
A QCT View of the Interplay between Hydrogen Bonds and Aromaticity in Small CHON Derivatives

 , , , and
, , , and
Abstract
: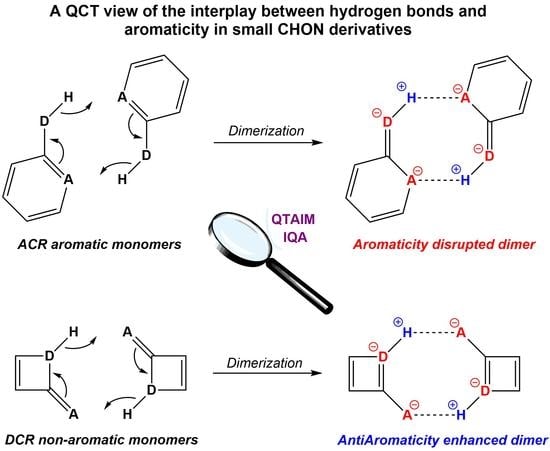
1. Introduction
2. Theoretical Framework
2.1. Real Space Wavefunction Analyses
2.2. Aromaticity
3. Computational Details
4. Results and Discussion
4.1. General Energetic Changes Induced by HB Formation
4.2. Quantum Chemical Topology Analyses
4.3. Perturbation of the Aromaticity of the π Skeleton
4.4. The Peculiar Case of the AZH (DCR) Dimer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Moore, T.S.; Winmill, T.F. CLXXVII—The state of amines in aqueous solution. J. Chem. Soc. Trans. 1912, 101, 1635–1676. [Google Scholar] [CrossRef]
- Luo, X.Y.; Fan, X.; Shi, G.L.; Li, H.R.; Wang, C.M. Decreasing the Viscosity in CO2 Capture by Amino-Functionalized Ionic Liquids through the Formation of Intramolecular Hydrogen Bond. J. Phys. Chem. B 2016, 120, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Cantu, D.C.; Koech, P.K.; Heldebrant, D.J.; Karkamkar, A.; Zheng, F.; Bearden, M.D.; Rousseau, R.; Glezakou, V.A. Directed Hydrogen Bond Placement: Low Viscosity Amine Solvents for CO2 Capture. ACS Sustain. Chem. Eng. 2019, 7, 7535–7542. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of Hydrogen Bond Donors and Acceptors on CO2 Absorption by Deep Eutectic Solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Saeed, U.; Khan, A.L.; Gilani, M.A.; Bilad, M.R.; Khan, A.U. Supported liquid membranes comprising of choline chloride based deep eutectic solvents for CO2 capture: Influence of organic acids as hydrogen bond donor. J. Mol. Liq. 2021, 335, 116155. [Google Scholar] [CrossRef]
- Cao, L.; Li, D.; Soto, F.A.; Ponce, V.; Zhang, B.; Ma, L.; Deng, T.; Seminario, J.M.; Hu, E.; Yang, X.Q.; et al. Highly Reversible Aqueous Zinc Batteries enabled by Zincophilic–Zincophobic Interfacial Layers and Interrupted Hydrogen-Bond Electrolytes. Angew. Chem. Int. Ed. 2021, 60, 18845–18851. [Google Scholar] [CrossRef]
- Sun, T.; Zheng, S.; Nian, Q.; Tao, Z. Hydrogen Bond Shielding Effect for High-Performance Aqueous Zinc Ion Batteries. Small 2022, 18, 2107115. [Google Scholar] [CrossRef]
- Xiong, Q.; Li, C.; Li, Z.; Liang, Y.; Li, J.; Yan, J.; Huang, G.; Zhang, X. Hydrogen-Bond-Assisted Solution Discharge in Aprotic Li–O2 Batteries. Adv. Mater. 2022, 34, 2110416. [Google Scholar] [CrossRef]
- Lu, X.; Cao, L.; Du, X.; Lin, H.; Zheng, C.; Chen, Z.; Sun, B.; Tao, S. Hydrogen-Bond-Induced High Performance Semitransparent Ternary Organic Solar Cells with 14% Efficiency and Enhanced Stability. Adv. Opt. Mater. 2021, 9, 2100064. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Lu, X.; Cao, L.; Du, X.; Lin, H.; Zheng, C.; Tao, S. Hydrogen bond induced high-performance quaternary organic solar cells with efficiency up to 17.48% and superior thermal stability. Mater. Chem. Front. 2021, 5, 3850–3858. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, J.; Urriolabeitia, A.; Polo, V.; Fernández-Buenestado, M.; Iglesias, M.; Fernández-Alvarez, F.J. Dehydrogenation of formic acid using iridium-NSi species as catalyst precursors. Dalton Trans. 2022, 51, 4386–4393. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cruzan, J.D.; Saykally, R.J. Water Clusters. Science 1996, 271, 929–933. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Chávez-Calvillo, R.; García-Revilla, M.; Hernández-Trujillo, J.; Christiansen, O.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Hydrogen-Bond Cooperative Effects in Small Cyclic Water Clusters as Revealed by the Interacting Quantum Atoms Approach. Chem. Eur. J. 2013, 19, 14304–14315. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Gómez, V.A.M.; Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Hydrogen bond cooperativity and anticooperativity within the water hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566. [Google Scholar] [CrossRef]
- Castor-Villegas, V.M.; Guevara-Vela, J.M.; Narváez, W.E.V.; Martín Pendás, Á.; Rocha-Rinza, T.; Fernández-Alarcón, A. On the strength of hydrogen bonding within water clusters on the coordination limit. J. Comput. Chem. 2020, 41, 2266–2277. [Google Scholar] [CrossRef]
- Dalrymple, S.A.; Shimizu, G.K.H. Crystal Engineering of a Permanently Porous Network Sustained Exclusively by Charge-Assisted Hydrogen Bonds. J. Am. Chem. Soc. 2007, 129, 12114–12116. [Google Scholar] [CrossRef]
- Wang, H.; Gurau, G.; Shamshina, J.; Cojocaru, O.A.; Janikowski, J.; MacFarlane, D.R.; Davis, J.H.; Rogers, R.D. Simultaneous membrane transport of two active pharmaceutical ingredients by charge assisted hydrogen bond complex formation. Chem. Sci. 2014, 5, 3449. [Google Scholar] [CrossRef]
- Pedzisa, L.; Hay, B.P. Aliphatic C-H Anion Hydrogen Bonds: Weak Contacts or Strong Interactions? J. Org. Chem. 2009, 74, 2554–2560. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef] [PubMed]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. Cooperative and anticooperative effects in resonance assisted hydrogen bonds in merged structures of malondialdehyde. Phys. Chem. Chem. Phys. 2017, 19, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Gallegos, M.; Valentín-Rodríguez, M.A.; Costales, A.; Rocha-Rinza, T.; Martín Pendás, Á. On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules 2021, 26, 4196. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; del Río-Lima, A.; Martín Pendás, Á.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Hydrogen-Bond Weakening through π Systems: Resonance-Impaired Hydrogen Bonds (RIHB). Chem. Eur. J. 2017, 23, 16605–16611. [Google Scholar] [CrossRef]
- Nowroozi, A.; Sarhadinia, S.; Masumian, E.; Nakhaei, E. A comprehensive theoretical study of tautomeric and conformeric preferences, intramolecular hydrogen bonding, and π-electron delocalisation in β-selenoaminoacrolein with its thio and oxo analogs. Struc. Chem. 2014, 25, 1359–1368. [Google Scholar] [CrossRef]
- Masumian, E.; Nowroozi, A. Computational investigation on the intramolecular resonance-inhibited hydrogen bonding: A new type of interaction versus the RAHB model. Theor. Chem. Acc. 2015, 134. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the .beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1, 3-diaryl-1, 3-propanedione enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. [Google Scholar] [CrossRef]
- Bertolasi, V.; Pretto, L.; Gilli, G.; Gilli, P. π-Bond cooperativity and anticooperativity effects in resonance-assisted hydrogen bonds (RAHBs). Acta Crystallogr. B Struct. Sci. 2006, 62, 850–863. [Google Scholar] [CrossRef]
- Wu, J.I.; Jackson, J.E.; von Ragué Schleyer, P. Reciprocal Hydrogen Bonding–Aromaticity Relationships. J. Am. Chem. Soc. 2014, 136, 13526–13529. [Google Scholar] [CrossRef]
- Kakeshpour, T.; Wu, J.I.; Jackson, J.E. AMHB: (Anti)aromaticity-Modulated Hydrogen Bonding. J. Am. Chem. Soc. 2016, 138, 3427–3432. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J.; Evans, C.; Lentz, D.; Roemer, M. The QTAIM Approach to Chemical Bonding Between Transition Metals and Carbocyclic Rings: A Combined Experimental and Theoretical Study of (η5-C5H5)Mn(CO)3, (η6-C6H6)Cr(CO)3, and (E)-(η5-C5H4)CF=CF(η5-C5H4)(η5-C5H5)2Fe. J. Am. Chem. Soc. 2008, 131, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.; Mosquera, R.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Driving Forces in the Sharpless Epoxidation Reaction: A Coupled AIMD/QTAIM Study. Inorg. Chem. 2017, 56, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Hooper, T.N.; Garçon, M.; White, A.J.P.; Crimmin, M.R. Room temperature catalytic carbon–hydrogen bond alumination of unactivated arenes: Mechanism and selectivity. Chem. Sci. 2018, 9, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Escofet, I.; Armengol-Relats, H.; Bruss, H.; Besora, M.; Echavarren, A.M. On the Structure of Intermediates in Enyne Gold(I)-Catalyzed Cyclizations: Formation of trans-Fused Bicyclo[5.1.0]octanes as a Case Study. Chem. Eur. J. 2020, 26, 15738–15745. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Guevara-Vela, J.M.; Crespo, D.M.; Costales, A.; Francisco, E. An unexpected bridge between chemical bonding indicators and electrical conductivity through the localization tensor. Phys. Chem. Chem. Phys. 2017, 19, 1790–1797. [Google Scholar] [CrossRef]
- Astakhov, A.A.; Tsirelson, V.G. Spatially resolved characterisation of electron localization and delocalisation in molecules: Extending the Kohn-Resta approach. Int. J. Quantum Chem. 2018, 118, e25600. [Google Scholar] [CrossRef]
- Gil-Guerrero, S.; Ramos-Berdullas, N.; Martín Pendás, Á.; Francisco, E.; Mandado, M. Anti-ohmic single molecule electron transport: Is it feasible? Nanoscale Adv. 2019, 1, 1901–1913. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M.; Salvador, P.; Duran, M. Electron sharing indexes at the correlated level. Application to aromaticity calculations. Faraday Discuss. 2007, 135, 325–345. [Google Scholar] [CrossRef] [Green Version]
- Casademont-Reig, I.; Guerrero-Avilés, R.; Ramos-Cordoba, E.; Torrent-Sucarrat, M.; Matito, E. How Aromatic Are Molecular Nanorings? The Case of a Six-Porphyrin Nanoring. Angew. Chem. Int. Ed. 2021, 60, 24080–24088. [Google Scholar] [CrossRef] [PubMed]
- del Rosario Merino-García, M.; Soriano-Agueda, L.A.; de Dios Guzmán-Hernández, J.; Martínez-Otero, D.; Rivera, B.L.; Cortés-Guzmán, F.; Barquera-Lozada, J.E.; Jancik, V. Benzene and Borazine, so Different, yet so Similar: Insight from Experimental Charge Density Analysis. Inorg. Chem. 2022, 61, 6785–6798. [Google Scholar] [CrossRef] [PubMed]
- Francisco, E.; Martín Pendás, A.; Blanco, M.A. A Molecular Energy Decomposition Scheme for Atoms in Molecules. J. Chem. Theory Comput. 2005, 2, 90–102. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Solà, M.; Viglione, R.G.; Zanasi, R. Local Aromaticity of the Six-Membered Rings in Pyracylene. A Difficult Case for the NICS Indicator of Aromaticity. J. Org. Chem. 2004, 69, 7537–7542. [Google Scholar] [CrossRef]
- Aihara, J.-i. Incorrect NICS-Based Prediction on the Aromaticity of the Pentalene Dication. Bull. Chem. Soc. Jpn 2004, 77, 101–102. [Google Scholar] [CrossRef]
- Faglioni, F.; Ligabue, A.; Pelloni, S.; Soncini, A.; Viglione, R.G.; Ferraro, M.B.; Zanasi, R.; Lazzeretti, P. Why Downfield Proton Chemical Shifts Are Not Reliable Aromaticity Indicators. Org. Lett. 2005, 7, 3457–3460. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Manoharan, M.; Jiao, H.; Stahl, F. The Acenes: Is There a Relationship between Aromatic Stabilization and Reactivity? Org. Lett. 2001, 3, 3643–3646. [Google Scholar] [CrossRef]
- Kruszewski, J.; Krygowski, T. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G. Aromaticity in Metallabenzenes. Chem. Eur. J. 2007, 13, 5873–5884. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Fradera, X.; Duran, M.; Solà, M. The delocalisation Index as an Electronic Aromaticity Criterion: Application to a Series of Planar Polycyclic Aromatic Hydrocarbons. Chem. Eur. J. 2003, 9, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Matito, E.; Duran, M.; Solà, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalisation. J. Chem. Phys. 2005, 122, 014109. [Google Scholar] [CrossRef] [PubMed]
- Bultinck, P.; Ponec, R.; Damme, S.V. Multicenter bond indices as a new measure of aromaticity in polycyclic aromatic hydrocarbons. J. Phys. Org. Chem. 2005, 18, 706–718. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. On the performance of some aromaticity indices: A critical assessment using a test set. J. Comput. Chem. 2008, 29, 1543–1554. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Keith, A.T. TK Gristmill Software; AIMALL (Version 19.02.13); AIMALL: Overland Park, KS, USA, 2019. [Google Scholar]
- Martín Pendás, A.; Francisco, E. Promolden. A QTAIM/IQA code (Avaliable from the authors upon request).
- Maxwell, P.; Martín Pendás, Á.; Popelier, P.L.A. Extension of the interacting quantum atoms (IQA) approach to B3LYP level density functional theory (DFT). Phys. Chem. Chem. Phys. 2016, 18, 20986–21000. [Google Scholar] [CrossRef] [PubMed]
- Matito, E. ESI-3D: Electron Sharing Indexes Program for 3D Molecular Space Partitioning. 2020. [Google Scholar]
- Feixas, F.; Matito, E.; Solà, M.; Poater, J. Patterns of π-electron delocalization in aromatic and antiaromatic organic compounds in the light of Hückel’s 4n + 2 rule. Phys. Chem. Chem. Phys. 2010, 12, 7126–7137. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | System | ||||
|---|---|---|---|---|---|
| 2HP (ACR) | 5.06 | 3.01 | 2HP (DCR) | −7.11 | −7.03 |
| NCO (ACR) | 0.00 | 0.00 | NCO (DCR) | 0.00 | 0.00 |
| AZH (ACR) | −2.82 | −3.34 | AZH (DCR) | 6.40 | 5.38 |
| 2AP (ACR) | 3.22 | 2.58 | 2AP (DCR) | −8.16 | −7.50 |
| NCN (ACR) | 0.00 | 0.00 | NCN (DCR) | 0.00 | 0.00 |
| AZA (ACR) | −10.05 | −9.58 | AZA (DCR) | 2.13 | 0.89 |
| ACR | ||||
| System | (D) | (H) | (A) | (C) |
| 2HP | −0.087 | +0.049 | −0.067 | +0.069 |
| NCO | −0.085 | +0.041 | −0.072 | +0.085 |
| AZH | −0.089 | +0.065 | −0.077 | +0.045 |
| 2AP | −0.040 | +0.070 | −0.021 | +0.019 |
| NCN | −0.051 | +0.088 | −0.024 | +0.008 |
| AZA | −0.061 | +0.113 | −0.063 | −0.002 |
| DCR | ||||
| System | (D) | (H) | (A) | (C) |
| 2HP | −0.070 | +0.105 | −0.024 | −0.040 |
| NCO | −0.052 | +0.085 | −0.025 | −0.007 |
| AZH | −0.037 | +0.064 | −0.024 | −0.011 |
| 2AP | −0.063 | +0.101 | −0.031 | −0.012 |
| NCN | −0.051 | +0.088 | −0.024 | +0.008 |
| AZA | −0.030 | +0.074 | −0.034 | −0.007 |
| ACR | ||||
| System | DI(D–H) | DI(D–C) | DI(C–A) | DI(H–A) |
| 2HP | −0.207 | +0.056 | −0.064 | +0.152 |
| NCO | −0.246 | +0.100 | −0.171 | +0.197 |
| AZH | −0.216 | +0.079 | −0.136 | +0.134 |
| 2AP | −0.151 | +0.051 | −0.034 | +0.103 |
| NCN | −0.181 | +0.073 | −0.076 | +0.117 |
| AZA | −0.262 | +0.131 | −0.167 | +0.165 |
| DCR | ||||
| System | DI(D–H) | DI(D–C) | DI(C–A) | DI(H–A) |
| 2HP | −0.209 | +0.065 | −0.091 | +0.132 |
| NCO | −0.162 | +0.069 | −0.070 | +0.099 |
| AZH | −0.103 | +0.034 | −0.043 | +0.066 |
| 2AP | −0.234 | +0.067 | −0.106 | +0.163 |
| NCN | −0.181 | +0.073 | −0.076 | +0.117 |
| AZA | −0.152 | +0.043 | −0.053 | +0.099 |
| System | MCI (a.u.) | FLU (a.u.) | System | MCI (a.u.) | FLU (a.u.) | ||
|---|---|---|---|---|---|---|---|
| 2HP (ACR) | −0.008 | +0.001 | − | 2HP (DCR) | +0.007 | −0.010 | + |
| AZH (ACR) | +0.001 | −0.017 | + | AZH (DCR) | −0.001 | −0.005 | − |
| 2AP (ACR) | −0.004 | +0.001 | − | 2AP (DCR) | +0.007 | −0.010 | + |
| AZA (ACR) | +0.004 | −0.021 | + | AZA (DCR) | −0.001 | −0.007 | − |
| Isomer | (kcal/mol) | FLU (a.u.) | MCI (a.u.) |
|---|---|---|---|
| bent–trans | +0.48 | +0.0006 | −0.0001 |
| bent–cis | 0.00 | 0.0000 | 0.0000 |
| planar | +7.38 | +0.0094 | −0.0038 |
| QTAIM charges | ||||
| System | Q(D) | (H) | (A) | (C) |
| bent–trans | −0.039 | +0.063 | −0.024 | −0.007 |
| bent–cis | −0.037 | +0.064 | −0.024 | −0.011 |
| planar | −0.185 | +0.127 | −0.030 | +0.065 |
| Delocalisation indices | ||||
| System | DI(D–H) | DI(D–C) | DI(C–A) | DI(H–A) |
| bent–trans | −0.099 | +0.035 | −0.043 | +0.064 |
| bent–cis | −0.103 | +0.034 | −0.043 | +0.066 |
| planar | −0.169 | +0.070 | −0.068 | +0.074 |
| IQA energy partition | ||||
| System | ||||
| bent–trans | −12.80 | −21.21 | +11.99 | −87.41 |
| bent–cis | −12.87 | −18.43 | +13.47 | −88.66 |
| planar | −40.98 | −121.17 | −1.07 | −106.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallegos, M.; Barrena-Espés, D.; Guevara-Vela, J.M.; Rocha-Rinza, T.; Pendás, Á.M. A QCT View of the Interplay between Hydrogen Bonds and Aromaticity in Small CHON Derivatives. Molecules 2022, 27, 6039. https://doi.org/10.3390/molecules27186039
Gallegos M, Barrena-Espés D, Guevara-Vela JM, Rocha-Rinza T, Pendás ÁM. A QCT View of the Interplay between Hydrogen Bonds and Aromaticity in Small CHON Derivatives. Molecules. 2022; 27(18):6039. https://doi.org/10.3390/molecules27186039
Chicago/Turabian StyleGallegos, Miguel, Daniel Barrena-Espés, José Manuel Guevara-Vela, Tomás Rocha-Rinza, and Ángel Martín Pendás. 2022. "A QCT View of the Interplay between Hydrogen Bonds and Aromaticity in Small CHON Derivatives" Molecules 27, no. 18: 6039. https://doi.org/10.3390/molecules27186039