
Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
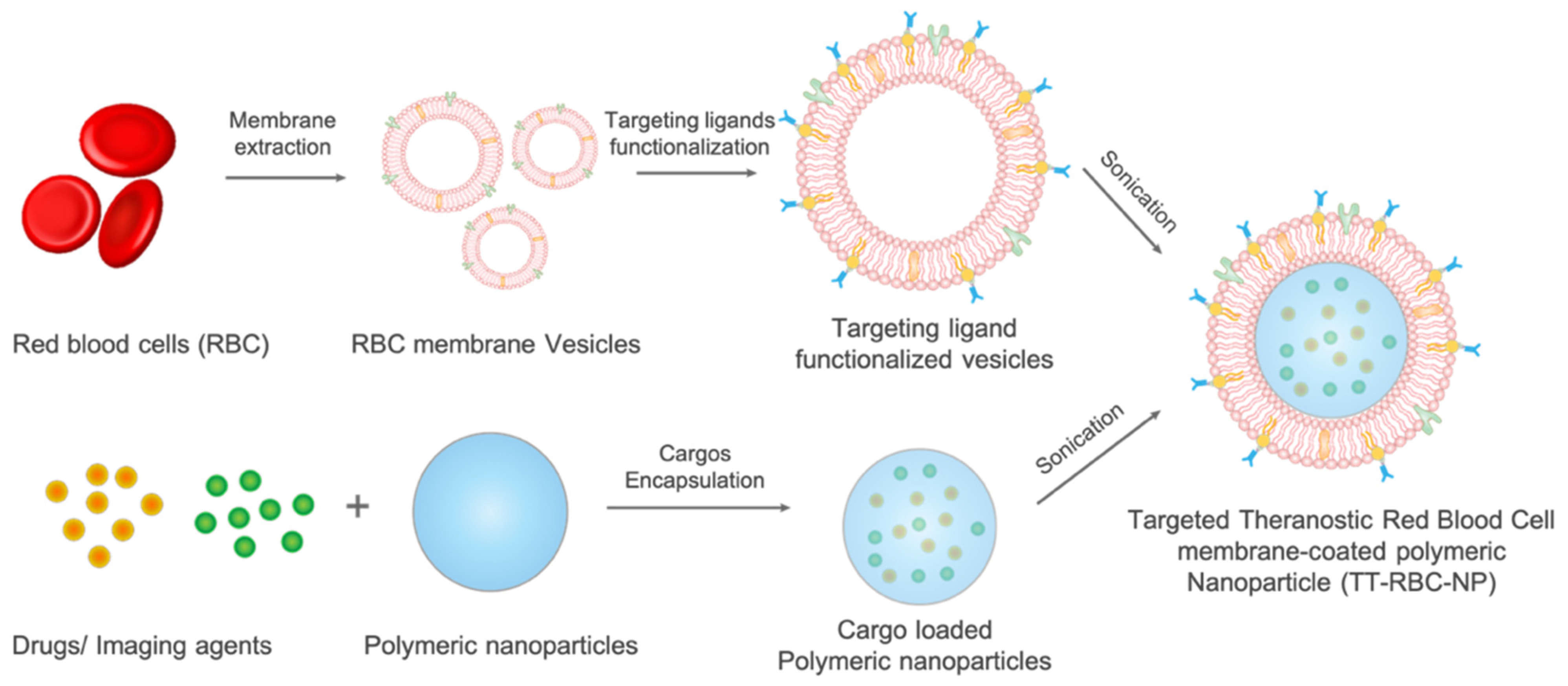
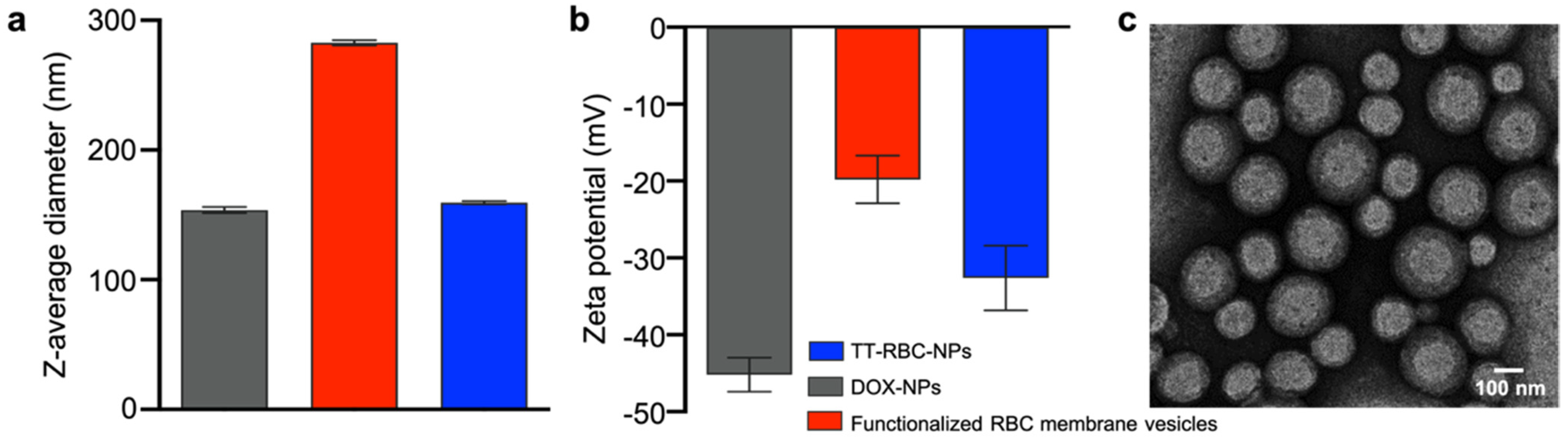
2.1. Synthesis and Characterization of TT-RBC-NPs
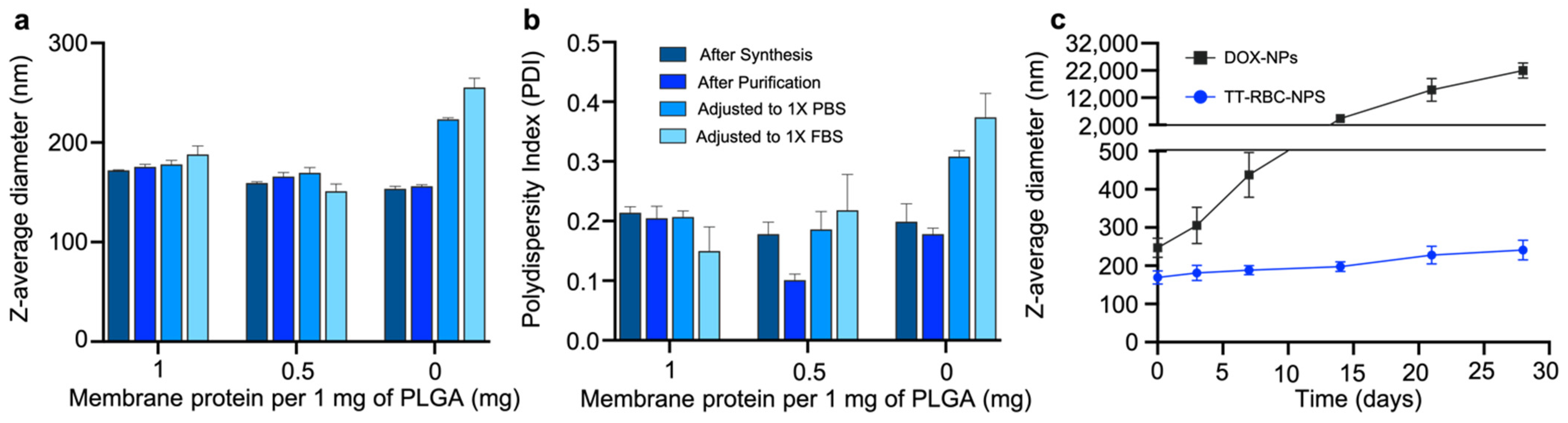
2.2. Optimization and Stability of TT-RBC-NPs
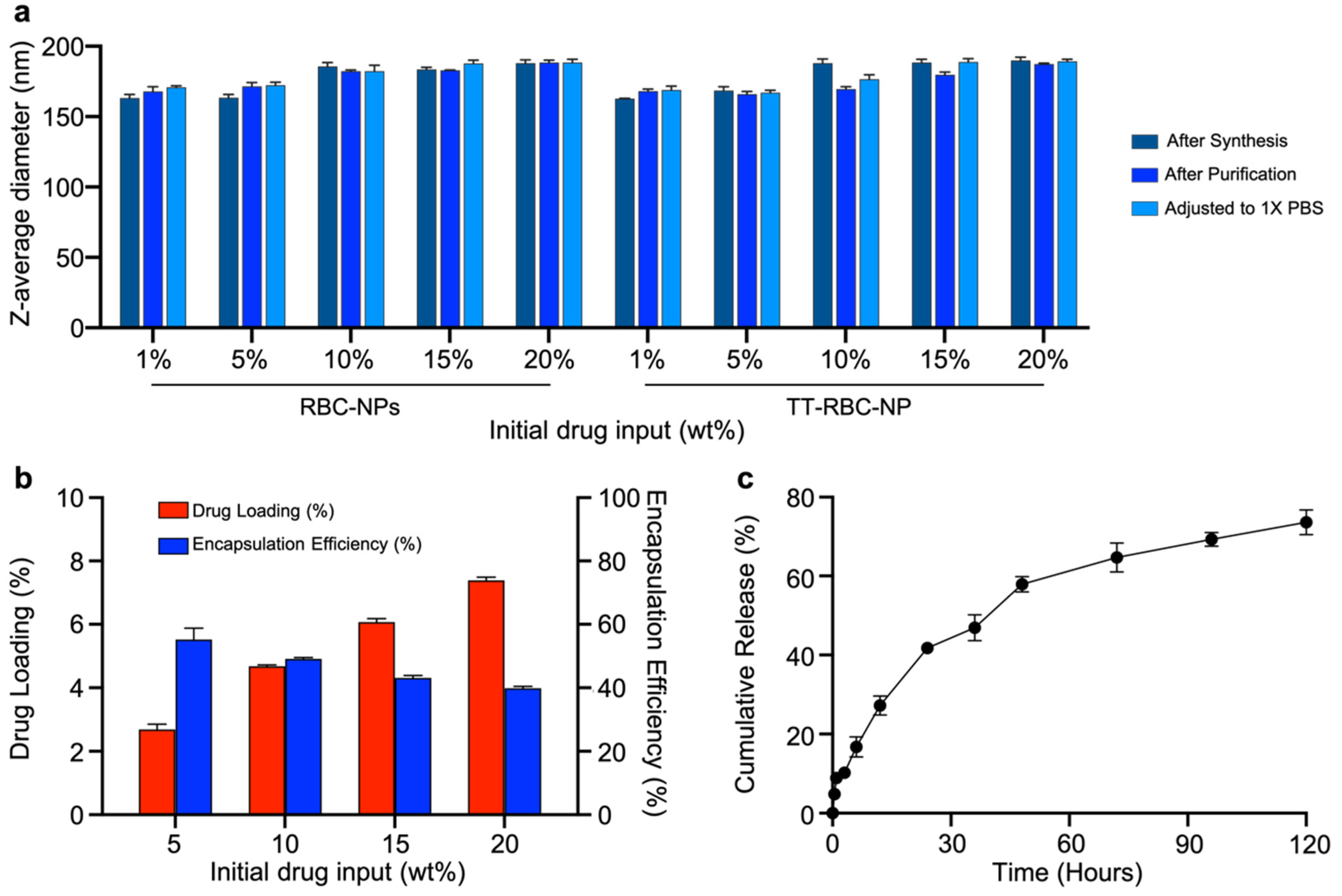
2.3. Drug Loading and Cumulative Release of TT-RBC-NPs
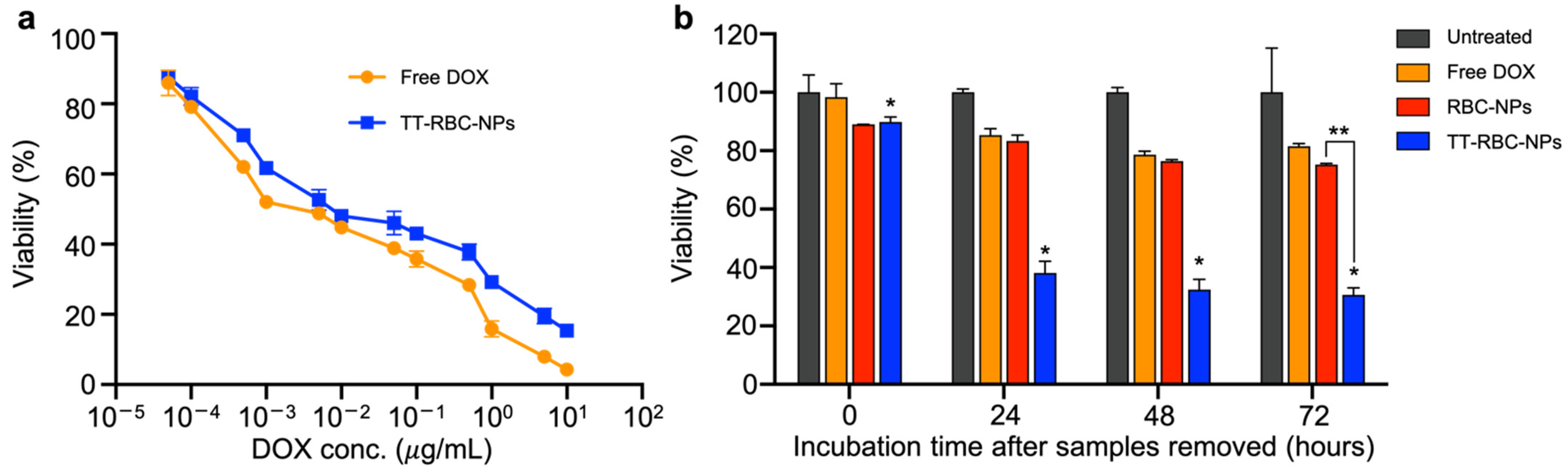
2.4. In Vitro 2D Therapeutic Efficacy
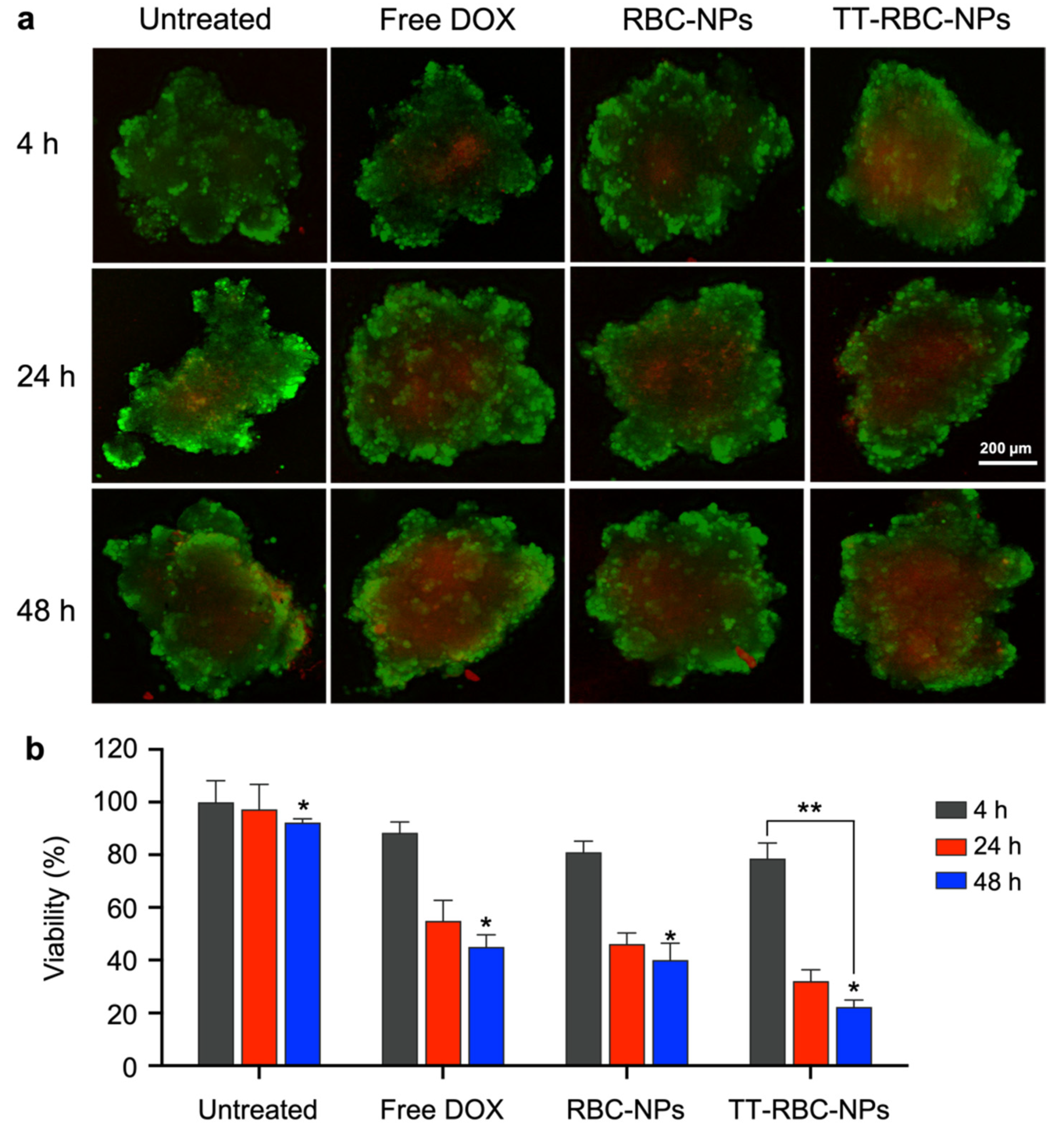
2.5. In Vitro Cellular Toxicity of TT-RBC-NPs in Three-Dimensional (3D) Spheroids
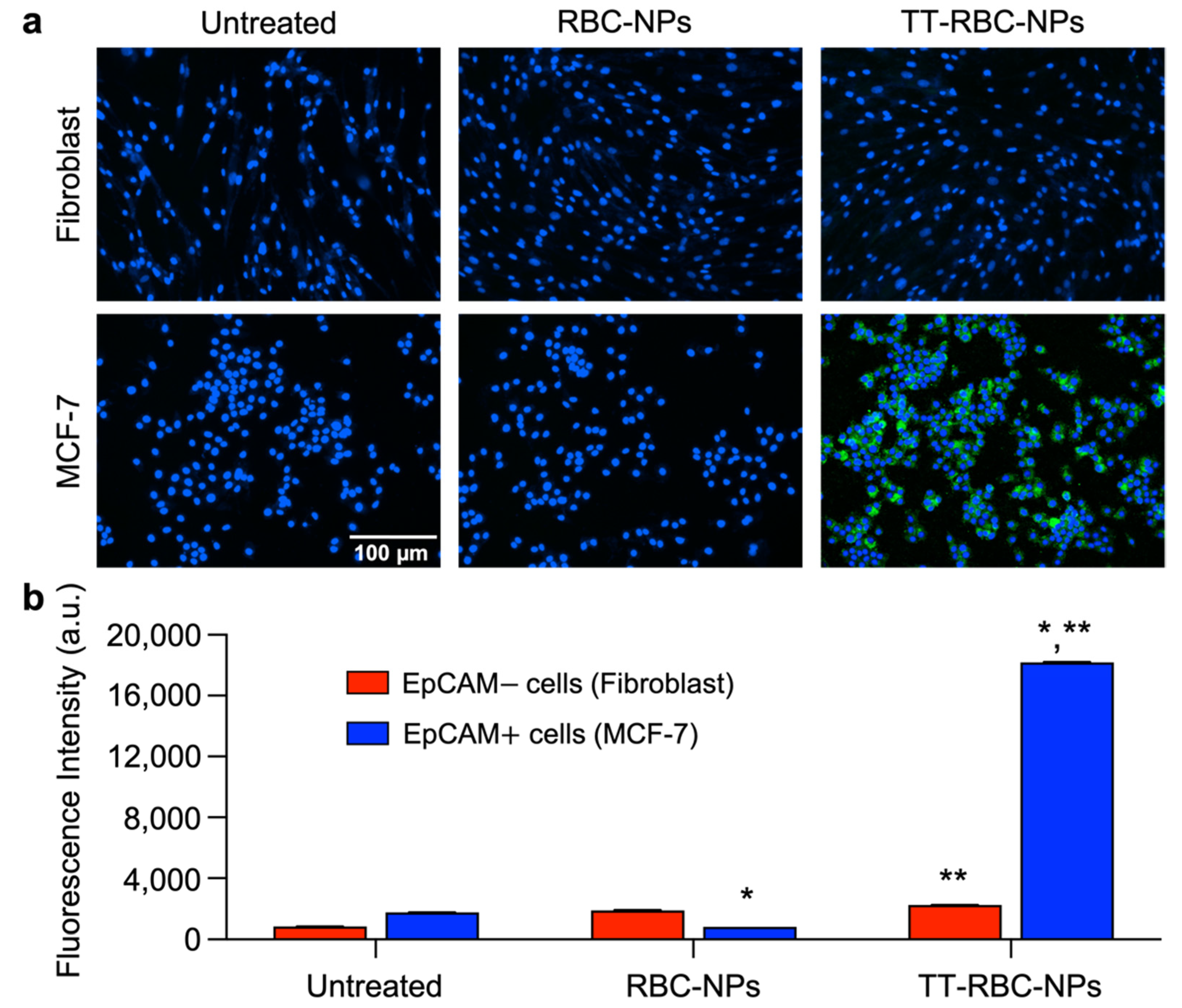
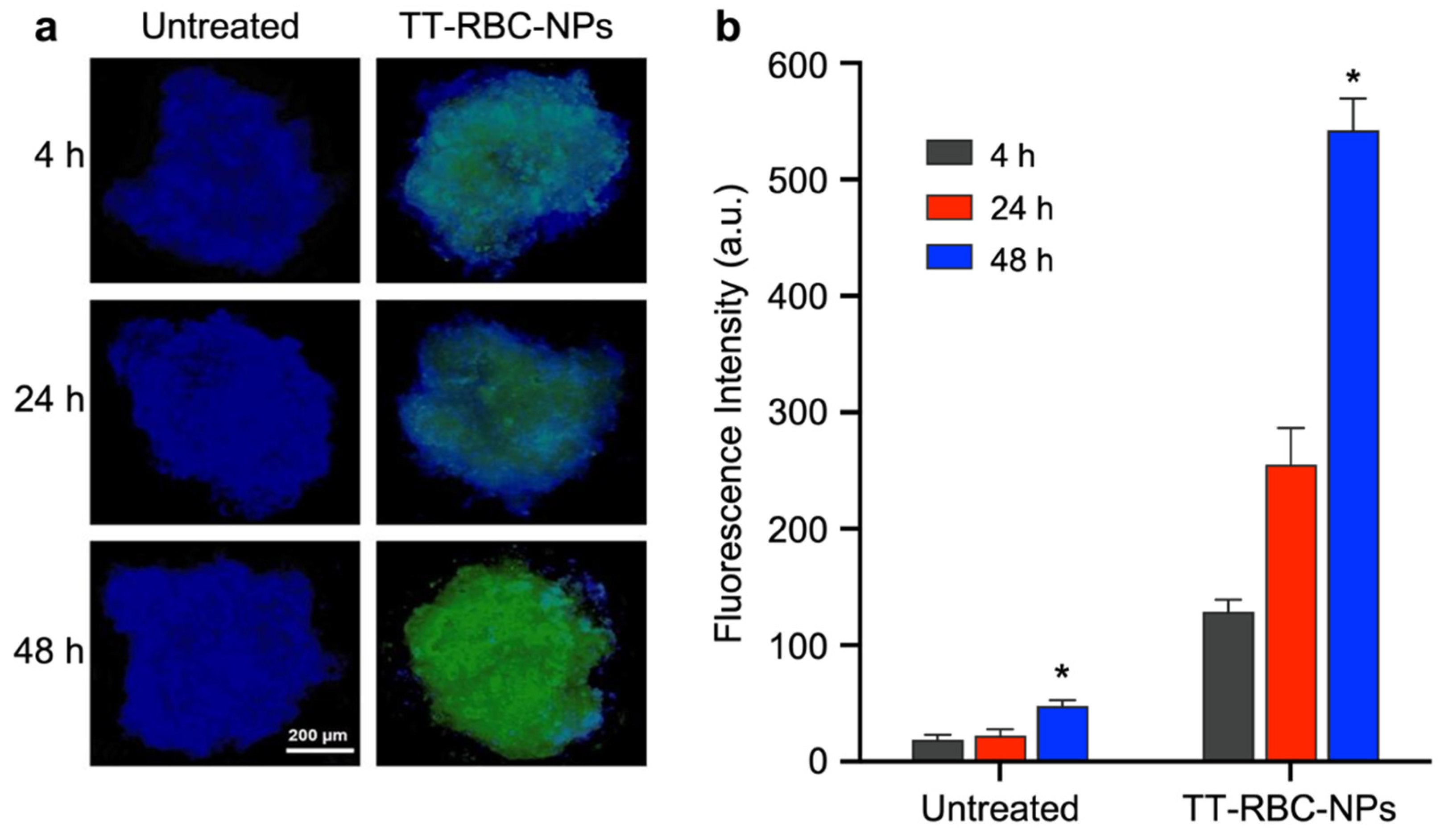
2.6. In Vitro Targeting Ability Study of TT-RBC-NPs
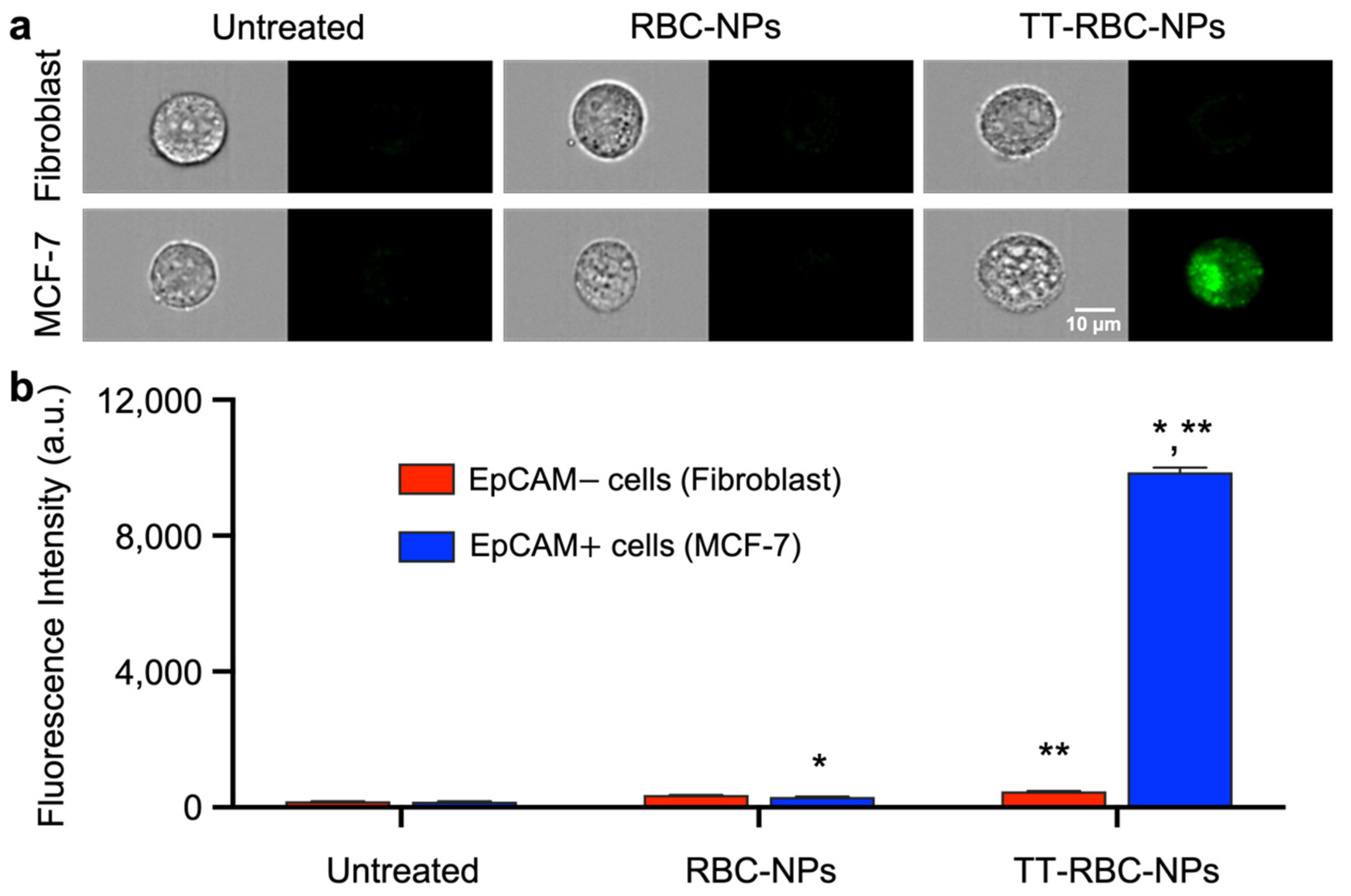
2.7. Nanoparticle Penetration and Uptake in Three-Dimensional (3D) Spheroids
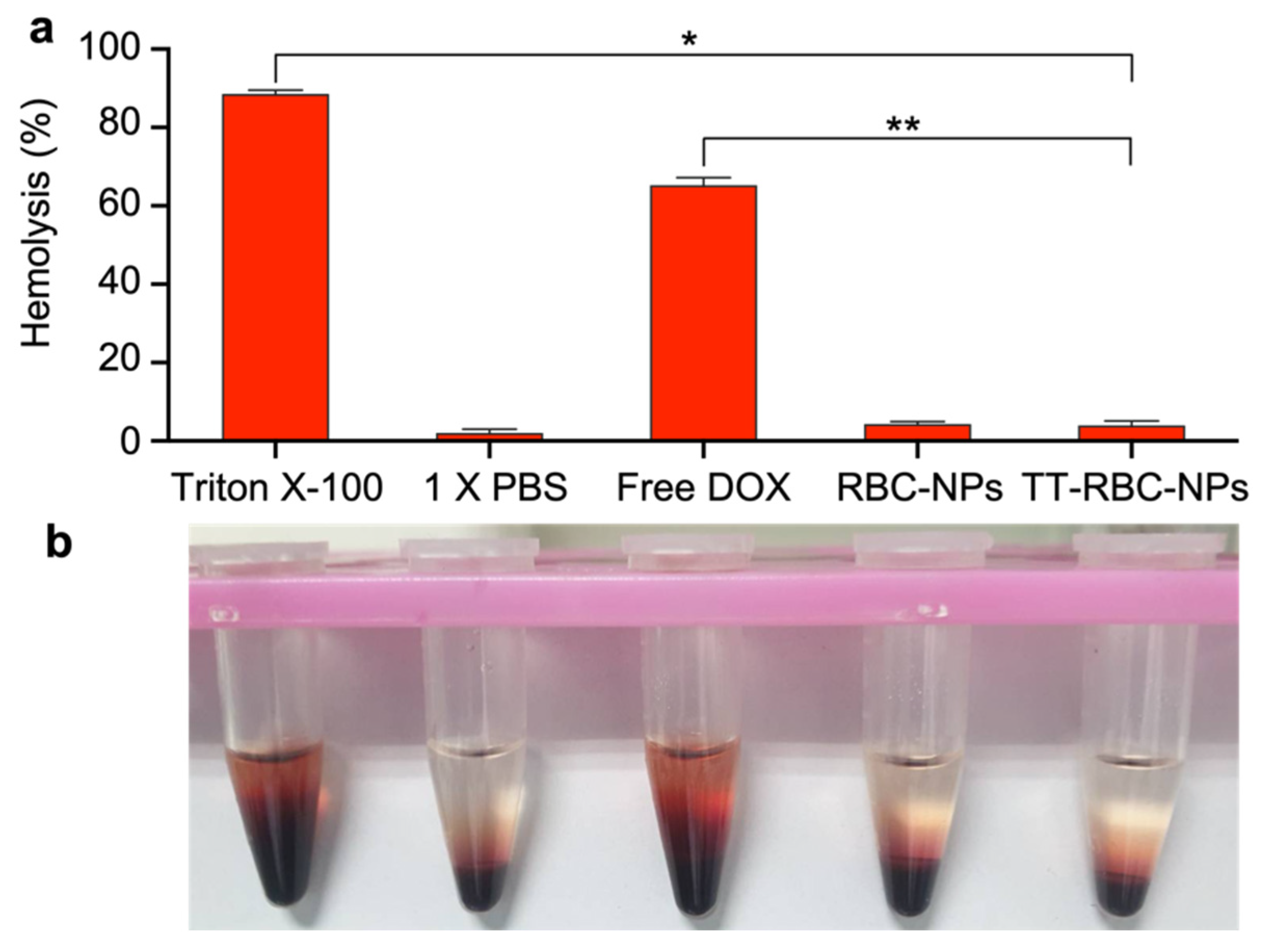
2.8. Blood Compatibility of TT-RBC-NPs
3. Materials and Methods
3.1. Materials
3.2. Preparation of Nanoparticles
3.3. Characterization of Nanoaparticles
3.4. In Vitro Therapeutic Efficacy on 2-Dimensional (2D) Cell Culture
3.5. Three-Dimensional (3D) In Vitro Live/Dead Cell Imaging and Cellular Cytotoxicity Assay
3.6. In Vitro Targeting Ability of TT-RBC-NPs
3.7. Nanoparticle Penetration in Three-Dimensional (3D) Spheroids
3.8. Blood Compatibility
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Matsumura, Y.; Hamaguchi, T.; Ura, T.; Muro, K.; Yamada, Y.; Shimada, Y.; Shirao, K.; Okusaka, T.; Ueno, H.; Ikeda, M. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br. J. Cancer 2004, 91, 1775–1781. [Google Scholar] [CrossRef]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Suter, T.; Plataniotis, G.; De Azambuja, E.; Sandri, M.T.; Criscitiello, C.; Goldhirsch, A.; Cipolla, C.; Roila, F. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann. Oncol. 2012, 23, vii155–vii166. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Warmuth, M.; Sellers, W.; Dorsch, M. Targeted cancer therapies in the twenty-first century: Lessons from imatinib. Clin. Pharmacol. Ther. 2010, 87, 543–552. [Google Scholar] [CrossRef]
- Rahman, A.M.; Yusuf, S.W.; Ewer, M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomed. 2007, 2, 567. [Google Scholar]
- Lao, J.; Madani, J.; Puértolas, T.; Álvarez, M.; Hernández, A.; Pazo-Cid, R.; Artal, Á.; Antón Torres, A. Liposomal doxorubicin in the treatment of breast cancer patients: A review. J. Drug Deliv. 2013, 2013. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, V.K.; Tewari, R.P.; Agarwal, V. Nanoparticle based drug delivery system: Advantages and applications. Indian J. Sci. Technol. 2011, 4, 177–180. [Google Scholar] [CrossRef]
- Videira, M.; Santos, A.; Botelho, M. Biodistribution of lipid nanoparticles: A comparative study of pulmonary versus intravenous administration in rats. Curr. Radiopharm. 2012, 5, 158–165. [Google Scholar] [CrossRef]
- Sadat, S.M.; Jahan, S.T.; Haddadi, A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J. Biomater. Nanobiotechnol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Zhu, G.; Xie, J.; Chen, X. Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2017, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Understanding the immunogenicity and antigenicity of nanomaterials: Past, present and future. Toxicol. Appl. Pharmacol. 2016, 299, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef]
- Thamphiwatana, S.; Angsantikul, P.; Escajadillo, T.; Zhang, Q.; Olson, J.; Luk, B.T.; Zhang, S.; Fang, R.H.; Gao, W.; Nizet, V. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl. Acad. Sci. USA 2017, 114, 11488–11493. [Google Scholar] [CrossRef] [PubMed]
- Luk, B.T.; Fang, R.H.; Hu, C.-M.J.; Copp, J.A.; Thamphiwatana, S.; Dehaini, D.; Gao, W.; Zhang, K.; Li, S.; Zhang, L. Safe and immunocompatible nanocarriers cloaked in RBC membranes for drug delivery to treat solid tumors. Theranostics 2016, 6, 1004. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef]
- Li, H.; Jin, K.; Luo, M.; Wang, X.; Zhu, X.; Liu, X.; Jiang, T.; Zhang, Q.; Wang, S.; Pang, Z. Size dependency of circulation and biodistribution of biomimetic nanoparticles: Red blood cell membrane-coated nanoparticles. Cells 2019, 8, 881. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Dumoga, S.; Singh, N. Red blood cells membrane-derived nanoparticles: Applications and key challenges in their clinical translation. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, e1776. [Google Scholar] [CrossRef]
- Wei, X.; Ying, M.; Dehaini, D.; Su, Y.; Kroll, A.V.; Zhou, J.; Gao, W.; Fang, R.H.; Chien, S.; Zhang, L. Nanoparticle functionalization with platelet membrane enables multifactored biological targeting and detection of atherosclerosis. ACS Nano 2018, 12, 109–116. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Fang, R.H.; Wang, K.-C.; Luk, B.T.; Thamphiwatana, S.; Dehaini, D.; Nguyen, P.; Angsantikul, P.; Wen, C.H.; Kroll, A.V. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 2015, 526, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Maiorano, G.; Gutiérrez-Millán, C.; Cortese, B.; Trapani, A.; D’Amone, S.; Gigli, G.; Palamà, I.E. Erythrocytes and Nanoparticles: New Therapeutic Systems. Appl. Sci. 2021, 11, 2173. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, K. Advances of biological-camouflaged nanoparticles delivery system. Nano Res. 2020, 13, 2617–2624. [Google Scholar] [CrossRef]
- Wu, M.; Le, W.; Mei, T.; Wang, Y.; Chen, B.; Liu, Z.; Xue, C. Cell membrane camouflaged nanoparticles: A new biomimetic platform for cancer photothermal therapy. Int. J. Nanomed. 2019, 14, 4431. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Jiang, G.; Chen, L.; Zhou, H.; Fourches, D.; Tropsha, A.; Yan, B. Chemical basis of interactions between engineered nanoparticles and biological systems. Chem. Rev. 2014, 114, 7740–7781. [Google Scholar] [CrossRef]
- Yang, W.; Wang, L.; Mettenbrink, E.M.; DeAngelis, P.L.; Wilhelm, S. Nanoparticle toxicology. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 269–289. [Google Scholar] [CrossRef]
- Lei, W.; Yang, C.; Wu, Y.; Ru, G.; He, X.; Tong, X.; Wang, S. Nanocarriers surface engineered with cell membranes for cancer targeted chemotherapy. J. Nanobiotechnol. 2022, 20, 1–21. [Google Scholar] [CrossRef]
- Thomsen, T.; Klok, H.-A. Chemical Cell Surface Modification and Analysis of Nanoparticle-Modified Living Cells. ACS Appl. Bio Mater. 2021, 4, 2293–2306. [Google Scholar] [CrossRef]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity assays: In vitro methods to measure dead cells. Assay Guid. Man. 2019. [Google Scholar]
- Unni, M.; Zhang, J.; George, T.J.; Segal, M.S.; Fan, Z.H.; Rinaldi, C. Engineering magnetic nanoparticles and their integration with microfluidics for cell isolation. J. Colloid Interface Sci. 2020, 564, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Wang, C.-P.J.; Lim, J.; Park, W.; Kwon, H.-K.; Kim, S.-N.; Kim, T.-H.; Park, C.G. Impact of the conjugation of antibodies to the surfaces of polymer nanoparticles on the immune cell targeting abilities. Nano Converg. 2021, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.-M.J.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.R.; Wei, X.Q.; Song, X.; Hao, L.Y.; Cai, X.X.; Zhang, Z.R.; Peng, Q.; Lin, Y.F. Independent effect of polymeric nanoparticle zeta potential/surface charge, on their cytotoxicity and affinity to cells. Cell Prolif. 2015, 48, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Ma, N.; Ma, C.; Li, C.; Wang, T.; Tang, Y.; Wang, H.; Mou, X.; Chen, Z.; He, N. Influence of nanoparticle shape, size, and surface functionalization on cellular uptake. J. Nanosci. Nanotechnol. 2013, 13, 6485–6498. [Google Scholar] [CrossRef]
- Zhang, S.; Li, J.; Lykotrafitis, G.; Bao, G.; Suresh, S. Size-dependent endocytosis of nanoparticles. Adv. Mater. 2009, 21, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, C.; Manzano, M.; Vallet-Regí, M. Nanoparticles coated with cell membranes for biomedical applications. Biology 2020, 9, 406. [Google Scholar] [CrossRef]
- Bidkar, A.P.; Sanpui, P.; Ghosh, S.S. Red blood cell-membrane-coated poly (lactic-co-glycolic acid) nanoparticles for enhanced chemo-and hypoxia-activated therapy. ACS Appl. Bio Mater. 2019, 2, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Hu, C.-M.J.; Fang, R.H.; Dehaini, D.; Carpenter, C.; Zhang, D.-E.; Zhang, L. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine 2013, 8, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Basto, P.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Zhang, L.; Pridgen, E.; Wang, A.Z.; Marein, S.L.; Westerhof, K.; Molnar, L.K. HER-2-Targeted Nanoparticle–Affibody Bioconjugates for Cancer Therapy. ChemMedChem Chem. Enabling Drug Discov. 2008, 3, 1839–1843. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.-G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.A.; Waite, J.; Sacristán, C.; Victora, G.D.; Zanin-Zhorov, A.; Dustin, M.L. Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 2009, 28, 79–105. [Google Scholar] [CrossRef]
- Lee, S.Y.; Nam, S.; Koo, J.S.; Kim, S.; Yang, M.; Jeong, D.I.; Hwang, C.; Park, J.; Cho, H.-J. Possible contribution of sialic acid to the enhanced tumor targeting efficiency of nanoparticles engineered with doxorubicin. Sci. Rep. 2020, 10, 1–15. [Google Scholar]
- Shi, L.; Zhang, J.; Zhao, M.; Tang, S.; Cheng, X.; Zhang, W.; Li, W.; Liu, X.; Peng, H.; Wang, Q. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale 2021, 13, 10748–10764. [Google Scholar] [CrossRef]
- Jain, A.; Cheng, K. The principles and applications of avidin-based nanoparticles in drug delivery and diagnosis. J. Control. Release 2017, 245, 27–40. [Google Scholar] [CrossRef]
- Huang, M.; Pu, Y.; Peng, Y.; Fu, Q.; Guo, L.; Wu, Y.; Zheng, Y. Biotin and glucose dual-targeting, ligand-modified liposomes promote breast tumor-specific drug delivery. Bioorg. Med. Chem. Lett. 2020, 30, 127151. [Google Scholar] [CrossRef]
- Neun, B.W.; Ilinskaya, A.N.; Dobrovolskaia, M.A. Updated method for in vitro analysis of nanoparticle hemolytic properties. Methods Mol. Biol. 2018, 1682, 91–102. [Google Scholar]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Method for analysis of nanoparticle hemolytic properties in vitro. Nano Lett. 2008, 8, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Krzyzaniak, J.F.; Yalkowsky, S.H. Lysis of human red blood cells 3: Effect of contact time on surfactant-induced hemolysis. PDA J. Pharm. Sci. Technol. 1998, 52, 66–69. [Google Scholar] [PubMed]
- Kim, D.; El-Shall, H.; Dennis, D.; Morey, T. Interaction of PLGA nanoparticles with human blood constituents. Colloids Surf. B Biointerfaces 2005, 40, 83–91. [Google Scholar] [CrossRef]
- Wang, F.; Gao, W.; Thamphiwatana, S.; Luk, B.T.; Angsantikul, P.; Zhang, Q.; Hu, C.M.J.; Fang, R.H.; Copp, J.A.; Pornpattananangkul, D. Hydrogel retaining toxin-absorbing nanosponges for local treatment of methicillin-Resistant Staphylococcus aureus infection. Adv. Mater. 2015, 27, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.-M.J.; Chen, K.N.; Luk, B.T.; Carpenter, C.W.; Gao, W.; Li, S.; Zhang, D.-E.; Lu, W.; Zhang, L. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale 2013, 5, 8884–8888. [Google Scholar] [CrossRef]
- Copp, J.A.; Fang, R.H.; Luk, B.T.; Hu, C.-M.J.; Gao, W.; Zhang, K.; Zhang, L. Clearance of pathological antibodies using biomimetic nanoparticles. Proc. Natl. Acad. Sci. USA 2014, 111, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, C.; Baguley, B.C.; Zhou, F.; Zhou, W.; Shaw, J.P.; Wang, Z.; Wu, Z.; Liu, J. Optimization of the formation of embedded multicellular spheroids of MCF-7 cells: How to reliably produce a biomimetic 3D model. Anal. Biochem. 2016, 515, 47–54. [Google Scholar] [CrossRef]
- Gao, S.; Chen, S.; Lu, Q. Cell-imprinted biomimetic interface for intelligent recognition and efficient capture of CTCs. Biomater. Sci. 2019, 7, 4027–4035. [Google Scholar] [CrossRef]
- Madsen, N.H.; Nielsen, B.S.; Nhat, S.L.; Skov, S.; Gad, M.; Larsen, J. Monocyte Infiltration and Differentiation in 3D Multicellular Spheroid Cancer Models. Pathogens 2021, 10, 969. [Google Scholar] [CrossRef]
- Freitag, I.; Matthäus, C.; Csaki, A.; Clement, J.H.; Cialla-May, D.; Weber, K.; Krafft, C.; Popp, J. Differentiation of MCF-7 tumor cells from leukocytes and fibroblast cells using epithelial cell adhesion molecule targeted multicore surface-enhanced Raman spectroscopy labels. J. Biomed. Opt. 2015, 20, 055002. [Google Scholar] [CrossRef]
- Jenkins, S.V.; Nima, Z.A.; Vang, K.B.; Kannarpady, G.; Nedosekin, D.A.; Zharov, V.P.; Griffin, R.J.; Biris, A.S.; Dings, R.P. Triple-negative breast cancer targeting and killing by EpCAM-directed, plasmonically active nanodrug systems. NPJ Precis. Oncol. 2017, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, S.K.; Angsantikul, P.; Pang, Z.; Nasongkla, N.; Hussen, R.S.D.; Thamphiwatana, S.D. Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment. Molecules 2022, 27, 6473. https://doi.org/10.3390/molecules27196473
Marshall SK, Angsantikul P, Pang Z, Nasongkla N, Hussen RSD, Thamphiwatana SD. Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment. Molecules. 2022; 27(19):6473. https://doi.org/10.3390/molecules27196473
Chicago/Turabian StyleMarshall, Suphalak Khamruang, Pavimol Angsantikul, Zhiqing Pang, Norased Nasongkla, Rusnah Syahila Duali Hussen, and Soracha D. Thamphiwatana. 2022. "Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment" Molecules 27, no. 19: 6473. https://doi.org/10.3390/molecules27196473
APA StyleMarshall, S. K., Angsantikul, P., Pang, Z., Nasongkla, N., Hussen, R. S. D., & Thamphiwatana, S. D. (2022). Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment. Molecules, 27(19), 6473. https://doi.org/10.3390/molecules27196473