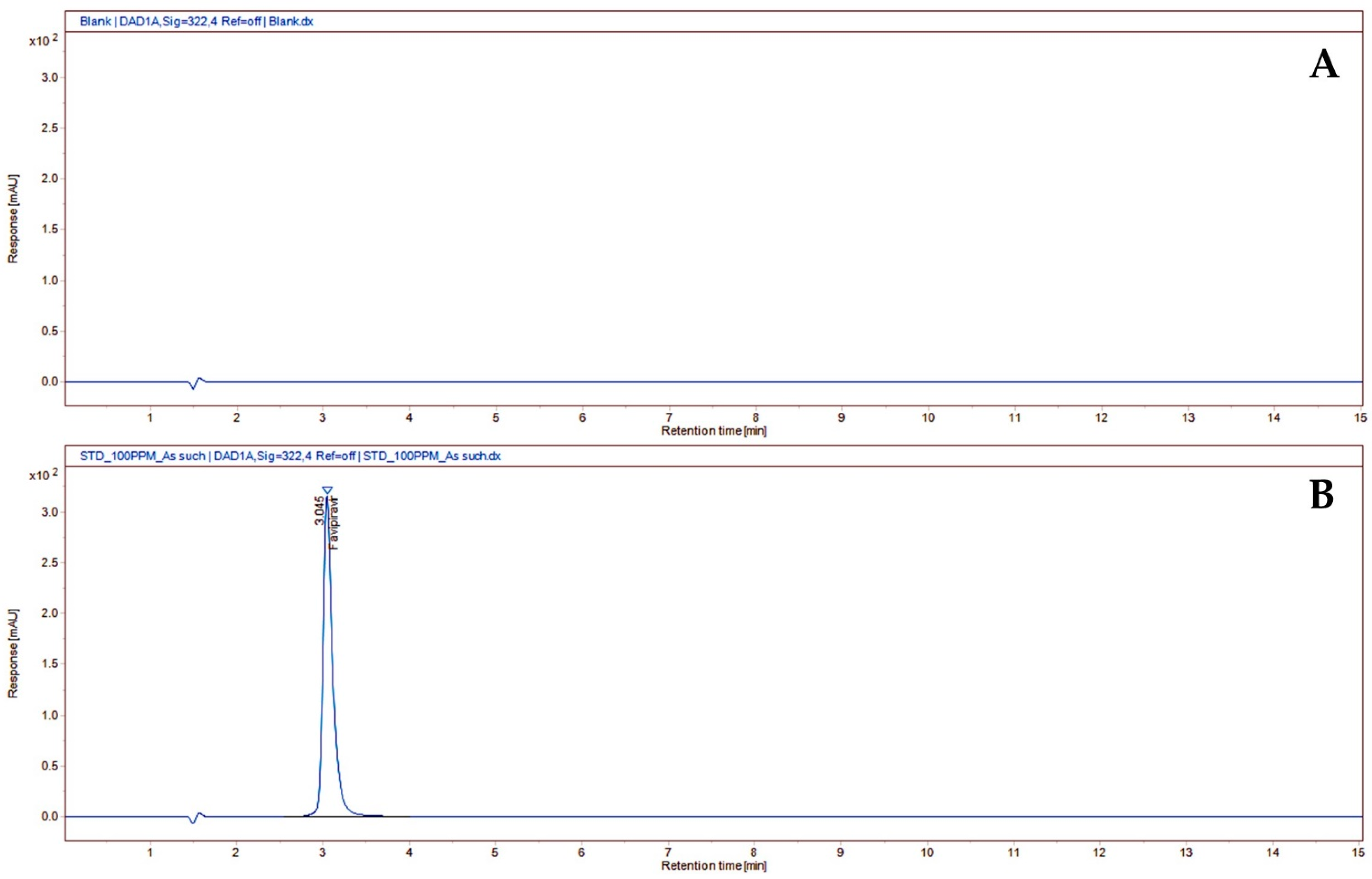
3.2.1. System Suitability
3.2.2. Degradation Behavior of Favipiravir in an Oral Suspension
3.2.3. Linearity
3.2.4. Accuracy
3.2.5. Precision
3.2.6. Limit of Detection and Quantification (LOD and LOQ)
3.2.7. Robustness
3.2.8. Solution Stability
3.3. Isolation and Purification of Degradation Product
3.4. Characterization of Alkali Degradation Products of Favipiravir Using UPLC-ESI-TQ-MS/MS and NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, J.; Parikh, S.; Patel, S.; Patel, R. A Comprehensive Overview On The Most Recent Trends In COVID-19 Diagnosis, Treatment Options And Vaccine Development Status: Comprehensive Overview On The Most Recent Trends In COVID-19. J. Pharm. Sci. Med. Res. 2021, 1, 1–20. [Google Scholar]
- National Center for Biotechnology Information PubChem Compound Summary for CID 492405, Favipiravir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Favipiravir (accessed on 1 August 2022).
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y.; et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering 2020, 6, 1192–1198. [Google Scholar] [CrossRef]
- Coomes, E.A.; Haghbayan, H. Favipiravir, an antiviral for COVID-19? J. Antimicrob. Chemother. 2020, 75, 2013–2014. [Google Scholar] [CrossRef]
- Acquavia, M.A.; Foti, L.; Pascale, R.; Nicolò, A.; Brancaleone, V.; Cataldi, T.R.; Martelli, G.; Scrano, L.; Bianco, G. Detection and quantification of Covid-19 antiviral drugs in biological fluids and tissues. Talanta 2020, 224, 121862. [Google Scholar] [CrossRef]
- Du, Y.; Chen, X. Favipiravir: Pharmacokinetics and Concerns About Clinical Trials for 2019-nCoV Infection. Clin. Pharmacol. Ther. 2020, 108, 242–247. [Google Scholar] [CrossRef]
- Ramachandran, R.; Junnuthula, V.R.; Gowd, G.S.; Ashokan, A.; Thomas, J.; Peethambaran, R.; Thomas, A.; Unni, A.K.K.; Panikar, D.; Nair, S.V.; et al. Theranostic 3-Dimensional nano brain-implant for prolonged and localized treatment of recurrent glioma. Sci. Rep. 2017, 7, srep43271. [Google Scholar] [CrossRef]
- Jain, A.S.; Pawar, P.S.; Sarkar, A.; Junnuthula, V.; Dyawanapelly, S. Bionanofactories for Green Synthesis of Silver Nanoparticles: Toward Antimicrobial Applications. Int. J. Mol. Sci. 2021, 22, 11993. [Google Scholar] [CrossRef]
- Ridolfo, R.; Tavakoli, S.; Junnuthula, V.; Williams, D.S.; Urtti, A.; Van Hest, J.C.M. Exploring the Impact of Morphology on the Properties of Biodegradable Nanoparticles and Their Diffusion in Complex Biological Medium. Biomacromolecules 2020, 22, 126–133. [Google Scholar] [CrossRef]
- Sarkar, A.; Junnuthula, V.; Dyawanapelly, S. Ocular Therapeutics and Molecular Delivery Strategies for Neovascular Age-Related Macular Degeneration (nAMD). Int. J. Mol. Sci. 2021, 22, 10594. [Google Scholar] [CrossRef]
- Gera, S.; Sampathi, S.; Maddukuri, S.; Dodoala, S.; Junnuthula, V.; Dyawanapelly, S. Therapeutic Potential of Naringenin Nanosuspension: In Vitro and In Vivo Anti-Osteoporotic Studies. Pharmaceutics 2022, 14, 1449. [Google Scholar] [CrossRef]
- Devassy, G.; Ramachandran, R.; Jeena, K.; Junnuthula, V.R.; Gopinatha, V.K.; Manju, C.A.; Manohar, M.; Nair, S.V.; Raghavan, S.C.; Koyakutty, M.; et al. Simultaneous release of two drugs from polymer nano-implant inhibits recurrence in glioblastoma spheroids. Precis. Nanomed. 2019, 2, 218–229. [Google Scholar] [CrossRef]
- Pailla, S.R.; Sampathi, S.; Junnuthula, V.; Maddukuri, S.; Dodoala, S.; Dyawanapelly, S. Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics 2022, 14, 978. [Google Scholar] [CrossRef]
- Sarkar, A.; Sodha, S.J.; Junnuthula, V.; Kolimi, P.; Dyawanapelly, S. Novel and investigational therapies for wet and dry age-related macular degeneration. Drug Discov. Today 2022, 27, 2322–2332. [Google Scholar] [CrossRef]
- Junnuthula, V.; Boroujeni, A.S.; Cao, S.; Tavakoli, S.; Ridolfo, R.; Toropainen, E.; Ruponen, M.; van Hest, J.; Urtti, A. Intravitreal Polymeric Nanocarriers with Long Ocular Retention and Targeted Delivery to the Retina and Optic Nerve Head Region. Pharmaceutics 2021, 13, 445. [Google Scholar] [CrossRef]
- Dyawanapelly, S.; Junnuthula, V.R.; Singh, A. The Holy Grail of Polymer Therapeutics for Cancer Therapy: An Overview on the Pharmacokinetics and Bio Distribution. Curr. Drug Metab. 2015, 16, 522–537. [Google Scholar] [CrossRef]
- Khatik, R.; Dwivedi, P.; Junnuthula, V.R.; Sharma, K.; Chuttani, K.; Mishra, A.K.; Dwivedi, A.K. Potential in vitro and in vivo colon specific anticancer activity in a HCT-116 xenograft nude mice model: Targeted delivery using enteric coated folate modified nanoparticles. RSC Adv. 2015, 5, 16507–16520. [Google Scholar] [CrossRef]
- COVID-19: FDC Launches First Favipiravir Oral Suspension Favenza in India. Available online: https://www.fdcindia.com/news/India’s%20First%20Favipiravir%20Oral%20Suspension-Favenza.pdf (accessed on 1 August 2022).
- Megahed, S.M.; Habib, A.A.; Hammad, S.F.; Kamal, A.H. Experimental design approach for development of spectrofluorimetric method for determination of favipiravir; a potential therapeutic agent against COVID-19 virus: Application to spiked human plasma. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 249, 119241. [Google Scholar] [CrossRef]
- Curley, P.; Neary, M.; Arshad, U.; Tatham, L.; Pertinez, H.; Box, H.; Rajoli, R.K.; Valentijn, A.; Sharp, J.; Rannard, S.P.; et al. Development of a Highly Sensitive Bioanalytical Assay for the Quantification of Favipiravir. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rezk, M.R.; Badr, K.A.; Abdel-Naby, N.S.; Ayyad, M.M. A novel, rapid and simple UPLC-MS/MS method for quantification of favipiravir in human plasma: Application to a bioequivalence study. Biomed. Chromatogr. 2021, 35, e5098. [Google Scholar] [CrossRef]
- Morsy, M.I.; Nouman, E.G.; Abdallah, Y.M.; Zainelabdeen, M.A.; Darwish, M.M.; Hassan, A.Y.; Gouda, A.S.; Rezk, M.R.; Abdel-Megied, A.M.; Marzouk, H.M. A novel LC-MS/MS method for determination of the potential antiviral candidate favipiravir for the emergency treatment of SARS-CoV-2 virus in human plasma: Application to a bioequivalence study in Egyptian human volunteers. J. Pharm. Biomed. Anal. 2021, 199, 114057. [Google Scholar] [CrossRef]
- Onmaz, D.E.; Abusoglu, S.; Onmaz, M.; Yerlikaya, F.H.; Unlu, A. Development and validation of a sensitive, fast and simple LC-MS/MS method for the quantitation of favipiravir in human serum. J. Chromatogr. B 2021, 1176, 122768. [Google Scholar] [CrossRef]
- Bulduk, I. HPLC-UV method for quantification of favipiravir in pharmaceutical formulations. Acta Chromatogr. 2021, 33, 209–215. [Google Scholar] [CrossRef]
- Mikhail, I.E.; Elmansi, H.; Belal, F.; Ibrahim, A.E. Green micellar solvent-free HPLC and spectrofluorimetric determination of favipiravir as one of COVID-19 antiviral regimens. Microchem. J. 2021, 165, 106189. [Google Scholar] [CrossRef]
- Marzouk, H.M.; Rezk, M.R.; Gouda, A.S.; Abdel-Megied, A.M. A novel stability-indicating HPLC-DAD method for determination of favipiravir, a potential antiviral drug for COVID-19 treatment; application to degradation kinetic studies and in-vitro dissolution profiling. Microchem. J. 2022, 172, 106917. [Google Scholar] [CrossRef]
- ICH Guideline. Validation of Analytical Procedures: Text and Methodology Q2 (R1)(2005). 2005. Available online: http//www.ich.org/cache/compo/363-272-1.html2005 (accessed on 1 August 2022).
- Glass, B.D.; Haywood, A. Stability considerations in liquid dosage forms extemporaneously prepared from commercially available products. J. Pharm. Pharm. Sci. 2006, 9, 398–426. [Google Scholar]
- Borman, P.; Elder, D. Q2(R1) Validation of Analytical Procedures. ICH Qual. Guidel. 2017, 127–166. [Google Scholar] [CrossRef]
- Solanki, R.V.; Patel, R.B.; Patel, R.K.; Sheth, R.A. Development and Validation of Fast and Robust Stability Indicating RP-HPLC Method for Simultaneous Estimation of Tolperisone Hydrochloride and Etoricoxib in Pharmaceutical Dosage Form. Int. J. Pharm. Investig. 2022, 12, 56–61. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Degradation | Condition | Degradation Products | Rt (Min.) | Assay (%) | % Degradation | Peak Purity |
|---|---|---|---|---|---|---|
| Sample as such | No treatment | - | - | 98.23 | - | 999.97 |
| Acid | 1 N HCl–1 mL/80 °C/60 min | 97.42 | - | 999.95 | ||
| 2 N HCl–1 mL/80 °C/60 min | - | - | 97.35 | - | 999.96 | |
| 5 N HCl–1 mL/80 °C/60 min | - | - | 96.25 | 1.98 | 999.90 | |
| Alkali | 1 N NaOH–1 mL/80 °C/60 min | DP1 | 1.98 | 87.78 | 10.45 | 999.95 |
| 2 N NaOH–1 mL/80 °C/60 min | DP1 | 1.98 | 81.76 | 16.47 | 999.92 | |
| 5 N NaOH–1 mL/80 °C/60 min | DP1 | 1.98 | 69.72 | 28.5 | 999.95 | |
| Oxidative | 10% H2O2–1 mL/80 °C/60 min | - | - | 97.90 | - | 999.96 |
| 30% H2O2–1 mL/80 °C/60 min | DP1 DP2 | 1.98 6.48 | 94.73 | 3.50 | 999.95 | |
| Thermal | 60 °C for 2 h | - | - | 97.51 | - | 999.96 |
| 80 °C for 1 h | - | - | 97.86 | - | 999.95 | |
| Photolytic | UV light 254 nm | DP1 DP2 | 1.98 6.48 | 96.20 | 2.03 | 999.93 |
| Recovery Levels | Amount Added Conc. (µg/mL) | Amount Recovered Conc. (µg/mL) | % Recovery |
|---|---|---|---|
| Level 1 (50%) | 50.8 | 50.5 | 99.4 |
| 50.2 | 49.8 | 99.2 | |
| 50.4 | 50.1 | 99.4 | |
| Level 2 (100%) | 100.3 | 100.1 | 99.8 |
| 100.6 | 99.98 | 99.3 | |
| 100.2 | 100.3 | 100.1 | |
| Level 3 (150%) | 150.2 | 149.6 | 99.4 |
| 150.8 | 150.2 | 99.6 | |
| 151.6 | 151.4 | 99.8 | |
| Mean | 99.55 | ||
| ±SD | 0.29 | ||
| %RSD | 0.29 |
| System Suitability | Intraday Precision (Set I) | Interday Precision (Set II) | |||
|---|---|---|---|---|---|
| Injection No. | Area Count (mAU) | Sample No. | Assay (% Label Claim) | Sample No. | Assay (% Label Claim) |
| 1 | 2416.372 | 1 | 97.63 | 1 | 97.96 |
| 2 | 2414.034 | 2 | 97.22 | 2 | 97.56 |
| 3 | 2428.586 | 3 | 96.60 | 3 | 97.16 |
| 4 | 2412.247 | 4 | 98.29 | 4 | 98.57 |
| 5 | 2411.316 | 5 | 98.78 | 5 | 98.78 |
| 6 | 98.42 | 6 | 96.34 | ||
| Mean | 2416.511 | Mean | 97.66 | Mean | 97.73 |
| ±SD | 7.02 | ±SD | 0.78 | ±SD | 0.91 |
| %RSD | 0.29 | %RSD | 0.79 | %RSD | 0.93 |
| Injection | 5 (µg/mL) | 2.5 (µg/mL) | 1.25 (µg/mL) | 0.625 (µg/mL) | 0.31 (µg/mL) | 0.15 (µg/mL) | 0.07 (µg/mL) |
|---|---|---|---|---|---|---|---|
| Area Count | |||||||
| 1 | 133.59 | 62.60 | 37.75 | 20.29 | 11.23 | 7.80 | ND |
| 2 | 132.42 | 65.49 | 34.55 | 20.97 | 11.26 | 7.51 | ND |
| 3 | 131.44 | 67.15 | 32.53 | 20.23 | 11.20 | 7.51 | ND |
| Mean | 132.48 | 65.15 | 34.94 | 20.50 | 11.23 | 7.61 | - |
| ±SD | 1.07 | 2.30 | 2.63 | 0.41 | 0.03 | 0.17 | - |
| %RSD | 0.81 | 3.54 | 7.53 | 2.02 | 0.26 | 2.23 | - |
| Method Parameters | Standard Solution | Assay (% Claim) | ||
|---|---|---|---|---|
| Theoretical Plate | Tailing Factor | %RSD | ||
| As such (Method precision) | 3437.76 | 1.31 | 0.30 | 97.66 |
| Variation in wavelength (320 nm) | 3439.83 | 1.30 | 0.34 | 97.45 |
| Variation in wavelength (324 nm) | 3438.75 | 1.30 | 0.32 | 97.56 |
| Variation in column oven temperature (20 °C) | 3436.56 | 1.31 | 0.34 | 97.53 |
| Variation in column oven temperature (30 °C) | 3437.65 | 1.34 | 0.32 | 97.63 |
| Variation in minor component in mobile phase (−5% of methanol) | 3439.83 | 1.32 | 0.30 | 97.87 |
| Variation in minor component in mobile phase (+5% of methanol) | 3537.78 | 1.30 | 0.36 | 98.02 |
| Variation in flow rate (0.8 mL/min) | 3683.70 | 1.28 | 0.32 | 97.43 |
| Variation in flow rate (1.2 mL/min) | 3627.70 | 1.34 | 0.35 | 96.45 |
| Variation in pH of Buffer solution (pH 4.3) | 3438.78 | 1.35 | 0.33 | 97.56 |
| Variation in pH of Buffer solution (pH 4.7) | 3435.43 | 1.32 | 0.36 | 97.45 |
| Time (Hrs.) | Standard Solution | Sample Solution | ||
|---|---|---|---|---|
| Area Count | % Deviation | Area Count | % Deviation | |
| Initial | 2416.71 | 0.0 | 2370.65 | 0.0 |
| 4 | 2412.78 | 0.1 | 2360.90 | 0.4 |
| 10 | 2425.56 | 0.3 | 2345.92 | 1.0 |
| 15 | 2414.25 | 0.2 | 2386.86 | 0.6 |
| 24 | 2428.51 | 0.4 | 2365.03 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, R.; Dube, A.; Solanki, R.; Khunt, D.; Parikh, S.; Junnuthula, V.; Dyawanapelly, S. Structural Elucidation of Alkali Degradation Impurities of Favipiravir from the Oral Suspension: UPLC-TQ-ESI-MS/MS and NMR. Molecules 2022, 27, 5606. https://doi.org/10.3390/molecules27175606
Patel R, Dube A, Solanki R, Khunt D, Parikh S, Junnuthula V, Dyawanapelly S. Structural Elucidation of Alkali Degradation Impurities of Favipiravir from the Oral Suspension: UPLC-TQ-ESI-MS/MS and NMR. Molecules. 2022; 27(17):5606. https://doi.org/10.3390/molecules27175606
Chicago/Turabian StylePatel, Ravi, Abhishek Dube, Ravisinh Solanki, Dignesh Khunt, Shalin Parikh, Vijayabhaskarreddy Junnuthula, and Sathish Dyawanapelly. 2022. "Structural Elucidation of Alkali Degradation Impurities of Favipiravir from the Oral Suspension: UPLC-TQ-ESI-MS/MS and NMR" Molecules 27, no. 17: 5606. https://doi.org/10.3390/molecules27175606