
The First 5′-Phosphorylated 1,2,3-Triazolyl Nucleoside Analogues with Uracil and Quinazoline-2,4-Dione Moieties: A Synthesis and Antiviral Evaluation
 , , and
, , and
Abstract
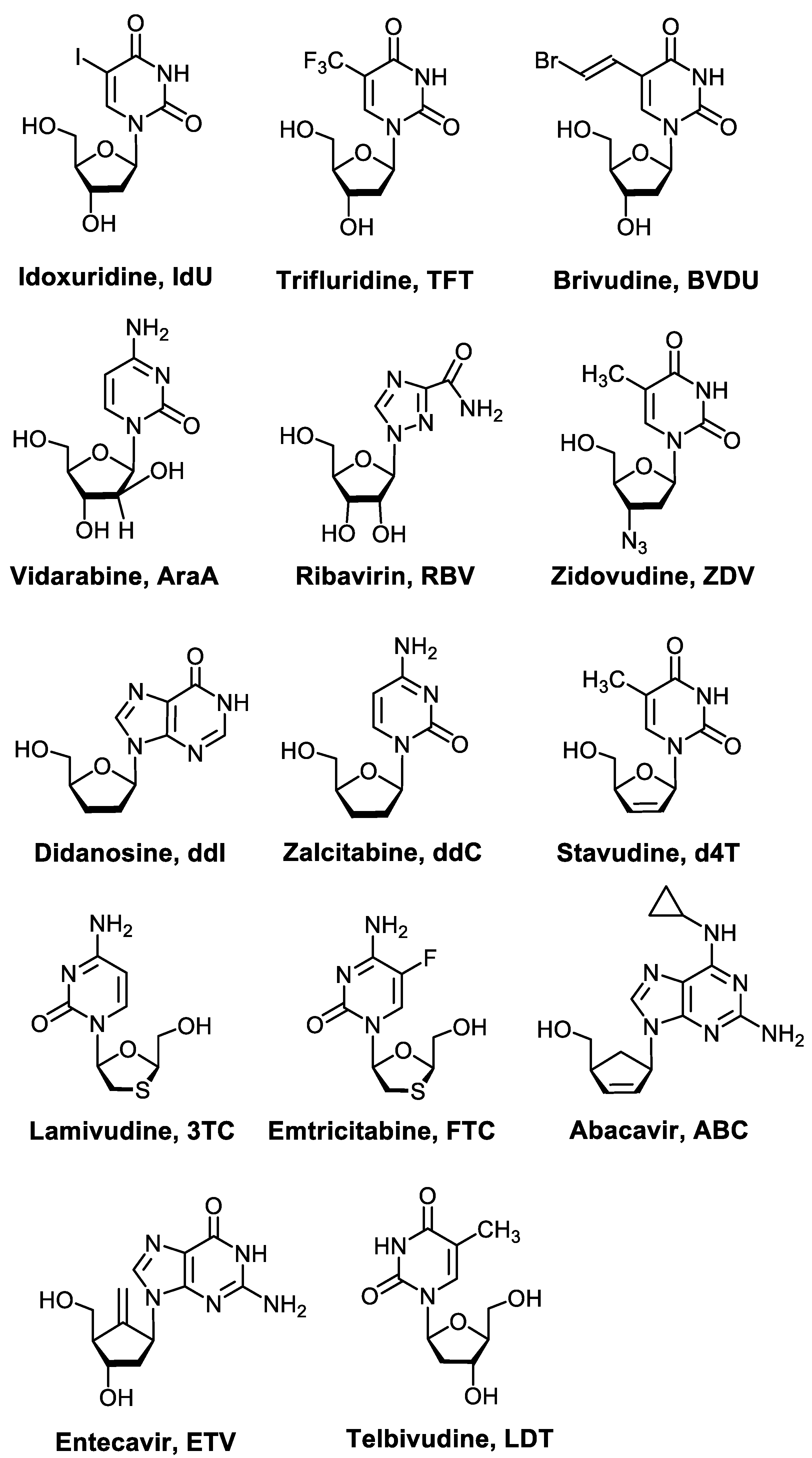
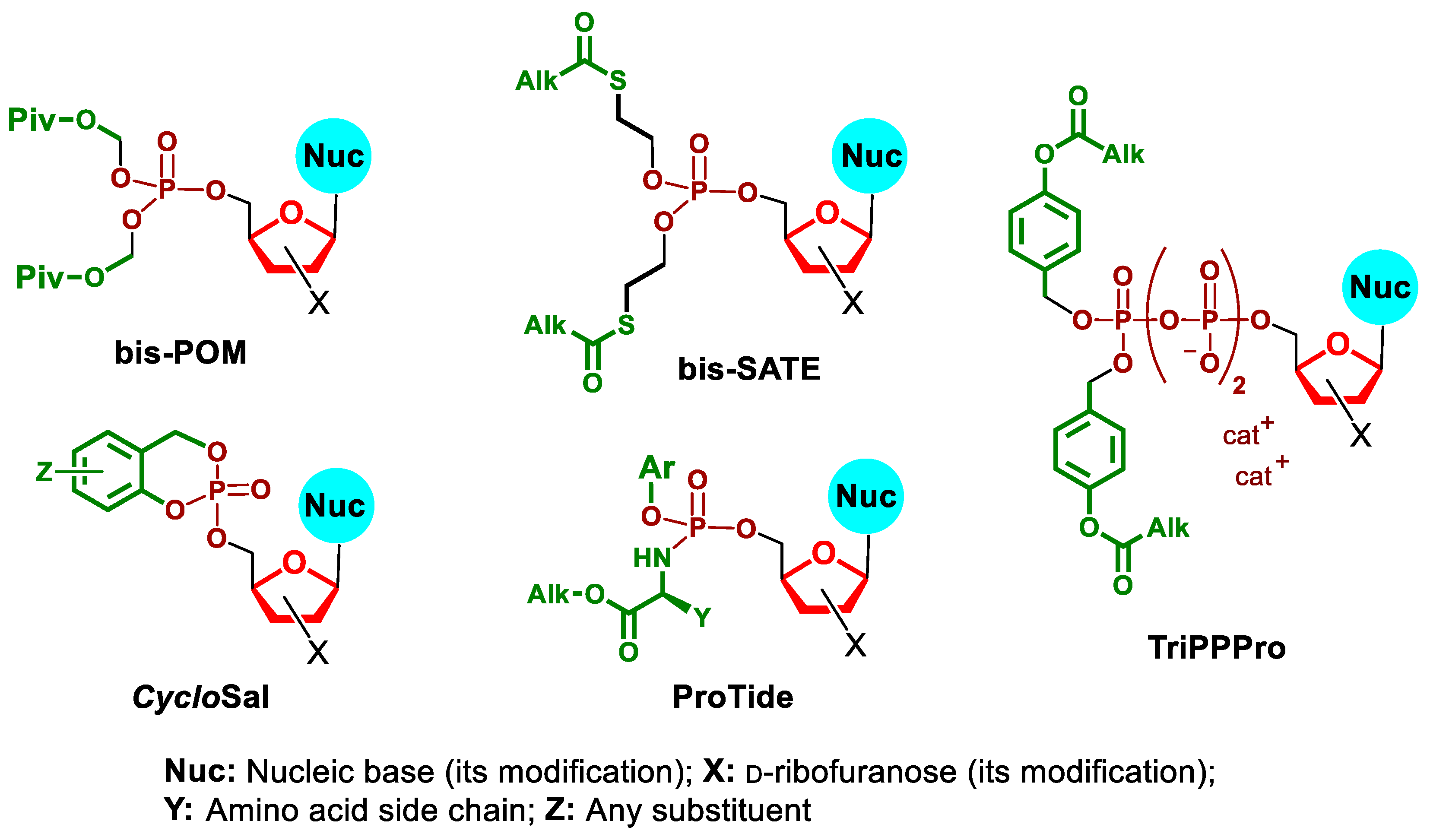
:1. Introduction
2. Results and Discussion
2.1. Chemistry
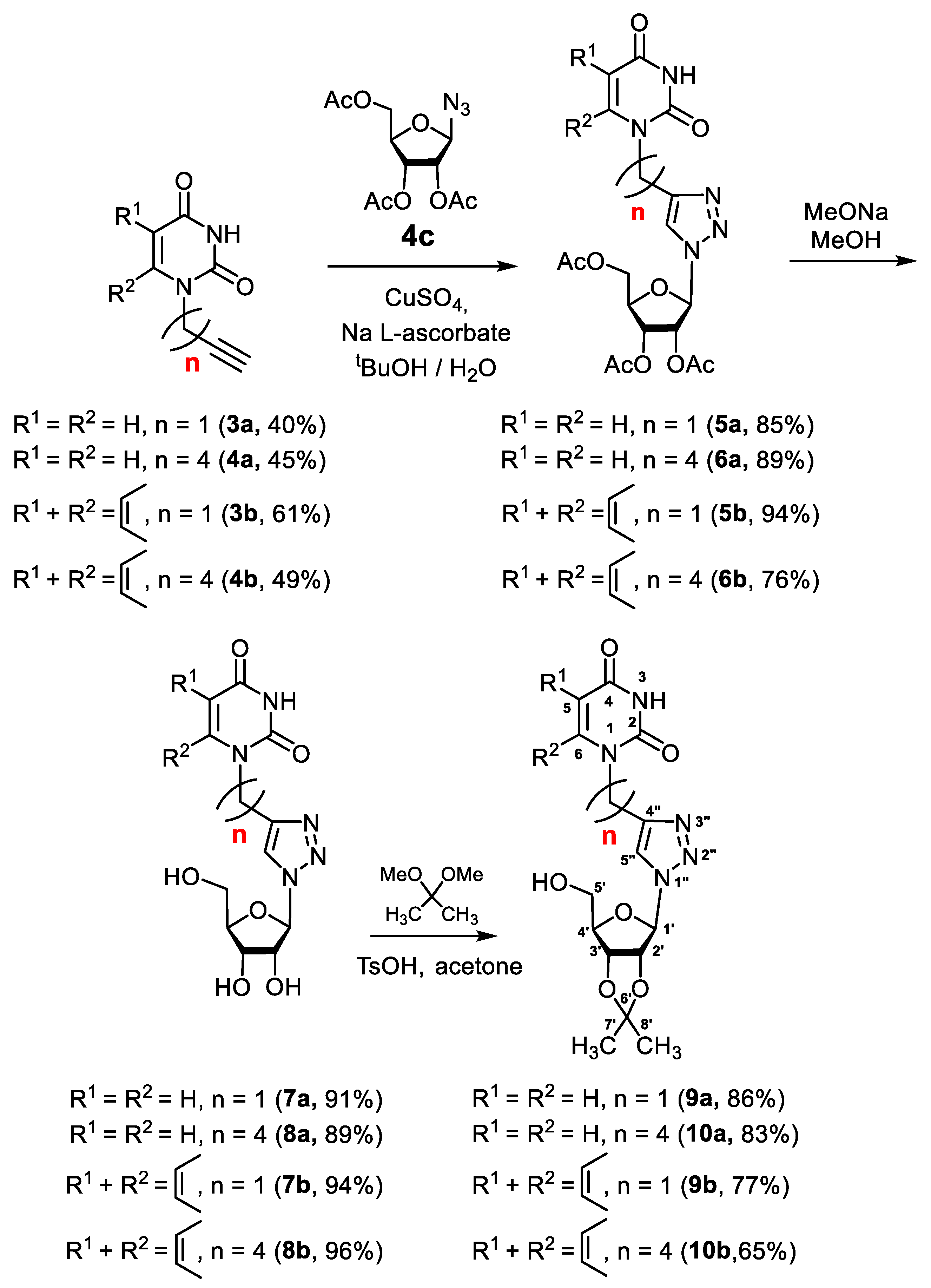
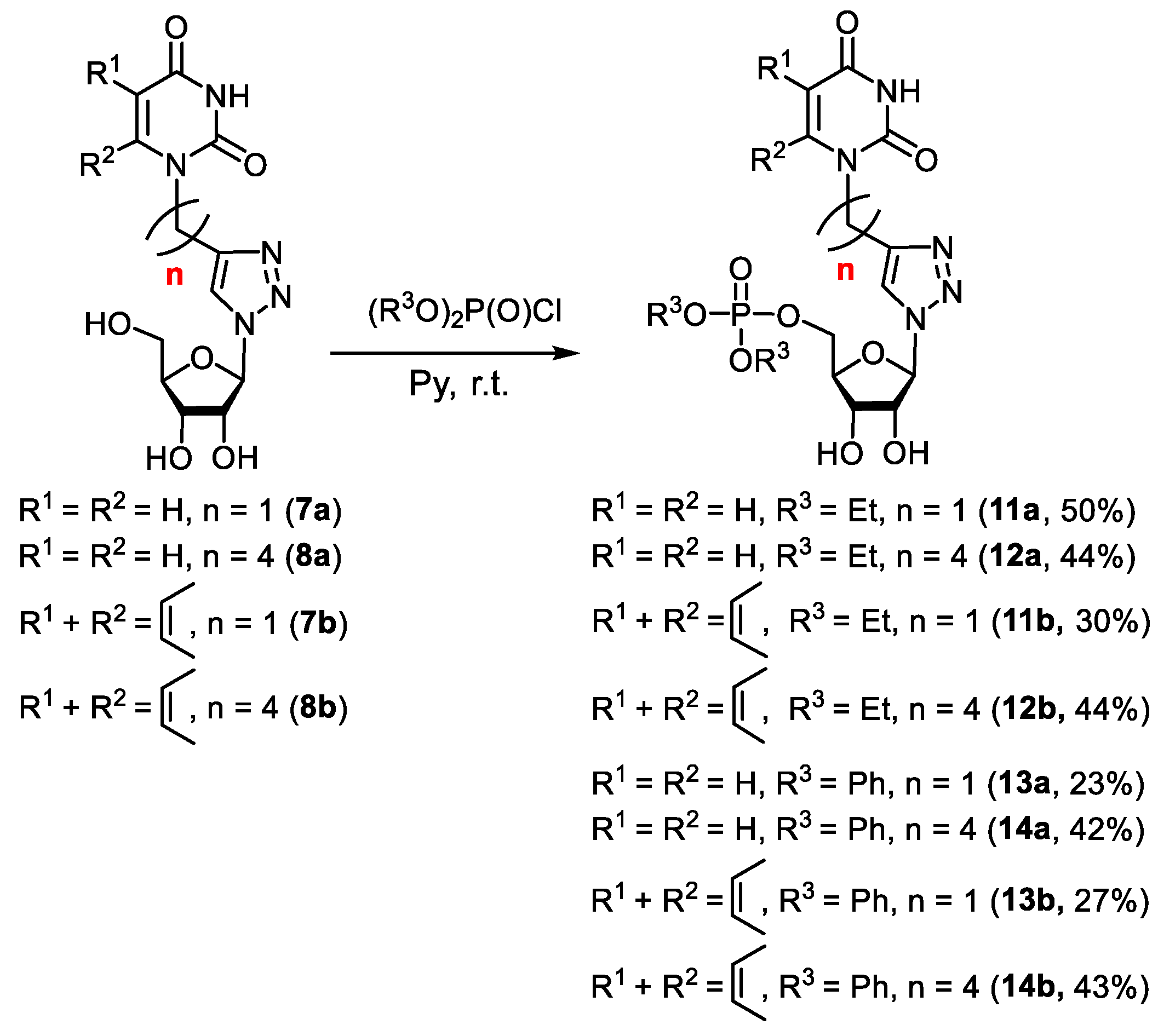
2.1.1. Synthesis of 1,2,3-Triazolyl Nucleoside Analogues
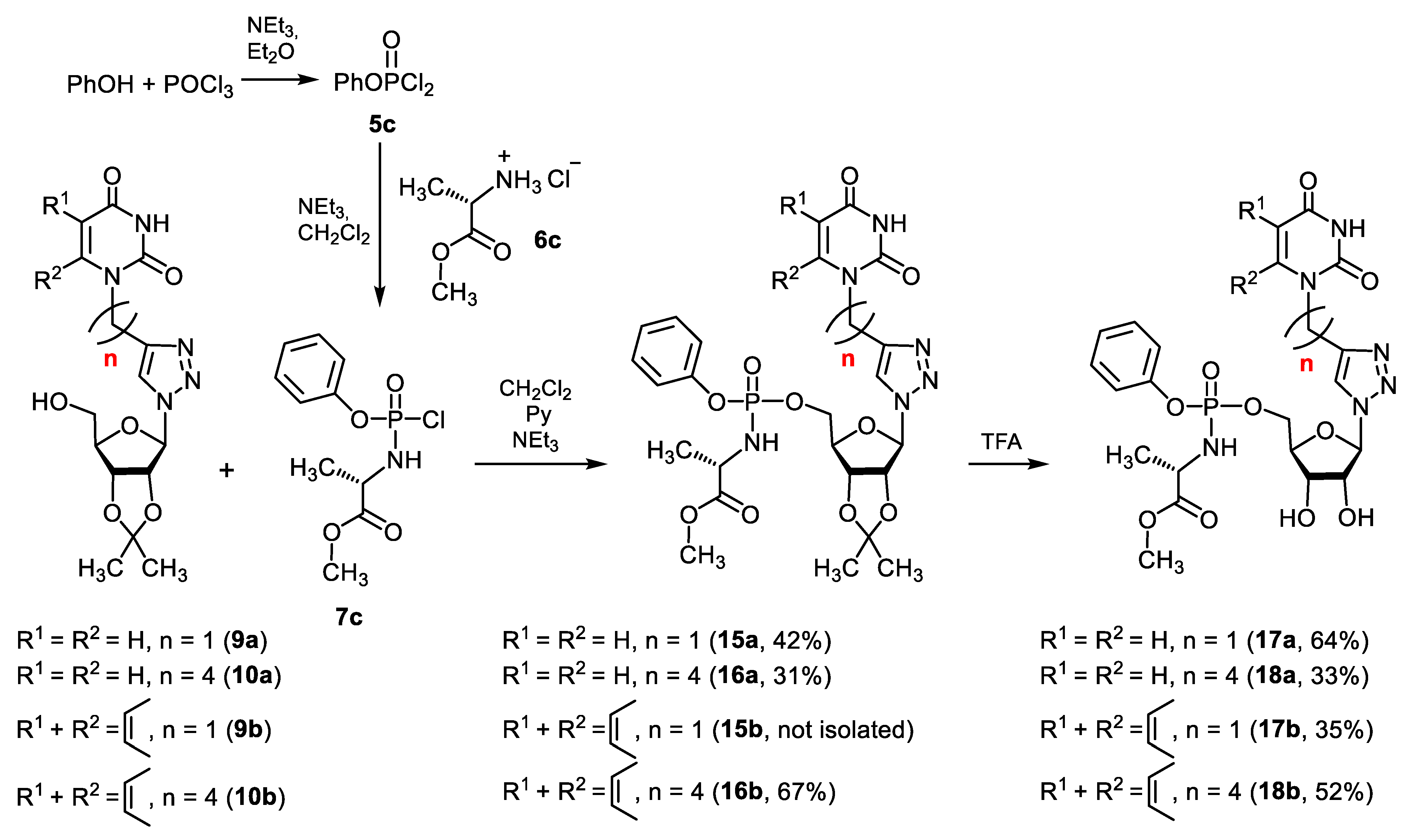
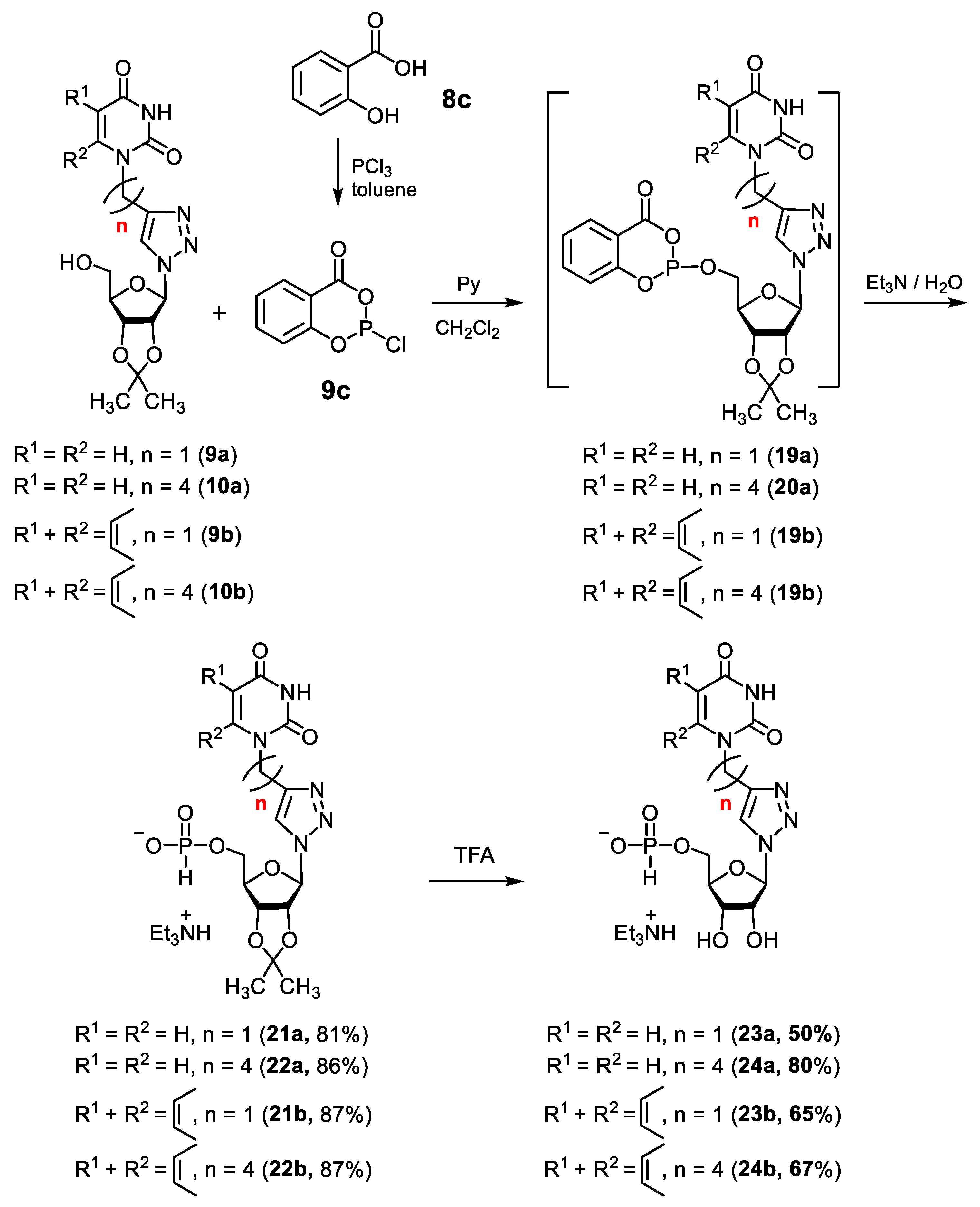
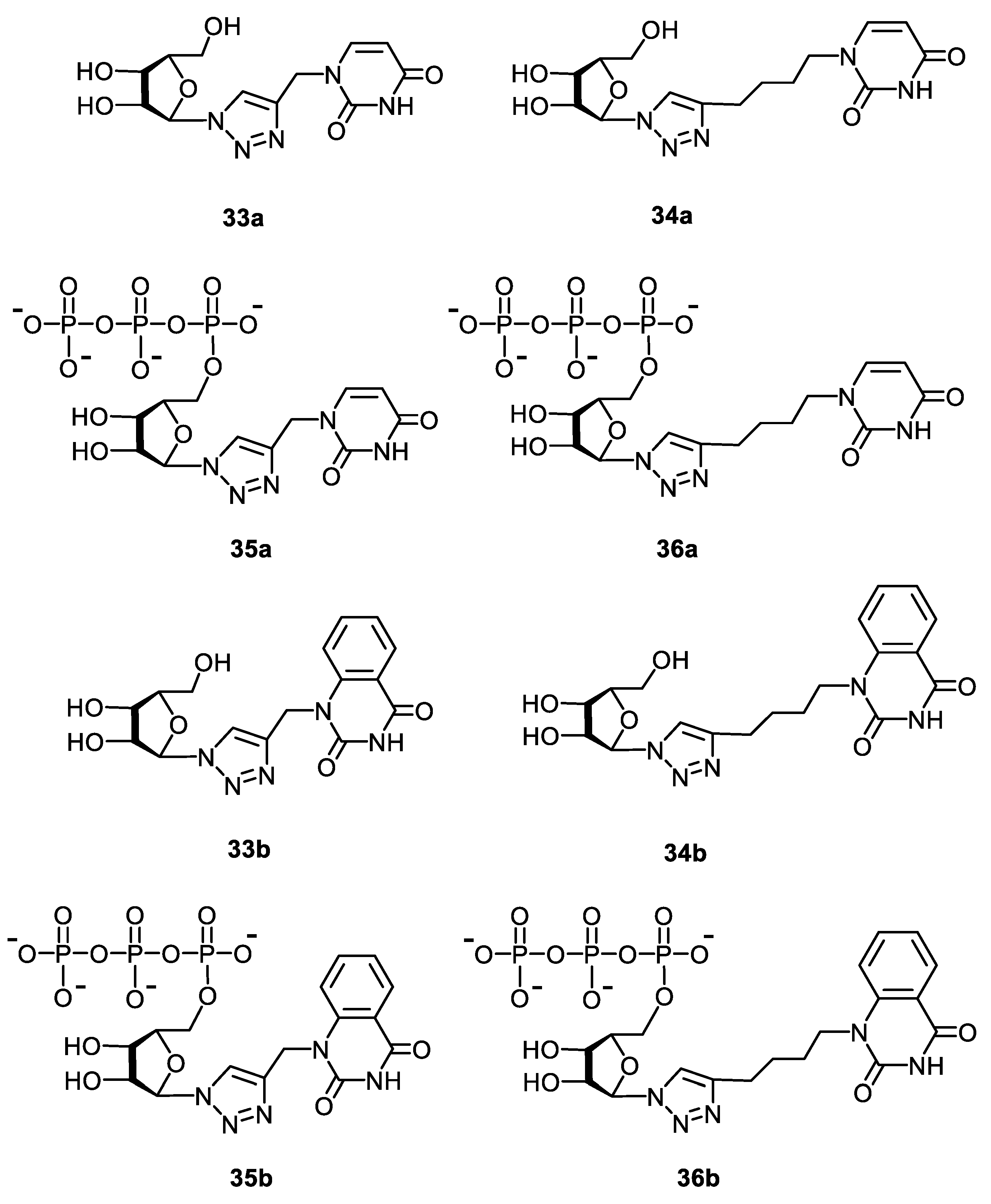
2.1.2. Synthesis of 5′-Phosphorylated Derivatives of 1,2,3-Triazolyl Nucleoside Analogues
2.2. Antiviral Evaluation
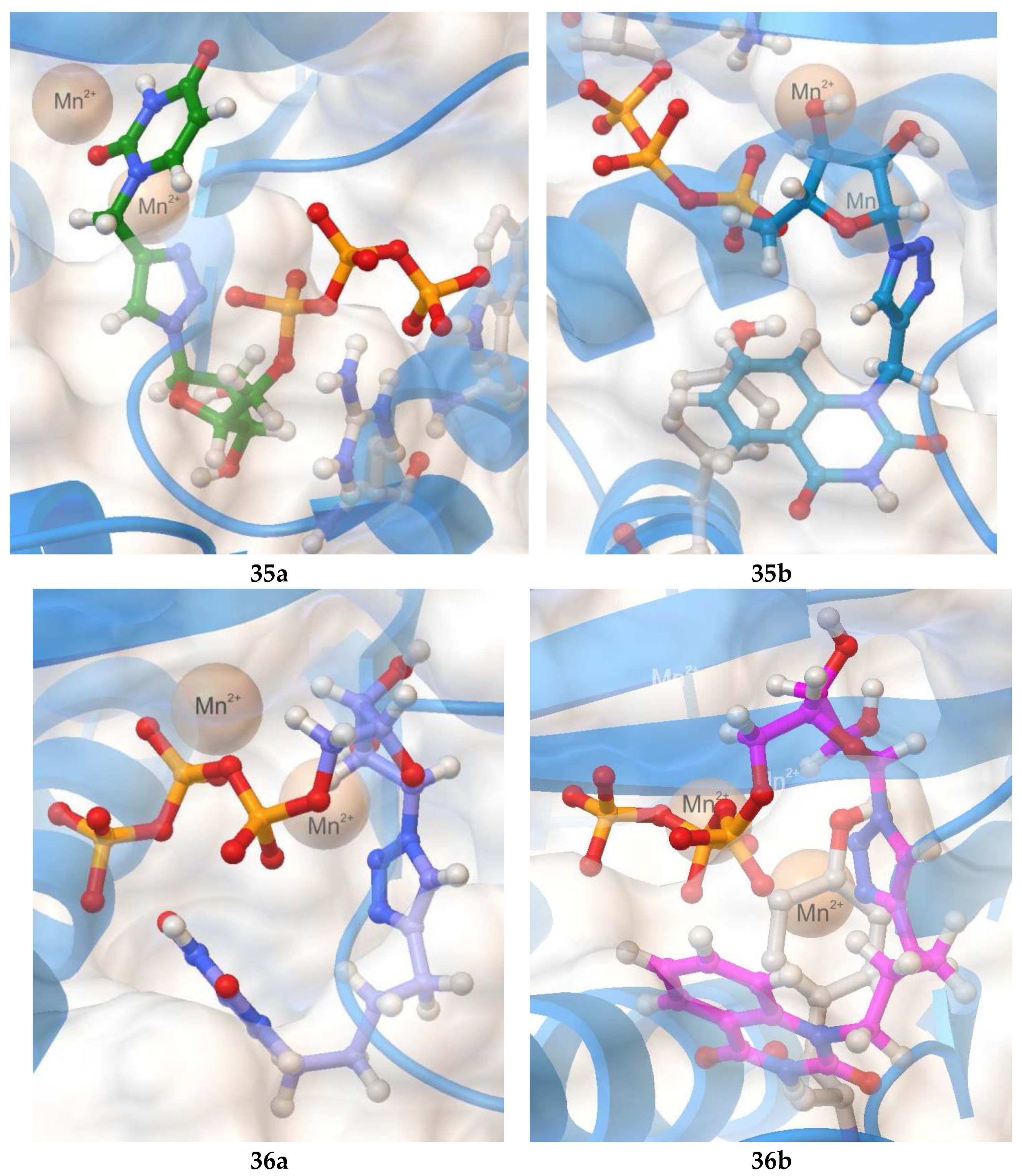
2.3. Molecular Docking
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pastuch-Gawołek, G.; Gillner, D.; Krol, E.; Walczak, K.; Wandzik, I. Selected nucleos(t)ide-based prescribed drugs and their multi-target activity. Eur. J. Pharm. 2019, 865, 172747. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Serpi, M.; Pertusati, F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 2018, 26, 2040206618775243. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef]
- De Clercq, E.; Descamps, J.; De Somer, P.; Barr, P.J.; Jones, A.S.; Walker, R.T. (E)-5-(2-Bromovinyl)-2’-deoxyuridine: A potent and selective anti-herpes agent. Proc. Natl. Acad. Sci. USA 1979, 76, 2947–2951. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Balzarini, J. Metabolism and mechanism of antiretroviral action of purine and pyrimidine derivatives. Pharm. World Sci. 1993, 16, 113–126. [Google Scholar] [CrossRef]
- Eyer, L.; Nencka, R.; De Clercq, E.; Seley-Radtke, K.; Ruzek, D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir. Chem. Chemother. 2018, 26, 2040206618761299. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Martin, J.C.; Hitchcock, M.J.M.; Kaul, S.; Dunkle, L.M.; Sterzycki, R.Z.; Mansuri, M.M.; Fridland, A.; Ghazzouli, I.; Kaul, S.; Dunkle, L.M.; et al. Comparative studies of 2′,3′-didehydro-2′3′-dideoxythymidine (d4T) with other pyrimidine nucleoside analogues. Ann. N. Y. Acad. Sci. 1990, 616, 22–28. [Google Scholar] [CrossRef]
- Gao, W.-Y.; Agbaria, R.; Driscoll, J.S.; Mitsuya, H. Divergent anti-human immunodeficiency virus activity and anabolic phosphorylation of 2′,3′-dideoxynucleoside analogs in resting and activated human cells. J. Biol. Chem. 1994, 269, 12633–12638. [Google Scholar] [CrossRef]
- Stein, D.S.; Moore, K.H. Phosphorylation of nucleoside analog antiretrovirals: A review for clinicians. Pharmacotherapy 2001, 21, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C., II; Widlanski, T.S. Overview of the Synthesis of Nucleoside Phosphates and Polyphosphates. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons: New York, NY, USA, 2003; Volume 15, pp. 1–31. [Google Scholar]
- Roy, R.; Depaix, A.; Périgaud, C.; Peyrottes, S. Recent trends in nucleotide synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.A.; Thomas, H.J.; Schaeffer, H.J. Synthesis of potential anticancer agents. XXVIII. Simple esters of 6-mercaptopurine ribonucleotide. J. Org. Chem. 1961, 26, 1929–1933. [Google Scholar] [CrossRef]
- Stella, V.J. Prodrugs and site-specific drug delivery. J. Med. Chem. 1980, 23, 1275–1285. [Google Scholar] [CrossRef]
- Farquhar, D.; Srivastva, D.N.; Kuttesch, N.J.; Saunders, P.P. Biologically reversible phosphate-protective group. J. Pharm. Sci. 1983, 72, 324–325. [Google Scholar] [CrossRef]
- Valette, G.; Pompon, A.; Girardet, J.-L.; Cappellacci, L.; Franchetti, P.; Grifantini, M.; La Colla, P.; Loi, A.G.; Perigaud, C.; Gosselin, G.; et al. Decomposition pathways and in vitro HIV inhibitory effects of isoddA pronucleotides: Toward a rational approach for intracellular delivery of nucleoside 5′-monophosphates. J. Med. Chem. 1996, 39, 1981–1990. [Google Scholar] [CrossRef]
- Meier, C. Pro-nucleotides—Recent advances in the design of efficient tools for the delivery of biologically active nucleoside monophosphates. Synlett 1998, 3, 233–242. [Google Scholar] [CrossRef]
- Meier, C. 2-Nucleos-5′-yl-4H-1,3,2-benzodioxaphosphinin-2-oxides—A new concept for lipophilic, potential prodrugs of biologically active nucleoside monophosphates. Angew. Chem. Int. Ed. Engl. 1996, 35, 70–72. [Google Scholar] [CrossRef]
- Meier, C.; Knispel, T.; Marquez, V.E.; Siddiqui, M.A.; De Clercq, E.; Balzarini, J. cycloSal-pronucleotides of 2′-Fluoro-ara- and 2′-Fluoro-ribo-2′,3′-dideoxyadenosine as a strategy to bypass a metabolic blockade. J. Med. Chem. 1999, 42, 1615–1624. [Google Scholar] [CrossRef]
- Meier, C.; Knispel, T.; De Clercq, E.; Balzarini, J. cycloSal-pronucleotides of 2′,3′-dideoxyadenosine and 2′,3′-dideoxy-2′,3′-didehydroadenosine: Synthesis and antiviral evaluation of a highly efficient nucleotide delivery system. J. Med. Chem. 1999, 42, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Haller-Meier, F.; De Clercq, E.; Meier, C. Antiviral activity of cyclosaligenyl prodrugs of acyclovir, carbovir and abacavir. Antivir. Chem. Chemother. 2002, 12, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. cycloSal-pronucleotides—Design of chemical trojan horses. Mini Rev. Med. Chem. 2002, 2, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Balzarini, J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antivir. Res. 2006, 71, 282–292. [Google Scholar] [CrossRef]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as Pro-Tides. Mini Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef]
- Yan, L.; Cao, R.; Zhang, H.; Li, Y.; Li, W.; Li, X.; Fan, S.; Li, S.; Zhong, W. Design, synthesis and evaluation of 2′-acetylene-7-deaza-adenosine phosphoamidate derivatives as anti-EV71 and anti-EV-D68 agents. Eur. J. Med. Chem. 2021, 226, 113852. [Google Scholar] [CrossRef]
- McGuigan, C.; Pathirana, R.N.; Mahmood, N.; Hay, A.J. Aryl phosphate derivatives of AZT inhibit HIV replication in cells where the nucleoside is poorly active. Bioorg. Med. Chem. Lett. 1992, 2, 701–704. [Google Scholar] [CrossRef]
- McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A.; et al. Design, synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [Google Scholar] [CrossRef]
- Singh, U.S.; Mulamoottil, V.A.; Chu, C.K. 2′-Fluoro-6′-methylene carbocyclic adenosine and its phosphoramidate prodrug: A novel anti-HBV agent, active against drug-resistant HBVmutants. Med. Res. Rev. 2018, 38, 977–1002. [Google Scholar] [CrossRef]
- Wang, G.; Lim, S.P.; Chen, Y.-L.; Hunziker, J.; Rao, R.; Gu, F.; She, C.C.; Ghafar, N.A.; Xu, H.; Chan, K.; et al. Structure-activity relationship of uridine-based nucleoside phosphoramidate prodrugs for inhibition of dengue virus RNA-dependent RNA polymerase. Bioorg. Med. Chem. Lett. 2018, 28, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The ProTide prodrug technology: From the concept to the clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Carlin, A.F.; Beadle, J.R.; Valiaeva, N.; Zhang, X.-Q.; Clark, A.E.; McMillan, R.E.; Leibel, S.L.; McVicar, R.N.; Xie, J.; et al. Rethinking Remdesivir: Synthesis, antiviral activity, and pharmacokinetics of oral lipid prodrugs. Antimicrob. Agents Chemother. 2021, 65, e01155-21. [Google Scholar] [CrossRef] [PubMed]
- Gollnest, T.; de Oliveira, T.D.; Schols, D.; Balzarini, J.; Meier, C. Lipophilic prodrugs of nucleoside triphosphates as biochemical probes and potential antivirals. Nat. Comm. 2015, 6, 8716–8730. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Schols, D.; Meier, C. Anti-HIV-active nucleoside triphosphate prodrugs. J. Med. Chem. 2020, 63, 6003–6027. [Google Scholar] [CrossRef] [PubMed]
- Gollnest, T.; de Oliveira, T.D.; Rath, A.; Hauber, I.; Schols, D.; Balzarini, J.; Meier, C. Membrane-permeable triphosphate prodrugs of nucleoside analogues. Angew. Chem. 2016, 55, 5255–5258. [Google Scholar] [CrossRef]
- Weising, S.; Sterrenberg, V.; Schols, D.; Meier, C. Synthesis and antiviral evaluation of TriPPPro-AbacavirTP, TriPPPro-CarbovirTP and their 1′,2′-cis-disubstituted analogues. ChemMedChem 2018, 13, 1771–1778. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.; Matyugina, E.; Shirokova, E. Anti-HIV therapy with AZT prodrugs: AZT phosphonate derivatives, current state and prospects. Expert Opin. Drug Metabol. Toxicol. 2010, 6, 701–714. [Google Scholar] [CrossRef]
- Malin, A.A.; Ostrovskii, V.A. Synthesis of thymidine derivatives as potential pharmaceuticals against HIV/AIDS infection. Russ. J. Org. Chem. 2001, 37, 759–780. [Google Scholar] [CrossRef]
- Li, Y.; Mao, S.; Hager, M.W.; Becnel, K.D.; Schinazi, R.F.; Liotta, D.C. Synthesis and evaluation of 2ʹ-substituted cyclobutyl nucleosides and nucleotides as potential anti-HIV agents. Bioorg. Med. Chem. Lett. 2007, 17, 3398–3401. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhou, L.; Zhang, H.; McBrayer, T.R.; Detorio, M.A.; Johns, M.; Bassit, L.; Powdrill, M.H.; Whitaker, T.; Coats, S.J.; et al. Synthesis and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleosides, their phosphoramidate prodrugs and 5′-triphosphates. Bioorg. Med. Chem. Lett. 2011, 21, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Toti, K.S.; Derudas, M.; Pertusati, F.; Sinnaeve, D.; Van den Broeck, F.; Margamuljana, L.; Martins, J.C.; Herdewijn, P.; Balzarini, J.; McGuigan, C.; et al. Synthesis of an apionucleoside family and discovery of a prodrug with anti-HIV activity. J. Org. Chem. 2014, 79, 5097–5112. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, D.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Cano-Soldado, P.; Pastor-Anglada, M. Transporters that translocate nucleosides and structural similar drugs: Structural requirements for substrate recognition. Med. Res. Rev. 2012, 32, 428–457. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, O.V.; Belenok, M.G.; Saifina, L.F.; Shulaeva, M.M.; Dobrynin, A.B.; Sharipova, R.R.; Voloshina, A.D.; Saifina, A.F.; Gubaidullin, A.T.; Khairutdinov, B.I.; et al. Synthesis of novel 1,2,3-triazolyl nucleoside analogues bearing uracil, 6-methyluracil, 3,6-dimethyluracil, thymine, and quinazoline-2,4-dione moieties. Tetrahedron Lett. 2019, 60, 151276. [Google Scholar] [CrossRef]
- Andreeva, O.V.; Garifullin, B.F.; Zarubaev, V.V.; Slita, A.V.; Yesaulkova, I.L.; ·Saifina, L.F.; Shulaeva, M.M.; Belenok, M.G.; Semenov, V.E.; Kataev, V.E. Synthesis of 1,2,3-triazolyl nucleoside analogues and their antiviral activity. Mol. Divers. 2020, 25, 473–490. [Google Scholar] [CrossRef]
- Fan, W.-Q.; Katritzky, A.R. Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Pergamon: Oxford, UK, 1997; Volume 4, pp. 11–14. [Google Scholar]
- Chatzileontiadou, D.S.M.; Parmenopoulou, V.; Manta, S.; Kantsadi, A.L.; Kylindri, P.; Griniezaki, M.; Kontopoulou, F.; Telopoulou, A.; Prokova, H.; Panagopoulos, D.; et al. Triazole double-headed ribonucleosides as inhibitors of eosinophil derived neurotoxin. Bioorg. Chem. 2015, 63, 152–165. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Efremenkova, O.V.; Andronova, V.L.; Galegov, G.A.; Solyev, P.N.; Karpenko, I.L.; Kochetkov, S.N. 5-(4-Alkyl-1,2,3-Triazol-1-yl)methyl Derivatives of 2′-Deoxyuridine as Inhibitors of Viral and Bacterial Growth. Russ. J. Bioorg. Chem. 2016, 42, 677–684. [Google Scholar] [CrossRef]
- Chatzileontiadou, D.S.M.; Tsika, A.C.; Diamantopoulou, Z.; Delbé, J.; Badet, J.; Courty, J.; Skamnaki, V.T.; Parmenopoulou, V.; Komiotis, D.; Hayes, J.M.; et al. Evidence for Novel Action at the Cell-Binding Site of Human Angiogenin Revealed by Heteronuclear NMR Spectroscopy, in silico and in vivo Studies. ChemMedChem 2018, 13, 259–269. [Google Scholar] [CrossRef]
- Nisic, F.; Speciale, G.; Bernardi, A. Stereoselective Synthesis of α- and β-Glycofuranosyl Amides by Traceless Ligation of Glycofuranosyl Azides. Chem. Eur. J. 2012, 18, 6895–6906. [Google Scholar] [CrossRef]
- Colin, B.; Jones, N.M.; McGuigan, C.; Riley, P.A. Synthesis and biological evaluation of some phosphate triester derivatives of the anti-cancer drug AraC. Nucl. Acids Res. 1989, 17, 7195–7201. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Edathil, J.P.; Wu, R.; Smidansky, E.D.; Cameron, C.E.; Peterson, B.R. One-Pot Synthesis of Nucleoside 5′-Triphosphates from Nucleoside 5′-H-Phosphonates. Org. Lett. 2008, 10, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Young, R.W. A Re-examination of the Reaction Between Phosphorus Trichloride and Salicylic Acid. J. Am. Chem. Soc. 1952, 74, 1672–1673. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A Novel Method for Phosphorylation of Nucleosides to 5′-Nucleotides. Tetrahedron Lett. 1967, 8, 5065–5068. [Google Scholar] [CrossRef]
- Ikemoto, T.; Haze, A.; Hatano, H.; Kitamoto, Y.; Ishida, M.; Nara, K. Phosphorylation of Nucleosides with Phosphorus Oxychloride in Trialkyl Phosphate. Chem. Pharm. Bull. 1995, 43, 210–215. [Google Scholar] [CrossRef]
- Rachwalak, M.; Rozniewska, M.; Golebiewska, J.; Jakubowski, T.; Sobkowski, M.; Romanowska, J. A practical synthesis of nucleoside 5′-diphosphates from nucleoside 5′-H-phosphonate monoesters. Synth. Commun. 2020, 50, 3836–3844. [Google Scholar] [CrossRef]
- Kowalinski, E.; Zubieta, C.; Wolkerstorfer, A.; Szolar, O.H.J.; Ruigrok, R.W.H.; Cusack, S. Structural Analysis of Specific Metal Chelating Inhibitor Binding to the Endonuclease Domain of Influenza pH1N1 (2009) Polymerase. PLoS Pathog. 2012, 8, e1002831. [Google Scholar] [CrossRef]
- Jockusch, S.; Tao, C.; Li, X.; Anderson, T.K.; Chien, M.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. A Library of Nucleotide Analogues Terminate RNA Synthesis Catalyzed by Polymerases of Coronaviruses that Cause SARS and COVID-19. Antivir. Res. 2020, 180, 104857. [Google Scholar] [CrossRef]
- Jácome, R.; Campillo-Balderas, J.A.; Ponce de León, S.; Becerra, A.; Lazcano, A. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci. Rep. 2020, 10, 9294. [Google Scholar] [CrossRef]
- Das, K.; Aramini, J.M.; Ma, L.-C.; Krug, R.M.; Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. SMB 2010, 17, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Fudo, S.; Yamamoto, N.; Nukaga, M.; Odagiri, T.; Tashiro, M.; Hoshino, T. Two distinctive binding modes of endonuclease inhibitors to the N-terminal region of influenza virus polymerase acidic subunit. Biochemistry 2016, 55, 2646–2660. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.S.; Chung, K.; Chu, C.K. Synthesis of 2′-deoxy-2′-fluoro-2′-C-methyl spiro cyclopentyl carbocyclic uridine analog as potential inhibitors of HCV NS5B polymerase. Nucl. Nucl. Nucl. Acids 2020, 30, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, T.; Arai, Y.; Yoshii, E. The Reaction of o-Phenylenediamine with Phenyl Phosphorodichloridate. Synthesis and Reactions of 2-Phenoxy-1,3-Dihydro-2-H-1,3,2-Benzodiazaphosphole-2-Oxide and Related Compounds. Chem. Pharm. Bull. 1973, 21, 202–206. [Google Scholar] [CrossRef]
- McGuigan, C.; Pathirana, R.N.; Balzarini, J.; De Clercq, E. Intracellular Delivery of Bioactive AZT Nucleotides by Aryl Phosphate Derivatives of AZT. J. Med. Chem. 1993, 36, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Research 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- HyperChem Professional 8.0 (2007). Hypercube, Inc. Available online: http://www.hyper.com/?tabid=360. (accessed on 26 July 2022).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]

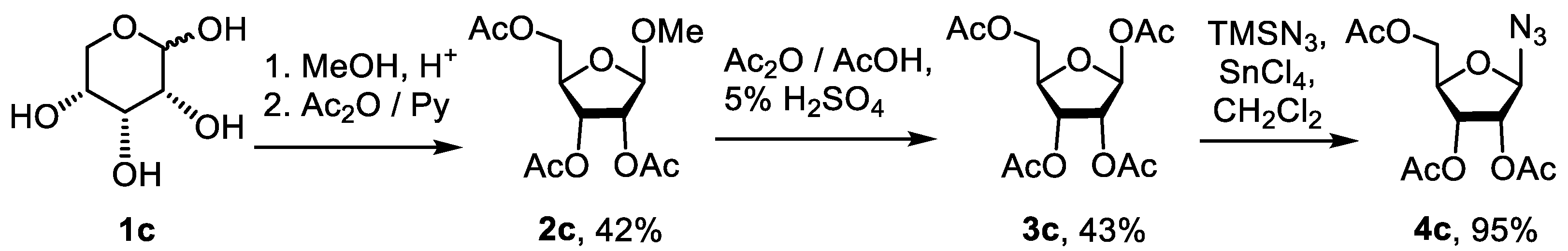



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CC50 a (μM) | IC50 b (μM) | SI c | Compound | CC50 a (μM) | IC50 b (μM) | SI c |
|---|---|---|---|---|---|---|---|
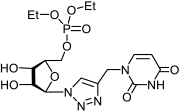 11a | >650 | 434 ± 39 | 2 |  12a | >596 | 250 ± 22 | 2 |
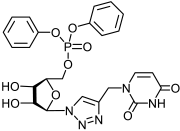 13a | >538 | 121 ± 7 | 4 | 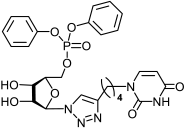 14a | >500 | 183 ± 15 | 3 |
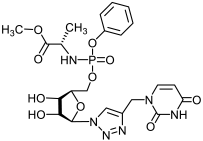 17a | >530 | 25 ± 4 | 21 |  18a | >463 | >463 | 1 |
 23a | >612 | 204 ± 28 | 3 | 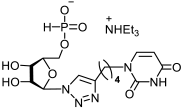 24a | >564 | 218 ± 32 | 3 |
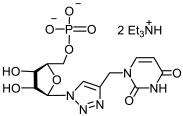 25a | >494 | >494 | 1 | 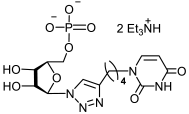 26a | >463 | 195 ± 26 | 2 |
 11b | >587 | 411 ± 51 | 1 | 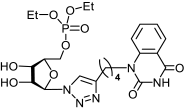 12b | >542 | >542 | 1 |
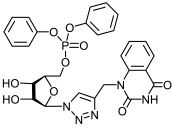 13b | >494 | 17.9 ± 3 | 28 | 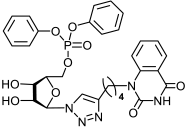 14b | 98 ± 6 | 51 ± 4 | 2 |
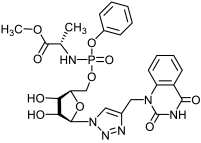 17b | >487 | >487 | 1 |  18b | >456 | >456 | 1 |
 23b | >555 | 176 ± 21 | 3 | 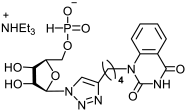 24b | >516 | >516 | 1 |
 25b | >458 | 159 ± 19 | 3 | 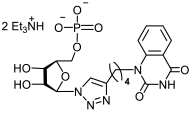 26b | >430 | >430 | 1 |
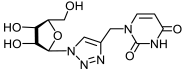 33a | >880 d | >880 d | 1 | 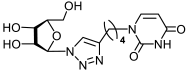 34a | >817 d | 343 ± 41 | 2 |
 33b | 132 ± 9 d | 42 ± 5 d | 3 |  34b | >719 d | 30 ± 4 d | 24 |
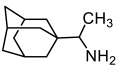 | |||||||
| Rimantadine | 340 ± 16 | 77 ± 8 | 4 | ||||
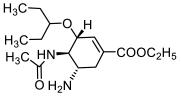 Ribavirin | 94 ± 48 | 31 ± 9 | 3 | ||||
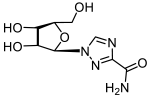 Oseltamivir carboxylate | >200 | 0.3 ± 0.06 | >667 |
| Compound | −Ebind (kcal/mol) | Ligand-Protein Interactions |
|---|---|---|
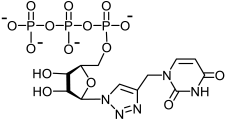 35a | 9.1 | H bonding: Arg84, Trp88 |
 35b | 9.8 | H bonding: Val122, Lys134 π-π: Tyr24 |
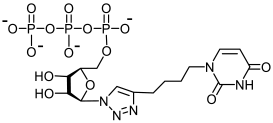 36a | 7.8 | no H bonding |
 36b | 8.9 | H bonding: Glu80, Val122 π-π: Tyr24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatarinov, D.A.; Garifullin, B.F.; Belenok, M.G.; Andreeva, O.V.; Strobykina, I.Y.; Shepelina, A.V.; Zarubaev, V.V.; Slita, A.V.; Volobueva, A.S.; Saifina, L.F.; et al. The First 5′-Phosphorylated 1,2,3-Triazolyl Nucleoside Analogues with Uracil and Quinazoline-2,4-Dione Moieties: A Synthesis and Antiviral Evaluation. Molecules 2022, 27, 6214. https://doi.org/10.3390/molecules27196214
Tatarinov DA, Garifullin BF, Belenok MG, Andreeva OV, Strobykina IY, Shepelina AV, Zarubaev VV, Slita AV, Volobueva AS, Saifina LF, et al. The First 5′-Phosphorylated 1,2,3-Triazolyl Nucleoside Analogues with Uracil and Quinazoline-2,4-Dione Moieties: A Synthesis and Antiviral Evaluation. Molecules. 2022; 27(19):6214. https://doi.org/10.3390/molecules27196214
Chicago/Turabian StyleTatarinov, Dmitry A., Bulat F. Garifullin, Mayya G. Belenok, Olga V. Andreeva, Irina Yu Strobykina, Anna V. Shepelina, Vladimir V. Zarubaev, Alexander V. Slita, Alexandrina S. Volobueva, Liliya F. Saifina, and et al. 2022. "The First 5′-Phosphorylated 1,2,3-Triazolyl Nucleoside Analogues with Uracil and Quinazoline-2,4-Dione Moieties: A Synthesis and Antiviral Evaluation" Molecules 27, no. 19: 6214. https://doi.org/10.3390/molecules27196214