
Enzalutamide Induces Apoptotic Insults to Human Drug-Resistant and -Sensitive Glioblastoma Cells via an Intrinsic Bax-Mitochondrion-Cytochrome C Caspase Cascade Activation Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
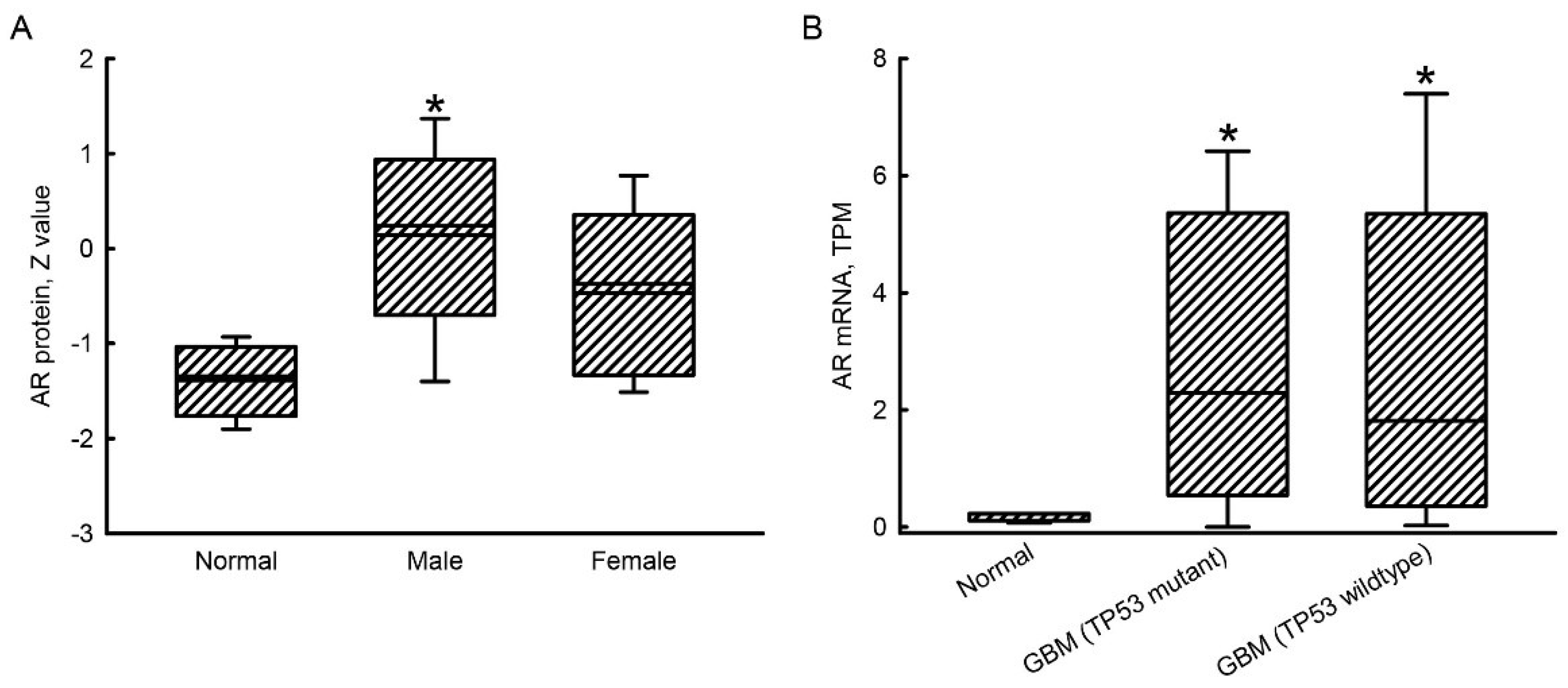
2.1. Upregulation of AR mRNA and Protein Expressions in GBM Patients
2.2. Exposure to Enzalutamide Decreased Viabilities of TMZ-Sensitive and -Resistant Glioblastoma Cells
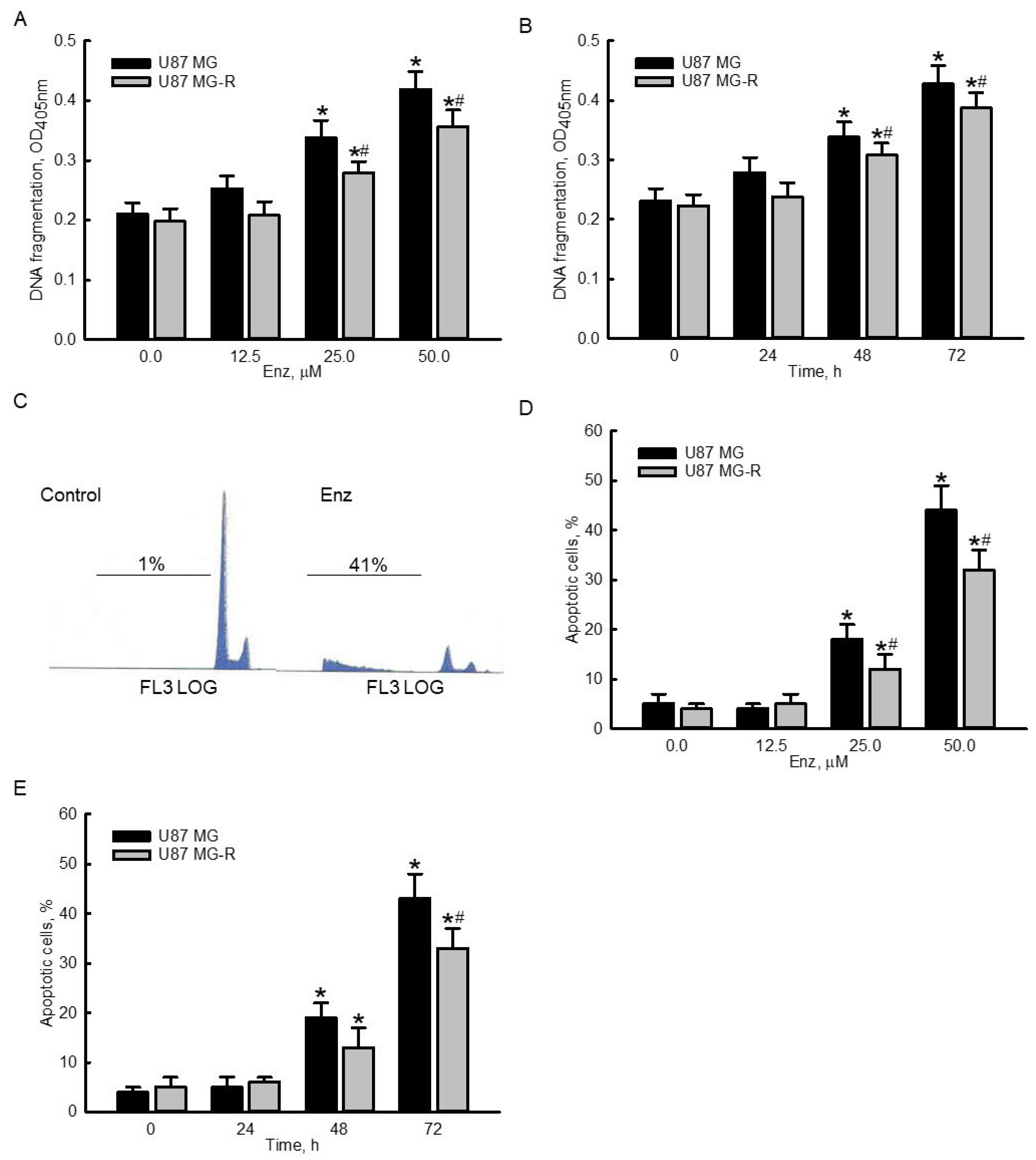
2.3. Enzalutamide Significantly Induced DNA Frgmentation and Cell Apoptosis in Human TMZ-Sensitive and -Resistant Glioblastoma Cells
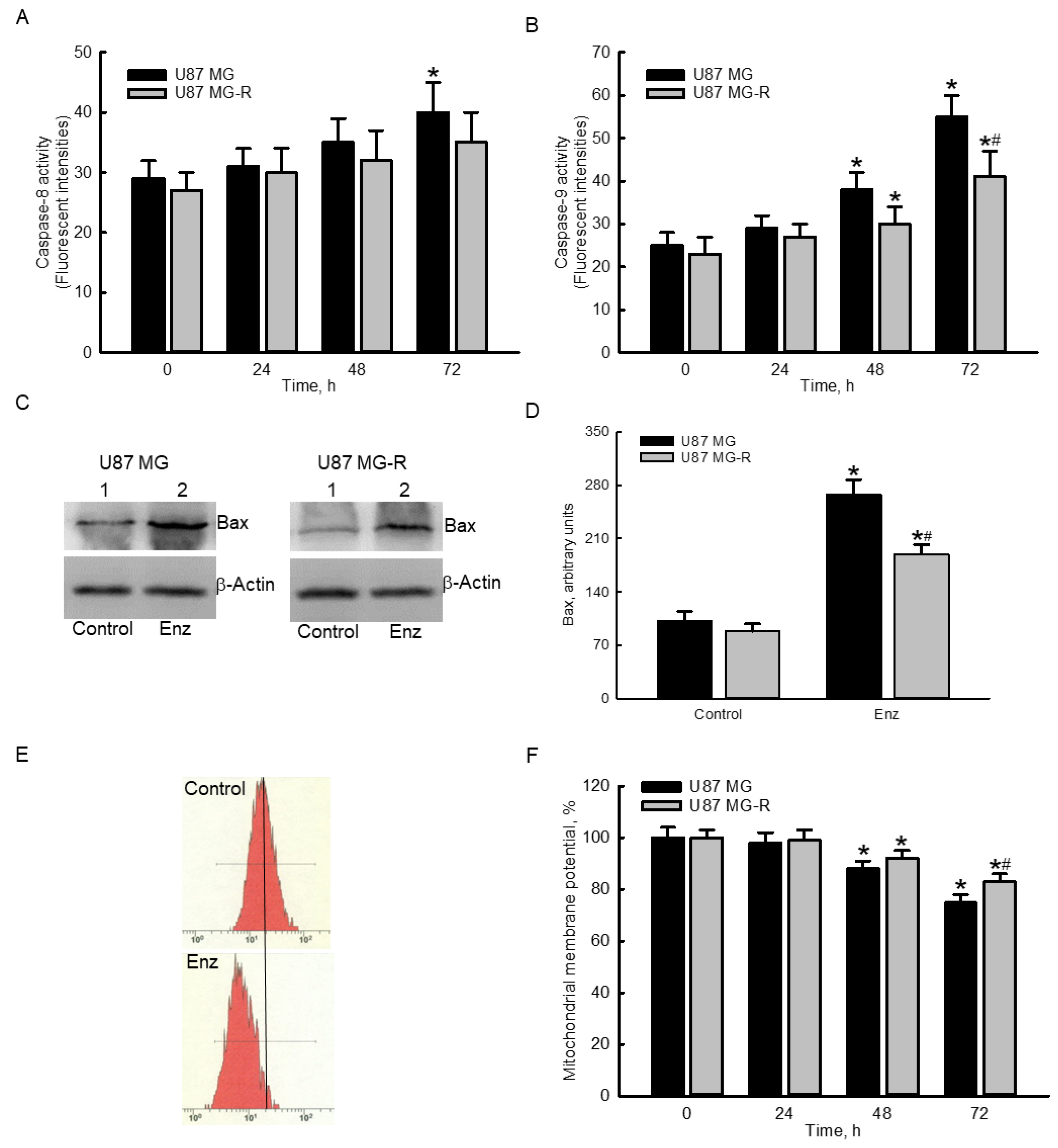
2.4. Enzalutamide Specifically Elevated Caspase-9 Activities and Proapoptotic Bcl-2-Associated X Protein (Bax) Levels but Decreased the Mitochondrial Membrane Potential (MMP) in Human TMZ-Sensitive and -Resistant Glioblastoma Cells
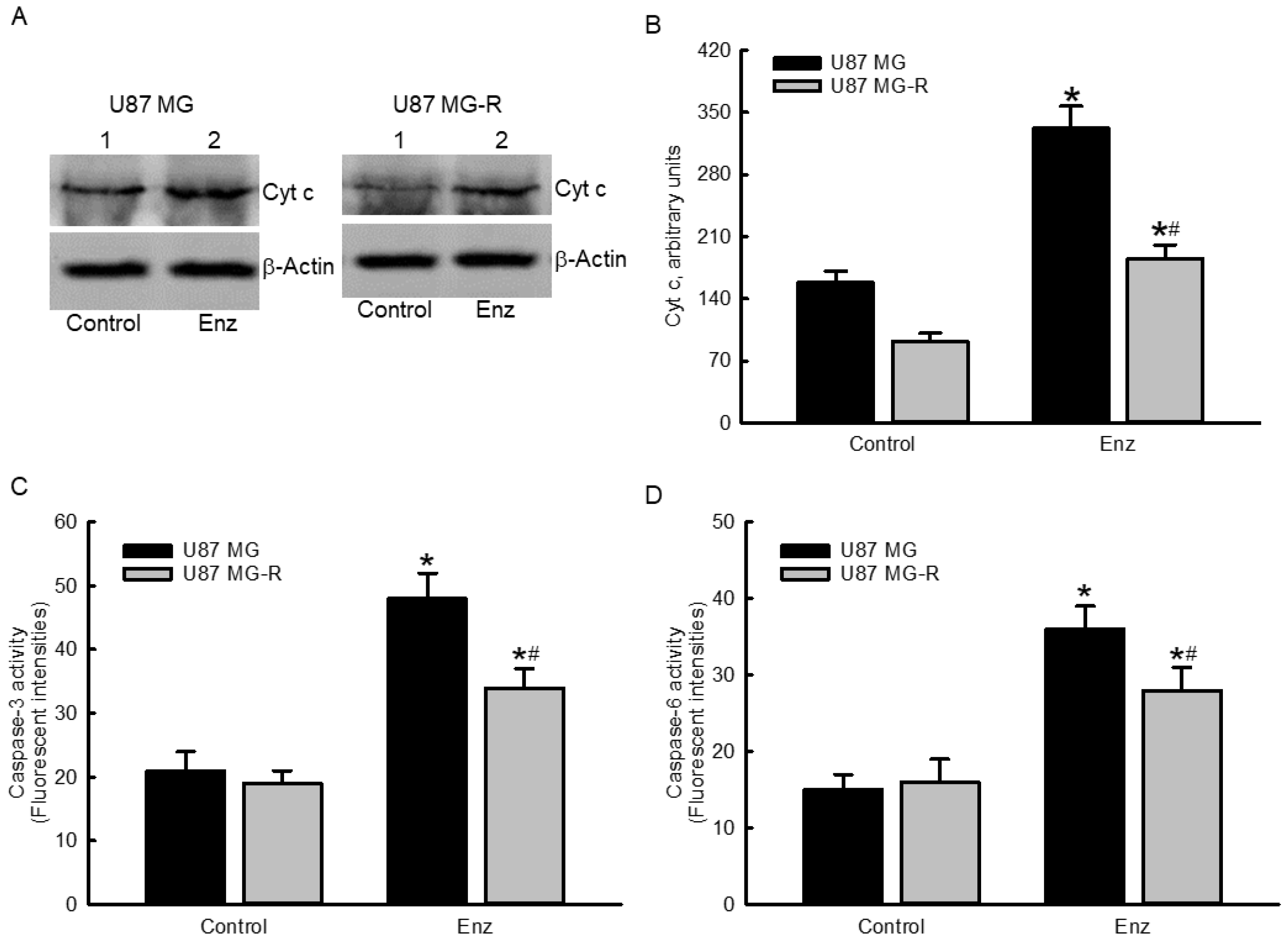
2.5. Enzalutamide Augmented Cytochrome c Levels and Subsequently Stimulated Cascade Activation of Caspases-3 and -6 in Human TMZ-Sensitive and TMZ-Resistant Glioblastoma Cells
2.6. Suppressing Caspase-6 Activity Concurrently Attenuated Enzalutamide-Induced Morphological Changes, DNA Fragmentation, and Cell Apoptosis in Human TMZ-Sensitive and -Resistant Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Data Mining
4.2. Culture of Human Normal Astrocytes and Glioblastoma Cells
4.3. Preparation of Human Drug-Resistant Glioblastoma Cells
4.4. Drug Treatment
4.5. Assays of Cell Morphology and Cell Viability
4.6. Quantification of DNA Fragmentation
4.7. Analysis of Apoptotic Cells
4.8. Determination of the Intrinsic or Extrinsic Apoptotic Pathway
4.9. Immunodetection of AR, Bax and Cytochrome c Proteins
4.10. Quantification of the Mitochondrial Membrane Potential (MMP)
4.11. Assay of Caspase-3 and -6 Activities
4.12. Suppression of Caspase-6 Activation
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef] [PubMed]
- Testa, E.; Palazzo, C.; Mastrantonio, R.; Viscomi, M.T. Dynamic interactions between tumor cells and brain microvascular endothelial cells in glioblastoma. Cancers 2022, 14, 3128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Peet, N.P.; Zhang, P.; Jiang, Y.; Rong, L. Current development of glioblastoma therapeutic agents. Mol. Cancer Ther. 2021, 20, 1521–1532. [Google Scholar] [CrossRef]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the mechanisms of temozolomide resistance in gliomas and the strategies to overcome the resistance. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188616. [Google Scholar] [CrossRef]
- Sun, D.P.; Lee, Y.W.; Chen, J.T.; Lin, Y.W.; Chen, R.M. The bradykinin-BDKRB1 axis regulates aquaporin 4 gene expression and consequential migration and invasion of malignant glioblastoma cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers 2020, 12, 667. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen receptor structure, function and biology: From bench to bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Lozano, D.C.; Piña-Medina, A.G.; Hansberg-Pastor, V.; Bello-Alvarez, C.; Camacho-Arroyo, I. Testosterone promotes glioblastoma cell proliferation, migration, and invasion through androgen receptor activation. Front. Endocrinol. 2019, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef]
- Antonarakis, E.S. AR Signaling in human malignancies: Prostate cancer and beyond. Cancers 2018, 10, 22. [Google Scholar] [CrossRef]
- Daswani, B.; Khan, Y. Insights into the role of estrogens and androgens in glial tumorigenesis. J. Carcinog. 2021, 20, 10. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Spratt, D.E.; Pierce, L.J.; Speers, C.W. ARe we there yet? Understanding androgen receptor signaling in breast cancer. NPJ Breast Cancer 2020, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Saad, F. Evidence for the efficacy of enzalutamide in postchemotherapy metastatic castrate-resistant prostate cancer. Ther. Adv. Urol. 2013, 5, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devos, G.; Devlies, W.; de Meerleer, G.; Baldewijns, M.; Gevaert, T.; Moris, L.; Milonas, D.; van Poppel, H.; Berghen, C.; Everaerts, W.; et al. Neoadjuvant hormonal therapy before radical prostatectomy in high-risk prostate cancer. Nat. Rev. Urol. 2021, 18, 739–762. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J. Castration—Recurrent prostate cancer is not androgen independent. Adv. Exp. Med. Biol. 2008, 617, 223–234. [Google Scholar] [PubMed]
- Rodríguez-Lozano, D.C.; Velázquez-Vázquez, D.E.; del Moral-Morales, A.; Camacho-Arroyo, I. Dihydrotestosterone induces proliferation, migration, and invasion of human glioblastoma cell lines. OncoTargets Ther. 2020, 13, 8813–8823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, F.; Ahmed, S.; Liu, K.; Zhang, C.; Cathcart, S.J.; DiMaio, D.J.; Punsoni, M.; Guan, B.; Zhou, P.; et al. Androgen receptor, although not a specific marker for, is a novel target to suppress glioma stem cells as a therapeutic strategy for glioblastoma. Front. Oncol. 2021, 11, 616625. [Google Scholar] [CrossRef]
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Guerrero Cazares, H. Sex-specific differences in glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef]
- Yang, J.D.; Chen, J.T.; Liu, S.H.; Chen, R.M. Contribution of the testosterone-androgen receptor-PARD3B signaling axis to tumorigenesis and malignance of glioblastoma multiforme through stimulating cell proliferation and colony formation. J. Clin. Med. 2022, 11, 4818. [Google Scholar] [CrossRef]
- Li, Z.; Sun, C.; Tao, S.; Osunkoya, A.O.; Arnold, R.S.; Petros, J.A.; Zu, X.; Moreno, C.S. The JNK inhibitor AS602801 synergizes with enzalutamide to kill prostate cancer cells in vitro and in vivo and inhibit androgen receptor expression. Transl. Oncol. 2020, 13, 100751. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Cheng, C.; Lan, X.; Xing, R.; Chen, Z.; Zhao, H.; Sun, J.; Wang, Y.; Niu, C.; Zhang, B.; et al. Regulation of p53wt glioma cell proliferation by androgen receptor-mediated inhibition of small VCP/p97-interacting protein expression. Oncotarget 2017, 8, 23142–23154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.M.; Chen, L.; Chang, W.C.; Su, S.Y.; Hung, Y.C.; Ma, W.L. Androgen/androgen receptor signaling in ovarian cancer: Molecular regulation and therapeutic potentials. Int. J. Mol. Sci. 2021, 22, 7748. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Mizokami, A. Suppressive role of androgen/androgen receptor signaling via chemokines on prostate cancer cells. J. Clin. Med. 2019, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, Z.J.; Mirabal, J.R.; Mazur, D.J.; Kohn, T.P.; Lipshultz, L.I.; Pastuszak, A.W. Selective androgen receptor modulators: Current knowledge and clinical applications. Sex. Med. Rev. 2019, 7, 84–94. [Google Scholar] [CrossRef]
- Orozco, M.; Valdez, R.A.; Ramos, L.; Cabeza, M.; Segovia, J.; Romano, M.C. Dutasteride combined with androgen receptor antagonists inhibit glioblastoma U87 cell metabolism, proliferation, and invasion capacity: Androgen regulation. Steroids 2020, 164, 108733. [Google Scholar] [CrossRef]
- Zalcman, N.; Canello, T.; Ovadia, H.; Charbit, H.; Zelikovitch, B.; Mordechai, A.; Fellig, Y.; Rabani, S.; Shahar, T.; Lossos, A.; et al. Androgen receptor: A potential therapeutic target for glioblastoma. Oncotarget 2018, 9, 19980–19993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Chang, Y.A.; Lin, Y.L.; Chio, C.C.; Chen, R.M. Preclinical effects of honokiol on treating glioblastoma multiforme via G1 phase arrest and cell apoptosis. Phytomedicine 2016, 23, 517–527. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling pathways in glioblastoma and therapeutic implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [Green Version]
- Abazid, A.; Martin, B.; Choinowski, A.; McNeill, R.V.; Brandenburg, L.O.; Ziegler, P.; Zimmermann, U.; Burchardt, M.; Erb, H.; Stope, M.B. The androgen receptor antagonist enzalutamide induces apoptosis, dysregulates the heat shock protein system, and diminishes the androgen receptor and estrogen receptor β1 expression in prostate cancer cells. J. Cell. Biochem. 2019, 120, 16711–16722. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug resistance in glioblastoma: The two faces of oxidative stress. Front. Mol. Biosci. 2021, 7, 620677. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Yang, S.T.; Chen, R.M. Major contribution of caspase-9 to honokiol-induced apoptotic insults to human drug-resistant glioblastoma cells. Molecules 2020, 25, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Tu, V.C. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am. J. Cardiovas. Drugs 2002, 2, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N.; Krammer, P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012, 19, 36–41. [Google Scholar] [CrossRef]
- Pilling, A.B.; Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhang, J.; Wang, L.; Liu, H.; Wang, J.; Liu, J.; Liu, Z.; Zhu, Y.; Xu, Y.; Yang, W.; et al. Non-apoptotic function of caspase-8 confers prostate cancer enzalutamide resistance via NF-κB activation. Cell Death Dis. 2021, 12, 833. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak pores: Are we closing the circle? Trends Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a potentially clinical useful marker of mitochondrial and cellular damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.; Shi, Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Browne, G.; Melino, G.; Cohen, G.M. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 2009, 16, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Lee, Y.W.; Cherng, Y.G.; Yang, S.T.; Liu, S.H.; Chen, T.L.; Chen, R.M. Hypoxia induced by cobalt chloride triggers autophagic apoptosis of human and mouse drug-resistant glioblastoma cells through targeting the PI3K-Akt-mTOR signaling pathway. Oxidative Med. Cell. Long. 2021, 2021, 5558618. [Google Scholar] [CrossRef]
- Chio, C.C.; Chen, K.Y.; Chuang, J.Y.; Liu, C.C.; Liu, S.H.; Chen, R.M. Improved effects of honokiol on temozolomide-induced autophagy and apoptosis of drug-sensitive and -tolerant glioma cells. BMC Cancer 2018, 18, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, C.C.; Lin, J.W.; Cheng, H.A.; Chiu, W.T.; Wang, Y.H.; Wang, J.J.; Hsing, C.H.; Chen, R.M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013, 87, 458–468. [Google Scholar] [CrossRef]
- Lin, J.W.; Chen, J.T.; Hong, C.Y.; Lin, Y.L.; Wang, K.T.; Yao, C.J.; Lai, G.M.; Chen, R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic Bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro-Oncology 2012, 14, 302–314. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.M.; Lin, Y.L.; Chou, C.W. GATA-3 transduces survival signals in osteoblasts through upregulation of bcl-xL gene expression. J. Bone Min. Res. 2010, 25, 2193–2204. [Google Scholar] [CrossRef]
- Wu, G.J.; Chen, K.Y.; Yang, J.D.; Liu, S.H.; Chen, R.M. Naringin improves osteoblast mineralization and bone healing and strength through estrogen receptor alpha-dependent regulation of alkaline phosphatase gene expression. J Agric. Food Chem. 2021, 69, 13020–13033. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol improves temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondria-dependent pathway. Phytomedicine 2018, 49, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Chen, J.T.; Tsai, H.C.; Chen, T.L.; Liu, S.H.; Chen, R.M. Protection of dexmedetomidine against ischemia/reperfusion-induced apoptotic insults to neuronal cells occurs via an intrinsic mitochondria-dependent pathway. J. Cell. Biochem. 2017, 118, 2635–2644. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-Y.; Chen, J.-T.; Chen, T.-H.; Chen, R.-M. Enzalutamide Induces Apoptotic Insults to Human Drug-Resistant and -Sensitive Glioblastoma Cells via an Intrinsic Bax-Mitochondrion-Cytochrome C Caspase Cascade Activation Pathway. Molecules 2022, 27, 6666. https://doi.org/10.3390/molecules27196666
Chang C-Y, Chen J-T, Chen T-H, Chen R-M. Enzalutamide Induces Apoptotic Insults to Human Drug-Resistant and -Sensitive Glioblastoma Cells via an Intrinsic Bax-Mitochondrion-Cytochrome C Caspase Cascade Activation Pathway. Molecules. 2022; 27(19):6666. https://doi.org/10.3390/molecules27196666
Chicago/Turabian StyleChang, Chia-Yu, Jui-Tai Chen, Tso-Hsiao Chen, and Ruei-Ming Chen. 2022. "Enzalutamide Induces Apoptotic Insults to Human Drug-Resistant and -Sensitive Glioblastoma Cells via an Intrinsic Bax-Mitochondrion-Cytochrome C Caspase Cascade Activation Pathway" Molecules 27, no. 19: 6666. https://doi.org/10.3390/molecules27196666