
Asymmetric Conjugate Addition of Ketones to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide


Department of Organic Chemistry, Faculty of Sciences, Institute of Organic Synthesis (ISO), University of Alicante, Apdo. 99, 03080 Alicante, Spain
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(19), 6668; https://doi.org/10.3390/molecules27196668
Submission received: 26 August 2022
/
Revised: 29 September 2022
/
Accepted: 30 September 2022
/
Published: 7 October 2022
(This article belongs to the Section Organic Chemistry)
Abstract
:Enantioenriched substituted succinimides are interesting compounds, and their asymmetric organocatalytic synthesis by the conjugated addition of ketones to maleimides has been scarcely explored. This study shows the enantioselective conjugate addition of ketones to maleimides organocatalyzed by a simple primary amine-salicylamide derived from a chiral trans-cyclohexane-1,2-diamine, which provides the desired succinimides in good to excellent yields (up to 98%) and with moderate to excellent enantioselectivities (up to 99%).
1. Introduction
Succinimides are attractive targets in organic synthesis, as they are present in natural products and drug candidates [1,2,3,4,5,6,7] and can be transformed into other interesting compounds [8,9,10,11]. One of the most direct ways of preparing enantioenriched substituted succinimides is the organocatalytic enantioselective conjugate addition of carbon nucleophiles to maleimides [12]. Thus, using chiral organocatalysts containing tertiary amines, these carbon nucleophiles can be generated by the α-deprotonation of the acidic hydrogens of pro-nucleophiles, such as 1,3-dicarbonyl compounds [12]. The formed enolate can coordinate with the protonated amine and, if the organocatalyst bears an acidic moiety coordinating the maleimide employing a hydrogen bond, a close transition state can be produced, leading to an efficient enantioselective process. However, when aldehydes or ketones are used as pro-nucleophiles, α-deprotonation is difficult. Conjugate addition can occur by creating a transient nucleophilic enamine generated using primary or secondary amine-bearing chiral organocatalysts.
The organocatalytic enantioselective conjugate addition reaction of aldehydes to maleimides has been profusely studied [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. However, the same process involving ketones is challenging and has been explored by a limited number of researchers. Thus, the limited number of organocatalysts employed in this enantioselective reaction with ketones are the chiral sulfonamides 1 [46] and 2 [47] and the thiophosphoramide 3 [43] (Figure 1). In addition, the quinidine-derived thiourea 4 combined with an amino acid 5 [48], the diaminomethyleneindenedione 6 [49,50], O-tert-butyl-L-threonine (7) [44], and a tricomponent noncovalent organocatalytic system formed by L-isoleucine, thiourea, and potassium hydroxide [51] are used (Figure 1).
Recently, we have used the primary amine-salicylamide derived from chiral trans-cyclohexane-1,2-diamine 8 (Figure 2) as an effective organocatalyst for the enantioselective conjugate addition of aldehydes to maleimides [40,45]. We report here that this organocatalyst can be effective in the much less common asymmetric addition of ketones to maleimides.
2. Results and Discussion
The search for the most appropriate reaction conditions (Table 1) was carried out using the model conjugate addition reaction of cyclohexanone (9a) to N-phenylmaleimide (1a) (2/1 molar ratio). Thus, the reaction organocatalyzed by 8 (20 mol%) in toluene as a solvent at room temperature during 3 d afforded the corresponding substituted succinimide as a 76/24 mixture of diastereomers, the anti-major one 11aa in 94% ee. The absolute configuration of the succinimide was determined by comparing the elution order of the corresponding four isomers in chiral HPLC with those in the literature [46]. The use of other hydrocarbons as solvents did not provide better results (Table 1, entries 2 and 3). In addition, chlorinated solvents were employed (Table 1, entries 4–6), with dichloromethane providing the best diastereoselectivity (80/20) and enantioselectivity (99% ee) for the main diastereomer 11aa. On the other hand, ether solvents (Table 1, entries 7 and 8) provided considerably lower stereoselectivities, as did acetonitrile (Table 1, entry 9), alcohol solvents or water, and methanol had no reaction (Table 1, entries 10–12). Moreover, we were intrigued as to whether a longer reaction time would modify the final stereoselectivity for 11aa when using dichloromethane as the best solvent. However, when the reaction was performed in 5 d reaction time, the final diastereo- and enantio-selectivity remained unaltered (Table 1, compare entries 4 and 13).
Next, we explore the influence of adding additives to the reaction using dichloromethane as the best solvent. Thus, adding 10 mol% of different carboxylic acids as additives had a result detrimental to the stereoselectivity (Table 1, entries 14–17). Moreover, the use of a 10 mol% of organic bases such as 4-(dimethylamino)pyridine (DMAP), imidazole, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), or 2,6-lutidine proved ineffective (Table 1, entries 18–21). However, the use of 1,4-diazabicyclo[2.2.2]octane (DABCO) (10 mol%) as additive raised the anti/syn diastereomer ratio to 85/25, and the enantioselectivity for 11aa was 99% (Table 1, entry 22). The loading of DABCO was increased and lowered, however this was not beneficial in any case (Table 1, entries 23 and 24). Furthermore, the reaction temperature was reduced to 5 °C while keeping DABCO (10 mol%) as an additive in dichloromethane, resulting in a diastereomeric ratio of 93/7 favoring 11aa and a 99% ee (Table 1, entry 25; see Figures S1, S2 and S44 in Supplementary Materials).
With the optimized reaction conditions in hand [8 (20 mol%), DABCO (10 mol%), CH2Cl2, 5 °C], we extended the procedure to other substrates (see Figure 3 and Table 2; see Figures S3–S42 and Figures S45–S84 in Supplementary Materials). We first explored the reaction of cyclohexanone (9a) with other N-substituted maleimides (10b–h) (Figure 3 and Table 2). Thus, electron-releasing and electron-withdrawing groups on the aromatic ring of the N-arylated maleimide provided the corresponding major adducts 11ab–af (Figure 3) with good diastereoselectivities and with moderate enantioselectivities (Table 2, entries 2–6). In addition, when N-methyl or N-ethyl maleimide was used, the diastereoselectivity of the reaction for 11ag and 11ah was good, with similar enantioselectivities of 76 and 77% (Table 2, entries 7 and 8). Using the simple maleimide (10i) provided an excellent enantioselectivity for 11ai (Table 2, entry 9).
We extended this conjugate addition to other ketones. Thus, five- and seven-membered cyclic ketones 9b-d reacted with N-phenylmaleimide (10a) to provide the corresponding major succinimides 11ba-da (Figure 3), with rather a low diastereoselectivity but with an ee reaching 93% for 11ca using 2,2,dimethylcyclopentanone (9c) (Table 2, entries 10–12). In these three cases, the presence of an acid additive such as p-nitrobenzoic acid proved superior to DABCO, working at room temperature being necessary to avoid very slow reactions.
Heterocyclic ketones such as 1-Boc-piperidin-4-one (9e) and 1-propylpiperidin-4-one (9f) were used, obtaining low diastereoselectivities and moderate enantioselectivities of 11ea and 11fa, respectively (Table 2, entries 13 and 14). In contrast, tetrahydro-4H-pyran-4-one (9g) provided low diastereoselectivity and a very high enantioselectivity of 11ga (Table 2, entry 15). These results were obtained working at room temperature.
Acyclic ketones were employed in the reaction with N-phenylmaleimide. Thus, acetone yielded succinimide 11ha in a 72% ee (Table 2, entry 16), whereas 1-phenylpropan-2-one (9i) afforded 11ia with very low diastereoselectivity and high enantioselectivity for 11ia (Table 2, entry 17). In addition, the reactions with 1-methoxypropan-2-one (9j) and 1-phenoxypropan-2-one (9k) yielded moderate diastereoselectivity and adducts 11ja and 11ka in low and moderate enantioselectivities, respectively (Table 2, entries 18 and 19).
Finally, and considering the good results obtained using the simple maleimide 10i as an electrophile, we explored the reaction with other ketones such as cycloheptanone (9d) and tetrahydro-4H-pyran-4-one (9g), obtaining the corresponding adducts 11di and 11gi, again with high enantioselections (Table 2, entries 20 and 21).
We scaled up the reaction leading to 11aa using 7.4 mmol of 9a and 3.7 mmol of 10a instead of 0.4 mmol of 9a and 0.2 mmol of 10a (see Materials and Methods). The corresponding adduct 11aa was obtained with a 97% ee (Table 2, entry 20).
Based on previous DFT calculations concerning the favorable transition states in the conjugated addition reaction of aldehydes to maleimides when using 8 [40], a suggested approach justifying the formation of 11aa is depicted in Figure 4. Here, 8 would act as a bifunctional species, forming a transient enamine and at the same time coordinating one of the carbonyl groups of the maleimide through a hydrogen bond involving the amide N-H in the organocatalyst.
3. Materials and Methods
3.1. General Information
Commercially available reagents (Acros Organics, Alfa Aesar, Fluorochem, Sigma Aldrich, TCI Chemicals) were used without further purification. 1H NMR spectra were recorded on Bruker AV300 (300 MHz) and Bruker AV400 (400 MHz) spectrometers in proton coupled mode at room temperature. 13C NMR spectra were recorded on Bruker AV300 (75 MHz) and Bruker AV400 (101 MHz) spectrometers in proton decoupled mode at room temperature. Chemical shifts (δ) are given in parts per million (ppm). CDCl3 was used as solvent and tetramethylsilane (TMS) as internal standard. Coupling constants (J) are given in Hz. Infrared (IR) spectra were recorded with an ATR Jasco FT/IR-4100 from neat samples. Wavenumbers (υ) are given in cm−1 and the intensity is provided as very strong (vs), strong (s), weak (w), or broad (br). High resolution mass spectrometry data were obtained on an Agilent 7200 Q-TOF (EI-QTOF) and on an Agilent 1260 Chip-HPLC in line with a mass spectrometer 6500 series Q-TOF (ESI-QTOF). Thin layer chromatography (TLC) was carried out on Macherey–Nagel Alugram Sil G UV254 aluminum sheets coated with a 0.2 mm layer of silica gel. A UV light lamp (254 nm) was employed for detection. Flash column chromatography was performed using silica gel 60 of 40–63 µm (230–400 mesh) size. The ee’s were determined on an Agilent 1100 Series HPLC equipped with an Agilent G1311A quaternary pump and an Agilent G1315B diode array detector (DAD). The employed conditions (column, mobile phase, flow rate, wavelength) are shown in each case. Reference racemic samples of adducts 11 were obtained by performing the conjugate addition reactions using a racemic mixture of 8 and ent-8 as organocatalyst at room temperature.
3.2. Enantioselective Michael Addition of Ketones to Maleimides: General Procedure
A mixture of organocatalyst 8 (9.4 mg, 0.04 mmol), DABCO (4.5 mg, 0.02 mmol), and 10 (0.2 mmol) was dissolved in CH2Cl2 (1 mL) in a glass vial (16 mm diameter). Then, 9 (0.4 mmol) was added and the mixture was stirred for 4 or 5 days at 5 °C (see Table 2). After this time, the solvent was evaporated under reduced pressure (15 torr) and the crude reaction mixture was purified by column chromatography (hexanes/ethyl acetate gradients), yielding adduct 11.
3-(2-Oxocyclohexyl)-1-phenylpyrrolidine-2,5-dione (11aa) [46]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 98% global yield (anti/syn = 93/7, 1H NMR on the reaction crude). White solid, mp: 131–133 °C; 1H NMR (400 MHz, CDCl3): δ = 7.47 (tt, J = 8.7, 1.7 Hz, 2H), 7.41–7.28 (m, 3H), 3.34–3.13 (m, 1H), 3.11–2.78 (m, 2H), 2.67–2.51 (m, 1H), 2.40–2.28 (m, 2H), 2.15 (dddq, J = 20.2, 12.7, 6.7, 3.4 Hz, 2H), 1.98 (m, 1H), 1.82–1.55 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.4 (major), 210.2 (minor), 178.7 (minor), 178.5 (major), 175.9 (major), 175.8 (minor), 132.5 (major), 132.2 (minor), 129.2 (major), 128.7 (minor), 128.6 (major), 126.9 (major), 126.7 (minor), 52.3 (minor), 50.9 (major), 42.0 (minor), 41.8 (major), 41.4 (major), 40.2 (minor), 33.5 (minor), 32.1 (major), 31.9 (major), 30.4 (minor), 27.3 (minor), 27.1 (major), 25.2 (minor), 25.0 (major) ppm; HPLC: ChiralCel OD-H column, hexanes/i-PrOH (70/30), flow rate = 0.5 mL/min, λ = 230 nm, tR (anti) = 31.1 min (major enantiomer) 58.7 min (minor enantiomer), tR (syn) = 39.4 min (major enantiomer) 43.4 min (minor enantiomer).
1-(4-Methylphenyl)-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ab). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 91% global yield (anti/syn = 90/10, 1H NMR on the reaction crude). White solid, mp: 144–146 °C; 1H NMR (300 MHz, CDCl3): δ = 7.30–7.25 (m, 2H), 7.24–7.17 (m, 2H), 3.34–3.15 (m, 1H), 2.68–2.53 (m, 1H), 2.48–2.27 (m, 5H, CH3 included), 2.23–2.07 (m, 2H), 2.03–1.93 (m, 1H), 1.86–1.52 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 210.4 (major), 210.2 (minor), 178.8 (minor), 178.7 (major), 176.1 (major), 176.0 (minor), 138.7 (major), 129.9 (major), 129.8 (minor), 126.7 (major), 126.5 (minor), 52.2 (minor), 50.9 (major), 42.1 (minor), 41.9 (major), 41.4 (major), 40.2 (minor), 33.4 (minor), 32.2 (major), 31.9 (major), 30.2 (minor), 29.8 (minor), 27.3 (minor), 27.1 (major), 25.2 (minor), 25.1 (major), 21.4 (major) ppm; IR (neat): υ = 2939 (w), 2862 (w), 1778 (w), 1697 (vs), 1647 (w), 1516 (w), 1400 (w), 1200 (w), 1122 (w), 1041 (w), 818 (w), 768 (w), 663 (w) cm−1; MS (70 eV, EI): m/z (%): 67.2 (17), 107.2 (49), 133.1 (29), 150.1 (15), 189.1 (100), 228.1 (22), 285.1 (M+, 79); HRMS (EI-QTOF) calcd. for C17H19NO3 (M+): 285.1365. Found: 285.1357; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 1.0 mL/min, λ = 254 nm, tR (anti) = 20.70 min (major enantiomer) 17.44 min (minor enantiomer), tR (syn) = 22.49 min (major enantiomer) 19.42 min (minor enantiomer).
1-(4-Methoxyphenyl)-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ac). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 90% global yield (anti/syn = 85/15, 1H NMR on the reaction crude). Yellow solid, mp: 120–122 °C; 1H NMR (400 MHz, CDCl3): δ = 7.28–7.19 (m, 2H), 7.00–6.95 (m, 2H), 3.81 (s, 3H), 3.31–3.12 (m, 1H), 3.10–2.76 (m, 2H), 2.67–2.50 (m, 1H), 2.48–2.27 (m, 2H), 2.21–2.03 (m, 2H), 2.02–1.94 (m, 1H), 1.81–1.55 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.4 (major), 201.2 (minor), 178.9 (minor), 178.8 (major), 176.2 (major), 176.1 (minor), 159.6 (major), 128.1 (major), 127.9 (minor), 125.1 (major), 124.9 (minor), 114.6 (major), 55.6 (major), 52.3 (minor), 50.9 (major), 42.0 (minor), 41.8 (major), 41.3 (major), 40.1 (minor), 33.4 (minor), 32.1 (major), 31.9 (major), 30.3 (minor), 29.8 (minor), 27.3 (minor), 27.1 (major), 25.1 (minor), 25.0 (major) ppm; IR (neat): υ = 2931 (w), 2858 (w), 1778 (w), 1697 (vs), 1512 (s), 1396 (w), 1246 (s), 1184 (s), 1026 (w), 833 (w), 663 (w) cm−1; MS (70 eV, EI): m/z (%): 123.1 (46), 205.1 (45), 244.1 (22), 301.1 (M+, 100); HRMS (EI-QTOF) calcd. for C17H19NO4 (M+): 301.1314. Found: 301.1308; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 0.8 mL/min, λ = 240 nm, tR (anti) = 34.38 min (major enantiomer) 29.47 min (minor enantiomer), tR (syn) = 41.26 min (major enantiomer) 45.35 min (minor enantiomer).
1-(4-Acetylphenyl)-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ad). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 82% global yield (anti/syn = 88/12, 1H NMR on the reaction crude). White solid, mp: 128–130 °C; 1H NMR (400 MHz, CDCl3): δ = 8.12–8.00 (m, 2H), 7.56–7.42 (m, 2H), 3.33–3.13 (m, 1H), 3.12–2.79 (m, 2H), 2.76–2.53 (m, 4H, CH3 included), 2.52–2.30 (m, 2H), 2.25–2.08 (m, 2H), 2.06–1.96 (m, 1H), 1.85–1.56 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.5 (major), 210.3 (minor), 197.3 (major), 137.2 (minor), 178.2 (minor), 178.1 (major), 175.4 (major), 175.2 (minor), 136.7 (major), 136.6 (major), 136.4 (minor), 129.2 (major), 126.8 (major), 126.7 (minor), 52.6 (minor), 51.0 (major), 42.0 (minor), 41.8 (major), 41.4 (major), 40.4 (minor), 33.8 (minor), 32.2 (major), 31.9 (major), 30.9 (minor), 29.8 (minor), 27.3 (minor), 27.1 (major), 26.8 (major), 25.2 (minor), 25.0 (major) ppm; IR (neat): υ = 2931 (w), 2866 (w), 1782 (w), 1697 (vs), 1601 (w), 1512 (w), 1400 (s), 1257 (w), 1192 (s), 1126 (w), 957 (w), 837 (w), 744 (w), 663 (w) cm−1; MS (70 eV, EI): m/z (%): 97.1 (15), 120.1 (22), 146.1 (21), 202.1 (25), 217.1 (100), 298.1 (65), 313.1 (M+, 54); HRMS (EI-QTOF) calcd. for C18H19NO4 (M+): 313.1314. Found: 313.1308; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 0.8 mL/min, λ = 230 nm, tR (anti) = 58.77 min (major enantiomer) 53.11 min (minor enantiomer), tR (syn) = 82.26 min (major enantiomer) 181.91 min (minor enantiomer).
1-(4-Bromophenyl)-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ae) [46]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 95% global yield (anti/syn = 92/8, 1H NMR on the reaction crude). White solid, mp: 148–150 °C; 1H NMR (300 MHz, CDCl3): δ = 7.64–7.54 (m, 2H), 7.27–7.18 (m, 2H), 3.33–3.09 (m, 1H), 3.10–2.74 (m, 2H), 2.67–2.47 (m, 1H), 2.46–2.25 (m, 2H), 2.22–2.06 (m, 2H), 2.03–1.92 (m, 1H), 1.85–1.50 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 210.5 (major), 210.3 (minor), 178.3 (minor), 178.2 (major), 175.5 (major), 175.4 (minor), 132.4 (major), 131.5 (major), 131.3 (minor), 128.5 (major), 128.3 (minor), 122.5 (major), 52.6 (minor), 51.0 (major), 42.1 (minor), 41.8 (major), 41.4 (major), 40.3 (minor), 33.7 (minor), 32.1 (major), 31.9 (major), 30.9 (minor), 27.3 (minor), 27.1 (major), 25.2 (minor), 25.0 (major) ppm; HPLC: ChiralCel OD-H column, hexanes/i-PrOH (70/30), flow rate = 0.5 mL/min, λ = 230 nm, tR (anti) = 40.14 min (major enantiomer) 63.08 min (minor enantiomer), tR (syn) = 46.56 min (major enantiomer) 52.85 min (minor enantiomer).
1-(4-Nitrophenyl)-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11af). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 80% global yield (anti/syn = 84/16, 1H NMR on the reaction crude). Yellow solid, mp: 116–118 °C; 1H NMR (400 MHz, CDCl3): δ = 8.39–8.28 (m, 2H), 7.64–7.57 (m, 2H), 3.40–3.16 (m, 1H), 3.15–2.83 (m, 2H), 2.72–2.54 (m, 1H), 2.52–2.30 (m, 2H), 2.26–2.11 (m, 2H), 2.07–1.96 (m, 1H), 1.87–1.56 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.87 (major), 210.5 (minor), 177.8 (minor), 177.7 (major), 175.0 (major), 174.8 (minor), 147.4 (minor), 147.1 (major), 138.1 (major), 137.9 (minor), 127.4 (major), 127.3 (minor), 124.6 (minor), 124.4 (major), 53.0 (minor), 21.1 (major), 42.1 (minor), 41.7 (major), 41.4 (major), 40.5 (minor), 34.1 (minor), 32.1 (major), 31.8 (major), 31.5 (minor), 29.8 (major), 27.4 (minor), 27.0 (major), 25.3 (minor), 25.0 (major) ppm; IR (neat): υ = 2931 (w), 2862 (w), 1786 (w), 1705 (vs), 1647 (w), 1523 (s), 1389 (w), 1342 (s), 1184 (s), 849 (w), 690 (w) cm−1; MS (70 eV, EI): m/z (%): 55.1 (32), 68.1 (22), 97.1 (100), 220.1 (37), 316.1 (M+, 17); HRMS (EI-QTOF) calcd. for C16H16N2O5 (M+): 316.1059. Found: 316.1041; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (80/20), flow rate = 0.6 mL/min, λ = 254 nm, tR (anti) = 146.02 min (major enantiomer) 95.57 min (minor enantiomer), tR (syn) = 156.09 min (major enantiomer) 174.45 min (minor enantiomer).
1-Methyl-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ag) [43]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 94% global yield (anti/syn = 91/9, 1H NMR on the reaction crude). White solid, mp: 90–92 °C; 1H NMR (400 MHz, CDCl3): δ = 3.21–3.08 (m, 1H), 3.02 (s, 3H), 2.98–2.63 (m, 2H), 2.52–2.42 (m, 1H), 2.41–2.27 (m, 2H), 2.14 (dddq, J = 19.4, 12.1, 6.2, 3.1 Hz, 2H), 2.01–1.92 (m, 1H), 1.81–1.51 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.2 (major), 210.1 (minor), 179.7 (minor), 179.4 (major), 176.9 (major), 51.4 (minor), 50.4 (major), 42.0 (minor), 41.9 (major), 41.4 (major), 40.1 (minor), 32.7 (minor), 32.1 (major), 32.0 (major), 29.6 (minor), 27.3 (minor), 27.2 (major), 25.1 (major), 25.0 (minor), 25.0 (major), 24.9 (minor) ppm; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 1.0 mL/min, λ = 210 nm, tR (anti) = 28.24 min (major enantiomer) 34.68 min (minor enantiomer), tR (syn) = 114.20 min (major enantiomer) 82.43 min (minor enantiomer).
1-Ethyl-3-(2-oxocyclohexyl)pyrrolidine-2,5-dione (11ah) [43]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 93% global yield (anti/syn = 83/17, 1H NMR on the reaction crude). White solid, mp: 96–98 °C; 1H NMR (400 MHz, CDCl3): δ = 3.65–3.48 (m, 2H), 3.22–3.06 (m, 1H), 3.03–2.60 (m, 2H), 2.50–2.40 (m, 1H), 2.39–2.24 (m, 2H), 2.20–2.05 (m. 2H), 2.01–1.91 (m, 1H), 1.83–1.49 (m, 3H), 1.25–1.13 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.2 (major), 210.1 (minor), 179.4 (minor), 179.2 (major), 176.7 (major), 51.6 (minor), 50.5 (major), 42.0 (minor), 41.9 (major), 41.3 (major), 39.9 (minor), 33.9 (major), 33.8 (minor), 32.8 (minor), 32.0 (major), 31.9 (major), 29.5 (minor), 27.3 (minor), 27.1 (major), 25.1 (major), 13.0 (minor), 12.7 (major) ppm; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 1.0 mL/min, λ = 210 nm, tR (anti) = 19.81 min (major enantiomer) 27.62 min (minor enantiomer), tR (syn) = 32.04 min (major enantiomer) 78.12 min (minor enantiomer).
3-(2-Oxocyclohexyl)pyrrolidine-2,5-dione (11ai). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 92% global yield (anti/syn = 76/24, 1H NMR on the reaction crude). White solid, mp: 159–161 °C; 1H NMR (400 MHz, CDCl3): δ = 8.63 (br s, 1H), 3.22–3.09 (m, 1H), 3.01–2.66 (m, 2H), 2.59–2.45 (m, 1H), 2.44–2.28 (m, 2H), 2.19–2.07 (m, 2H), 2.00–1.86 (m, 1H), 1.83–1.47 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 210.3 (major), 210.1 (minor), 180.1 (minor), 179.7 (major), 177.2 (minor), 177.0 (major), 51.3 (minor), 50.3 (major), 42.6 (major), 42.0 (minor), 41.8 (major), 41.4 (minor), 33.8 (minor), 33.2 (major), 32.0 (major), 29.5 (minor), 27.3 (minor), 27.2 (major), 25.0 (major) ppm; IR (neat): υ = 3244 (br), 3039 (br), 2943 (w), 2873 (w), 1766 (w), 1693 (vs), 1354 (w), 1180 (w), 833 (w), 791 (w), 737 (w) cm−1; MS (70 eV, EI): m/z (%): 55.1 (17), 97.1 (100), 99.1 (65), 195.1 (M+, 27); HRMS (EI-QTOF) calcd. for C10H13NO3 (M+): 195.0895. Found: 195.0888; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 1.0 mL/min, λ = 254 nm, tR (anti) = 56.48 min, tR (syn) = 25.46 min.
3-(2-Oxocyclopentyl)-1-phenylpyrrolidine-2,5-dione (11ba) [50]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 89% global yield (anti/syn = 51/49, 1H NMR on the reaction crude). Brown solid, mp: 66–68 °C; 1H NMR (300 MHz, CDCl3): δ = 7.53–7.32 (m, 4H, major + minor), 7.31–7.20 (m, 1H, major + minor), 3.54–3.20 (m, 1H, major + minor), 3.09–2.79 (m, 2H, major + minor), 2.65–2.23 (m, 3H, major + minor), 2.22–2.06 (m, 2H, major + minor), 1.98–1.80 (m, 1H, major + minor), 1.76–1.59 (m, 1H, major + minor) ppm; 13C NMR (75 MHz, CDCl3): δ = 218.2 (minor), 217.7 (major), 178.2 (minor), 177.9 (major), 175.3 (major), 175.2 (minor), 132.1 (major), 131.9 (minor), 129.3 (major), 128.9 (minor), 128.8 (major), 126.7 (major), 126.6 (minor), 50.4 (major), 50.2 (minor), 39.6 (major), 38.8 (minor), 37.8 (minor), 37.7 (major), 32.5 (major), 32.4 (minor), 27.2 (major), 25.5 (minor), 20.8 (major), 20.5 (minor) ppm; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 0.5 mL/min, λ = 230 nm, tR (anti) = 36.08 min (major enantiomer) 33.94 min (minor enantiomer), tR (syn) = 39.87 min (major enantiomer) 33.40 min (minor enantiomer).
3-(3,3-Dimethyl-2-oxocyclopentyl)-1-phenylpyrrolidine-2,5-dione (11ca). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 65% global yield (anti/syn = 64/36, 1H NMR on the reaction crude). White solid, mp: 95–97 °C; 1H NMR (400 MHz, CDCl3): δ = 7.51–7.45 (m, 2H), 7.43–7.34 (m, 2H), 7.28–7.24 (m, 1H), 3.51–3.19 (m, 1H), 3.12–2.93 (m, 2H), 2.61–2.36 (m, 1H), 2.25–2.02 (m, 1H), 1.95–1.67 (m, 3H), 1.13 (d, J = 5.5 Hz, 3H), 1.02 (d, J = 9.3 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 222.0 (minor), 221.4 (major), 178.2 (minor), 177.9 (major), 175.4 (major), 175.3 (minor), 132.2 (minor), 132.0 (major), 129.4 (major), 128.9 (minor), 128.8 (major), 126.8 (major), 126.7 (minor), 50.4 (major), 50.0 (minor), 44.9 (minor), 44.8 (major), 39.6 (major), 39.0 (minor), 36.6 (major), 36.3 (minor), 32.6 (major), 25.1 (minor), 25.0 (major), 23.9 (minor), 23.8 (major), 23.7 (minor), 21.9 (major) ppm; IR (neat): υ = 2970 (s), 2904 (s), 1782 (w), 1705 (vs), 1500 (w), 1458 (w), 1389 (s), 1242 (w), 1184 (s), 1061 (vs), 879 (w), 760 (w), 694 (w) cm−1; MS (70 eV, EI): m/z (%): 54.1 (21), 111.1 (28), 119.1 (41), 175.1 (100), 201.1 (28), 285.1 (M+, 28); HRMS (EI-QTOF) calcd. for C17H19NO3 (M+): 285.1365. Found: 285.1363; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 0.7 mL/min, λ = 254 nm, tR (anti) = 60.81 min (major enantiomer) 44.23 min (minor enantiomer), tR (syn) = 65.94 min (major enantiomer) 55.30 min (minor enantiomer).
3-(2-Oxocycloheptyl)-1-phenylpyrrolidine-2,5-dione (11da) [47]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 92% global yield (anti/syn = 66/34, 1H NMR on the reaction crude). Grey solid, mp: 102–104 °C; 1H NMR (300 MHz, CDCl3): δ = 7.51–7.43 (m, 2H), 7.42–7.35 (m, 1H), 7.31–7.28 (m, 2H), 3.58–3.23 (m, 1H), 3.12–2.92 (m, 1H), 2.91–2.70 (m, 1H), 2.69–2.54 (m, 1H), 2.52–2.33 (m, 1H), 2.08–1.95 (m, 2H), 1.94–1.52 (m, 5H), 1.49–1.10 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ = 213.9 (major), 213.2 (minor), 178.6 (major), 176.0 (major), 175.9 (minor), 132.4 (major), 132.1 (minor), 129.2 (major), 128.7 (minor), 128.6 (major), 126.8 (major), 126.6 (minor), 52.8 (minor), 51.8 (major), 43.7 (major), 43.5 (minor), 43.4 (major), 42.3 (minor), 33.0 (minor), 31.9 (major), 30.1 (minor), 29.9 (major), 29.8 (major), 29.7 (minor), 29.4 (major), 28.7 (minor), 23.4 (major) ppm ; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 0.8 mL/min, λ = 230 nm, tR (anti) = 30.73 min (major enantiomer) 67.64 min (minor enantiomer), tR (syn) = 28.47 min (major enantiomer) 25.08 min (minor enantiomer).
3-(4-Oxo-1-tert-butoxycarbonylpiperidine-3-yl)-1-phenylpyrrolidin-2,5-dione (11ea). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 94% global yield (anti/syn = 56/44, 1H NMR on the reaction crude). Yellow solid, mp: 74–76 °C; 1H NMR (300 MHz, CDCl3): δ = 7.55–7.44 (m, 2H, major + minor), 7.43–7.37 (m, 1H, major + minor), 7.36–7.29 (m, 2H, major + minor), 4.66–4.20 (m, 2H, major + minor), 3.51–3.03 (m, 3H, major + minor), 2.98–2.81 (m, 2H, major + minor), 2.68–2.35 (m, 3H, major + minor), 1.51 (s, 9H, major + minor) ppm; 13C NMR (75 MHz, CDCl3): δ = 207.3 (minor), 207.0 (major), 178.1 (minor), 177.6 (major), 175.3 (major), 175.2 (minor), 154.8 (minor), 154.3 (major), 132.3 (major), 132.2 (minor), 129.3 (major), 128.8 (major), 126.8 (major), 81.2 (major), 81.0 (minor), 51.9 (minor), 50.1 (major), 41.3 (minor), 41.1 (major), 38.7 (major), 38.5 (minor), 31.9 (major), 28.5 (minor), 28.4 (major) ppm; IR (neat): υ = 2989 (w), 2935 (w), 1778 (w), 1701 (vs), 1500 (w), 1389 (s), 1242 (w), 1165 (s), 864 (w), 756 (w); 694 (w) cm−1; HRMS (ESI-QTOF) calcd. for C20H24N2O5Na [(M+Na)+]: 395.1583. Found: 395.1582; HPLC: ChiralPak IC column, hexanes/i-PrOH (70/30), flow rate = 1.0 mL/min, λ = 230 nm, tR (anti) = 166.10 min (major enantiomer) 106.81 min (minor enantiomer), tR (syn) = 178.57 min (major enantiomer) 42.19 min (minor enantiomer).
3-(4-Oxo-1-propylpiperidin-3-yl)-1-phenylpyrrolidine-2,5-dione (11fa). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 91% global yield (anti/syn = 65/35, 1H NMR on the reaction crude). Red solid, mp: 68–70 °C; 1H NMR (300 MHz, CDCl3): δ = 7.52–7.44 (m, 2H), 7.42–7.38 (m, 1H), 7.37–7.29 (m, 2H), 3.56–3.24 (m, 1H), 3.23–2.94 (m, 3H), 2.92–2.79 (m, 1H), 2.78–2.56 (m, 2H), 2.55–2.16 (m, 5H), 1.63–1.47 (m, 2H), 1.00–0.90 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 208.5 (minor), 208.4 (major), 178.2 (minor), 178.0 (major), 175.6 (major), 175.5 (minor), 132.4 (major), 132.2 (minor), 129.3 (major), 128.7 (major), 126.8 (major), 126.7 (minor), 59.1 (major), 57.1 (major), 56.6 (minor), 53.3 (major), 53.2 (minor), 51.4 (minor), 50.1 (major), 41.1 (minor), 41.0 (major), 39.3 (major), 39.0 (minor), 34.4 (minor), 32.5 (major), 20.8 (major), 12.0 (minor), 11.9 (major) ppm; IR (neat): υ = 2981 (w), 2924 (w), 1782 (w), 1705 (vs), 1647 (w), 1500 (w), 1385 (s), 1184 (s), 756 (w), 694 (s) cm−1; MS (70 eV, EI): m/z (%): 110.1 (<10), 140.1 (100), 256.1 (<10), 285.2 (M+, 26); HRMS (EI-QTOF) calcd. for C18H22N2O3 (M+): 314.1630. Found: 314.1619; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (70/30), flow rate = 0.8 mL/min, λ = 240 nm, tR (anti) = 16.20 min (major enantiomer) 26.53 min (minor enantiomer), tR (syn) = 17.44 min (major enantiomer) 13.43 min (minor enantiomer).
3-(4-Oxotetrahydro-2H-pyran-3-yl)-1-phenylpyrrolidine-2,5-dione (11ga) [46]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 93% global yield (anti/syn = 58/42, 1H NMR on the reaction crude). Yellow solid, mp: 109–111 °C; 1H NMR (300 MHz, CDCl3): δ = 7.55–7.44 (m, 2H, major + minor), 7.43–7.36 (m, 1H, major + minor), 7.35–7.28 (m, 2H, major + minor), 4.47–4.08 (m, 2H, major + minor), 3.79–3.60 (m, 1H, major + minor), 3.58–3.43 (m, 1H, major + minor), 3.18–2.80 (m, 2H, major + minor), 2.79–2.57 (m, 2H, major + minor), 2.56–2.28 (m, 2H, major + minor) ppm; 13C NMR (75 MHz, CDCl3): δ = 206.3 (minor), 205.9 (major), 178.0 (minor), 177.5 (major), 175.3 (major), 175.2 (minor), 132.2 (major), 132.1 (minor), 129.3 (major), 128.8 (minor), 126.8 (major), 70.9 (minor), 70.7 (major), 53.0 (minor), 51.5 (major), 42.6 (minor), 42.5 (major), 37.4 (major), 34.5 (minor), 32.2 (major); 29.8 (minor) ppm; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 0.6 mL/min, λ = 230 nm, tR (anti) = 197.01 min, tR (syn) = 177.10 min.
3-(2-Oxopropyl)-1-phenylpyrrolidine-2,5-dione (11ha) [49]. The product was obtained in 71% yield. White solid, mp: 101–103 °C; 1H NMR (300 MHz, CDCl3): δ = 7.52–7.37 (m, 3H), 7.35–7.29 (m, 2H), 3.22–3.03 (m, 4H), 2.63–2.51 (m, 1H), 2.22 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 205.7, 178.7, 175.6, 132.2, 129.3, 128.8, 126.7, 43.7, 35.7, 34.8, 29.9 ppm; HPLC: ChiralCel OD-H column, hexanes/i-PrOH (80/20), flow rate = 0.8 mL/min, λ = 230 nm, tR (S) = 45.40 min, tR (R) = 50.79 min.
3-(2-Oxo-1-phenylpropyl)-1-phenylpyrrolidine-2,5-dione (11ia). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 56% global yield (anti/syn = 52/48, 1H NMR on the reaction crude). Colorless oil; 1H NMR (300 MHz, CDCl3): δ = 7.52–7.29 (m, 14H, major + minor), 7.25–7.15 (m, 4H, major + minor), 7.08–6.99 (m, 2H, major + minor), 4.60 (s, J = 4.3 Hz, 1H, major), 4.48 (d, J = 4.9 Hz, 1H, minor), 3.77 (dt, J = 9.4, 5.1, Hz, 1H, major), 3.16 (ddd, J = 9.6, 5.9, 4.4 Hz, 1H, minor), 3.09–2.96 (m, 2H, major), 2.70–2.53 (m, 2H, minor), 2.20 (s, 3H, major), 2.11 (s, 3H, minor) ppm; 13C NMR (75 MHz, CDCl3): δ = 207.4 (minor), 206.3 (major), 178.1 (major), 175.7 (minor), 175.3 (major), 136.0 (major), 133.5 (minor), 132.3 (minor), 131.8 (major), 129.8 (major), 129.6 (minor), 129.5 (major), 129.4 (minor), 129.3 (major), 128.8 (major), 128.7 (major), 128.5 (minor), 126.7 (major), 126.5 (minor), 58.8 (minor), 58.3 (major), 43.8 (major), 41.9 (minor), 32.1 (major), 32.0 (minor), 29.2 (major), 29.1 (minor) ppm; IR (neat): υ = 2962 (w), 2908 (w), 1778 (w), 1705 (vs), 1500 (w), 1385 (w), 1257 (w), 1176 (w), 1080 (s), 1022 (vs), 795 (vs), 694 (s) cm−1; MS (70 eV, EI): m/z (%): 91.1 (14), 115.1 (19), 117.1 (24), 174.1 (16), 265.1 (100), 266.1 (19), 307.1 (M+, <10) ; HRMS (EI-QTOF) calcd. for C19H17NO3 (M+): 307.1208. Found: 307.1210; HPLC: ChiralPak AD-H column, hexanes/i-PrOH (90/10), flow rate = 1.0 mL/min, λ = 280 nm, tR (anti) = 42.99 min (major enantiomer) 55.30 min (minor enantiomer), tR (syn) = 46.98 min (major enantiomer) 38.04 min (minor enantiomer).
3-(1-Methoxy-2-oxopropyl)-1-phenylpyrrolidine-2,5-dione (11ja) [50]. The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 94% global yield (anti/syn = 62/38, 1H NMR on the reaction crude). Yellow solid, mp: 67–69 °C; 1H NMR (300 MHz, CDCl3): δ = 7.53–7.43 (m, 3H), 7.33–7.25 (m, 2H), 4.39 (d, J = 2.3 Hz, 1H), 3.51 (s, 3H), 3.50–3.44 (m, 1H), 2.84–2.77 (m, 2H), 2.29 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 207.8 (major), 176.9 (major), 175.8 (major), 132.0 (major), 129.4 (major), 129.3 (minor), 128.9 (major), 128.8 (minor), 126.7 (minor), 126.5 (major), 86.3 (minor), 84.5 (major), 60.3 (major), 60.0 (minor), 42.6 (major), 42.2 (minor), 31.8 (minor), 29.2 (major), 27.2 (minor), 27.1 (major) ppm; HPLC: ChiralPak IB column, hexanes/i-PrOH (80/20), flow rate = 0.8 mL/min, λ = 240 nm, tR (anti) = 23.87 min (major enantiomer) 22.10 min (minor enantiomer), tR (syn) = 24.79 min (major enantiomer) 26.65 min (minor enantiomer).
3-(1-Benzyloxy-2-oxopropyl)-1-phenylpyrrolidine-2,5-dione (11ka). The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 96% global yield (anti/syn = 75/25, 1H NMR on the reaction crude). Yellow solid, mp: 85–87 °C; 1H NMR (300 MHz, CDCl3): δ = 7.44–7.30 (m, 9H), 7.21–7.18 (m, 1H), 4.84–4.53 (m, 3H), 3.56–3.48 (m, 1H), 2.88–2.78 (, 2H), 2.25 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 208.9 (minor), 207.6 (major), 177.0 (major), 175.8 (minor), 175.4 (minor), 175.2 (major), 136.7 (major), 136.4 (minor), 132.0 (minor), 131.9 (major), 129.3 (major), 129.3 (minor), 128.9 (major), 128.8 (major), 128.8 (minor), 128.7 (major), 128.5 (minor), 128.3 (minor), 128.2 (major), 126.7 (minor), 126.5 (major), 83.6 (minor), 82.4 (major), 74.6 (major), 73.9 (minor), 42.7 (major), 42.4 (minor), 31.8 (minor), 29.4 (major), 27.4 (minor), 27.3 (major) ppm; IR (neat): υ = 2939 (w), 2870 (w); 1786 (w), 1705 (vs), 1647 (w), 1597 (w), 1539 (w), 1496 (w), 1454 (w), 1389 (s), 1308 (w), 1192 (s), 1122 (w), 1049 (w), 945 (w), 744 (s), 694 (s) cm−1; HRMS (ESI-QTOF) calcd. for C20H20NO4 [(M+H)+]: 338.1392. Found: 338.1393; HPLC: ChiralCel OD-H column, hexanes/i-PrOH (80/20), flow rate = 1.0 mL/min, λ = 230 nm, tR (anti) = 31.89 min (major enantiomer) 26.74 min (minor enantiomer), tR (syn) = 95.87 min (major enantiomer) 49.79 min (minor enantiomer).
3-(2-Oxocycloheptyl)pyrrolidine-2,5-dione (11di): The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 85% global yield (anti/syn = 60/40, 1H NMR on the reaction crude). Yellow solid, mp: 71–73 °C; 1H NMR (500 MHz, CDCl3): δ = 8.27 (br s, 1H), 3.30–3.11 (m, 1H), 2.91–2.79 (m, 1H), 2.71–2.47 (m, 2H), 2.45–2.28 (m, 1H), 1.97–1.73 (m, 3H), 1.71–1.19 (m, 6H) ppm; 13C NMR (125 MHz, CDCl3): δ = 213.7 (major), 213.1 (minor), 179.7 (minor), 179.5 (major), 176.9 (minor), 176.7 (major), 52.0 (minor), 51.2 (major), 44.7 (major), 43.8 (major), 43.6 (minor), 43.5 (minor), 33.5 (minor), 33.2 (major), 32.1 (minor), 30.1 (minor), 29.8 (major), 29.7 (major), 29.4 (major), 28.1 (minor), 23.5 (major), 22.8 (minor) ppm; IR (neat): υ = 3120 (br), 2927 (w), 2858 (w), 1774 (w), 1701 (vs), 1639 (w), 1593 (w), 1539 (w), 1496 (w), 1450 (w), 1350 (s), 1308 (w), 1180 (vs), 1068 (s), 1018 (s), 945 (w), 910 (w), 812 (w), 756 (s), 725 (s) cm−1; MS (70 eV, EI): m/z (%): 54.1 (24), 55.1 (50), 67.1 (23), 83.1 (20), 98.1 (49), 99.1 (80), 111.1 (100), 112.1 (44), 151.1 (14), 209.1 (M+, 12); HRMS (EI-QTOF) calcd. for C11H15NO3 (M+): 209.1052. Found: 209.1046; HPLC: ChiralPak IB column, hexanes/EtOH (90/10), flow rate = 0.8 mL/min, λ = 240 nm, tR (anti) = 28.57 min (minor enantiomer) 32.76 min (major enantiomer), tR (syn) = 26.67 min (minor enantiomer) 30.05 min (major enantiomer).
3-(4-Oxotetrahydro-2H-pyran-3-yl)pyrrolidine-2,5-dione (11gi): The major anti diastereomer was obtained combined with the minor syn diastereomer as an inseparable mixture in 90% global yield (anti/syn = 51/49, 1H NMR on the reaction crude). Yellow solid, mp: 52–54 °C; 1H NMR (500 MHz, CDCl3): δ = 4.31–4.10 (m, 1H, major + minor), 4.02–3.38 (m, 2H, major + minor), 3.37–2.80 (m, 1H, major + minor), 2.79–2.30 (m, 2H, major + minor), 1.91–1.20 (m, 4H, major + minor) ppm; 13C NMR (125 MHz, CDCl3): δ = 210.3 (major), 205.9 (major), 205.6 (minor), 178.0 (major), 175.6 (minor), 70.5 (minor), 69.3 (major), 68.8 (major). 68.5 (minor), 63.3 (major), 60.0 (minor), 52.3 (major), 51.2 (minor), 44.2 (major), 42.6 (minor), 38.8 (minor), 36.8 (major), 35.2 (minor), 34.8 (major), 33.4 (minor), 29.9 (major) ppm; IR (neat): υ = 3232 (br), 2931 (w), 2866 (w), 1774 (w), 1705 (vs), 1639 (w), 1593 (w), 1543 (w), 1493 (w), 1454 (w), 1362 (w), 1308 (w), 1184 (s), 1149 (w), 1088 (w), 964 (w), 849 (w), 756 (w), 694 (w) cm−1; MS (70 eV, EI): m/z (%): 54.1 (70), 68.1 (19), 82.1 (<10), 99.1 (100), 197.0 (M+, <10); HRMS (EI-QTOF) calcd. for C9H11NO4 (M+): 197.0688. Found: 197.0690; HPLC: ChiralPak IB column, hexanes/EtOH (90/10), flow rate = 0.8 mL/min, λ = 240 nm, tR (anti) = 28.36 min, tR (syn) = 31.05 min.
3.3. Scaled-Up Enantioselective Michael Addition Reaction of Cyclohexanone and N-Phenylmaleimide
A mixture of organocatalyst 8 (172.7 mg, 0.74 mmol), DABCO (41.3 mg, 0.37 mmol), and 10a (0.64 g, 3.7 mmol) was dissolved in CH2Cl2 (10 mL) in a glass vial (16 mm diameter). Then, 9a (0.76 mL, 7,4 mmol) was added and the mixture was stirred for four days at 5 °C. After this time, the solvent was evaporated under reduced pressure (15 torr) and the crude reaction mixture was purified by column chromatography (hexanes/ethyl acetate gradients), yielding the corresponding diastereomeric adducts (0.92 g, 92% yield).
4. Conclusions
We have demonstrated that a simple primary amine-salicylamide derived from chiral trans-cyclohexane-1,2-diamine acts as an appropriate organocatalyst for the diastereo- and enantio-selective conjugate addition of ketones to maleimides, with the presence of an organic base such as 1,4-diazabicyclo[2.2.2]octane (DABCO) generally improving the stereoselectivity. Cyclic and acyclic ketones are used in this addition reaction with N-aryl- and N-alkyl-maleimides, usually affording the final succinimides in good yields. The stereoselectivity of the process ranges from low to very good in the case of diastereoselectivity and from moderate to excellent concerning enantioselectivity, even when using simple N-unsubstituted maleimide. This asymmetric procedure is an interesting alternative to the short array of methodologies leading to these substituted ketone-containing succinimides.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27196668/s1, Figures S1–S42 NMR spectra of compounds 11, Figures S43–S84: HPLC chromatograms of compounds 11.
Author Contributions
Conceptualization, methodology, project administration, supervision and writing original draft, R.C.; and investigation and methodology, A.T.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministerio de Economía, Industria y Competitividad grant number PGC2018-096616-B-I00, the Spanish Ministerio de Ciencia, Innovación y Universidades (CTQ201788171-P) and the University of Alicante (VIGROB-173).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all compounds 11, except compound 11ia, are available from the authors.
References
- Barker, D.; Lin, D.H.S.; Carland, J.E.; Chu, C.P.Y.; Chebib, M.; Brimble, M.A.; Savage, G.P.; McLeod, M.D. Methyllycaconitine analogues have mixed antagonist effects at nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2005, 13, 4565–4575. [Google Scholar] [CrossRef] [PubMed]
- Uddin, J.; Ueda, K.; Siwu, E.R.O.; Kita, M.; Uemura, D. Cytotoxic labdane alkaloids from an ascidian Lissoclinum sp.: Isolation, structure elucidation, and structure-activity relationship. Bioorg. Med. Chem. 2006, 14, 6954–6961. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Rugseree, N.; Maithip, P.; Kongsaeree, P.; Prabpai, S.; Thebtaranonth, Y. Hirsutellones A-E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea body centered cubic 2594. Tetrahedron 2005, 61, 5577–5583. [Google Scholar] [CrossRef]
- Freiberg, C.; Brunner, N.A.; Schiffer, G.; Lampe, T.; Pohlmann, J.; Brands, M.; Raabe, M.; Haebich, D.; Ziegelbauer, K. Identification and characterization of the first class of potent bacterial Acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem. 2004, 279, 26066–26073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Fuse, E.; Figg, W.D. Thalidomide metabolism by the CYP2C subfamily. Clin. Cancer Res. 2002, 8, 1964–1973. [Google Scholar] [PubMed]
- Curtin, M.L.; Garland, R.B.; Heyman, H.R.; Frey, R.R.; Michaelides, M.R.; Li, J.; Pease, L.J.; Glaser, K.B.; Marcotte, P.A.; Davidsen, S.K. Succinimide hydroxamic acids as potent inhibitors of histone deacetylase (HDAC). Bioorg. Med. Chem. Lett. 2002, 12, 2919–2923. [Google Scholar] [CrossRef]
- Ahmed, S. Molecular modelling study of pyrrolidine-2,5-dione based aromatase inhibitors and other known inhibitors. Drug Des. Discov. 1996, 14, 77–89. [Google Scholar] [PubMed]
- Ballini, R.; Bosica, G.; Cioci, G.; Fiorini, D.; Petrini, M. Conjugate addition of nitroalkanes to N-substituted maleimides. Synthesis of 3-alkylsuccinimides and pyrrolidines. Tetrahedron 2003, 59, 3603–3608. [Google Scholar] [CrossRef]
- Vo-Hoang, Y.; Gasse, C.; Vidal, M.; Garbay, C.; Galons, H. Efficient synthesis of N-benzyl-3-aminopyrrolidine-2,5-dione and N-benzyl-3-aminopyrrolidin-2-one. Tetrahedron Lett. 2004, 45, 3603–3605. [Google Scholar] [CrossRef]
- Nöth, J.; Frankowski, K.J.; Neuenswander, B.; Aubé, J.; Reiser, O. Efficient synthesis of γ-lactams by a tandem reductive amination/lactamization sequence. J. Comb. Chem. 2008, 10, 456–459. [Google Scholar] [CrossRef]
- Fenster, E.; Hill, D.; Reiser, O.; Aube, J. Automated three-component synthesis of a library of γ-lactams. Beilstein J. Org. Chem. 2012, 8, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, P.; Kaur, J.; Chimni, S.S. Asymmetric organocatalytic addition reactions of maleimides: A promising approach towards the synthesis of chiral succinimide derivatives. Chem. Asian J. 2013, 8, 328–346. [Google Scholar] [CrossRef]
- Zhao, G.-L.; Xu, Y.; Sundén, H.; Eriksson, L.; Sayah, M.; Cordova, A. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides. Chem. Commun. 2007, 734–735. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.-F.; Peng, L.; Wang, L.-l.; Wang, L.-X.; Xu, X.-Y. Chiral primary amine thiourea promoted highly enantioselective Michael reactions of isobutylaldehyde with maleimides. Tetrahedron 2010, 66, 8928–8932. [Google Scholar] [CrossRef]
- Xue, F.; Liu, L.; Zhang, S.; Duan, W.; Wang, W. A simple primary amine thiourea catalyzed highly enantioselective conjugate addition of α,α-disubstituted aldehydes to maleimides. Chem. Eur. J. 2010, 16, 7979–7982. [Google Scholar] [CrossRef]
- Yu, F.; Jin, Z.; Huang, H.; Ye, T.; Liang, X.; Ye, J. A highly efficient asymmetric Michael addition of α,α-disubstituted aldehydes to maleimides catalyzed by primary amine thiourea salt. Org. Biomol. Chem. 2010, 8, 4767–4774. [Google Scholar] [CrossRef]
- Ma, Z.-w.; Liu, Y.-x.; Li, P.-l.; Ren, H.; Zhu, Y.; Tao, J.-c. A highly efficient large-scale asymmetric Michael addition of isobutyraldehyde to maleimides promoted by a novel multifunctional thiourea. Tetrahedron Asymmetry 2011, 22, 1740–1748. [Google Scholar] [CrossRef]
- Miura, T.; Masuda, A.; Ina, M.; Nakashima, K.; Nishida, S.; Tada, N.; Itoh, A. Asymmetric Michael reactions of α,α-disubstituted aldehydes with maleimides using a primary amine thiourea organocatalyst. Tetrahedron Asymmetry 2011, 22, 1605–1609. [Google Scholar] [CrossRef]
- Miura, T.; Nishida, S.; Masuda, A.; Tada, N.; Itoh, A. Asymmetric Michael additions of aldehydes to maleimides using a recyclable fluorous thiourea organocatalyst. Tetrahedron Lett. 2011, 52, 4158–4160. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Nájera, C. Enantioselective Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by chiral primary amine-guanidines. Tetrahedron Asymmetry 2012, 23, 1625–1627. [Google Scholar] [CrossRef]
- Nugent, T.C.; Sadiq, A.; Bibi, A.; Heine, T.; Zeonjuk, L.L.; Vankova, N.; Bassil, B.S. Noncovalent bifunctional organocatalysts: Powerful tools for contiguous quaternary-tertiary stereogenic carbon formation, scope, and origin of enantioselectivity. Chem. Eur. J. 2012, 18, 4088–4098. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael addition of aldehydes to maleimides organocatalyzed by chiral 1,2-diamines: An experimental and theoretical study. Tetrahedron Asymmetry 2013, 24, 1531–1535. [Google Scholar] [CrossRef]
- Kozma, V.; Szőllősi, G. Enantioselective Michael addition of aldehydes to maleimides catalyzed by surface-adsorbed natural amino acids. Catal. Sci. Technol. 2022, 12, 4709–4726. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Gómez-Bengoa, E.; Nájera, C. Enantioselective synthesis of succinimides by Michael addition of aldehydes to maleimides organocatalyzed by chiral primary amine-guanidines. Eur. J. Org. Chem. 2013, 2013, 5085–5092. [Google Scholar] [CrossRef]
- Durmaz, M.; Sirit, A. Calixarene-based chiral primary amine thiourea promoted highly enantioselective asymmetric Michael reactions of α,α-disubstituted aldehydes with maleimides. Tetrahedron Asymmetry 2013, 24, 1443–1448. [Google Scholar] [CrossRef]
- Kokotos, C.G. An asymmetric Michael addition of α,α-disubstituted aldehydes to maleimides leading to a one-pot enantioselective synthesis of lactones catalyzed by amino acids. Org. Lett. 2013, 15, 2406–2409. [Google Scholar] [CrossRef]
- Qiao, Y.; Headley, A.D. A simple and highly effective water-compatible organocatalytic system for asymmetric direct Michael reactions of linear aldehydes to maleimides. Green Chem. 2013, 15, 2690–2694. [Google Scholar] [CrossRef]
- Yang, W.; Jiang, K.-Z.; Lu, X.; Yang, H.-M.; Li, L.; Lu, Y.; Xu, L.-W. Molecular assembly of an achiral phosphine and a chiral primary amine: A highly efficient supramolecular catalyst for the enantioselective Michael reaction of aldehydes with maleimides. Chem. Asian J. 2013, 8, 1182–1190. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Solvent-dependent enantioswitching in the Michael addition of α,α-disubstituted aldehydes to maleimides organocatalyzed by mono-N-BOC-protected cyclohexa-1,2-diamines. Tetrahedron Asymmetry 2014, 25, 1091–1094. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.T.; Zhang, T.; Du, H.L.; Ma, Z.W.; Zhang, C.H.; Tao, J.C. Highly enantioselective Michael addition promoted by a new diterpene-derived bifunctional thiourea catalyst: A doubly stereocontrolled approach to chiral succinimide derivatives. Chirality 2014, 26, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ferrándiz, J.; Fiser, B.; Gómez-Bengoa, E.; Chinchilla, R. Solvent-induced reversal of enantioselectivity in the synthesis of succinimides by the addition of aldehydes to maleimides catalysed by carbamate-monoprotected 1,2-diamines. Eur. J. Org. Chem. 2015, 2015, 1218–1225. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; Díaz-Corona, L.; Jiménez-González, E.; Juaristi, E. Asymmetric Michael addition organocatalyzed by α,β-dipeptides under solvent-free reaction conditions. Molecules 2017, 22, 1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, O.; Boufroura, H.; Thomassigny, C.; Perato, S.; Gaucher, A.; Marrot, J.; Prim, D. Modular Urea-Based Catalytic Platforms Bearing Flexible Pyridylmethylamine and Rigid Pyridyl-Imidazolidine Fragments. Eur. J. Org. Chem. 2017, 2017, 746–752. [Google Scholar] [CrossRef]
- Flores-Ferrándiz, J.; Chinchilla, R. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides in deep eutectic solvents. Tetrahedron Asymmetry 2017, 28, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Kochetkov, S.V.; Kucherenko, A.S.; Zlotin, S.G. Asymmetric Michael addition of aldehydes to maleimides in primary amine-based aqueous ionic liquid-supported recyclable catalytic system. Mendeleev Commun. 2017, 27, 473–475. [Google Scholar] [CrossRef]
- Ma, Z.-W.; Liu, X.-F.; Liu, J.-T.; Liu, Z.-J.; Tao, J.-C. Highly enantioselective Michael addition of α,α-disubstituted aldehydes to maleimides catalyzed by new primary amine-squaramide bifunctional organocatalysts. Tetrahedron Lett. 2017, 58, 4487–4490. [Google Scholar] [CrossRef]
- De Simone, N.A.; Meninno, S.; Talotta, C.; Gaeta, C.; Neri, P.; Lattanzi, A. Solvent-free enantioselective Michael reactions catalyzed by a calixarene-based primary amine thiourea. J. Org. Chem. 2018, 83, 10318–10325. [Google Scholar] [CrossRef] [PubMed]
- Schiza, A.; Spiliopoulou, N.; Shahu, A.; Kokotos, C.G. Combining organocatalysis with photoorganocatalysis: Photocatalytic hydroacylation of asymmetric organocatalytic Michael addition products. New J. Chem. 2018, 42, 18844–18849. [Google Scholar] [CrossRef]
- Szollosi, G.; Kozma, V. Design of heterogeneous organocatalyst for the asymmetric Michael addition of aldehydes to maleimides. ChemCatChem 2018, 10, 4362–4368. [Google Scholar] [CrossRef]
- Torregrosa-Chinillach, A.; Moragues, A.; Pérez-Furundarena, H.; Chinchilla, R.; Gómez-Bengoa, E.; Guillena, G. Enantioselective Michael addition of aldehydes to maleimides organocatalyzed by a chiral primary amine-salicylamide. Molecules 2018, 23, 3299. [Google Scholar] [CrossRef]
- Landeros, J.M.; Suchy, L.; Avila-Ortiz, C.G.; Maulide, N.; Juaristi, E. Dendrimeric α,β-dipeptidic conjugates as organocatalysts in the asymmetric Michael addition reaction of isobutyraldehyde to N-phenylmaleimides. Monatsh. Chem. 2019, 150, 777–788. [Google Scholar] [CrossRef]
- Du, Z.-H.; Qin, W.-J.; Tao, B.-X.; Yuan, M.; Da, C.-S. N-Primary-amine tetrapeptide-catalyzed highly asymmetric Michael addition of aliphatic aldehydes to maleimides. Org. Biomol. Chem. 2020, 18, 6899–6904. [Google Scholar] [CrossRef] [PubMed]
- Kozma, V.; Fueloep, F.; Szollosi, G. 1,2-Diamine-derived (thio)phosphoramide organocatalysts in asymmetric Michael additions. Adv. Synth. Catal. 2020, 362, 2444–2458. [Google Scholar] [CrossRef]
- Sadiq, A.; Nugent, T.C. Catalytic Access to Succinimide Products Containing Stereogenic Quaternary Carbons. ChemistrySelect 2020, 5, 11934–11938. [Google Scholar] [CrossRef]
- Torregrosa-Chinillach, A.; Sanchez-Laó, A.; Santagostino, E.; Chinchilla, R. Organocatalytic asymmetric conjugate addition of aldehydes to maleimides and nitroalkenes in deep eutectic solvents. Molecules 2019, 24, 4058. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, M.-M.; Zhang, S.; Xu, Z.-A.; Li, H.; Yu, X.-H.; Wang, W. Chiral pyrrolidine sulfonamide catalyzed enantioselective Michael addition of cyclohexanones to maleimides. Synlett 2011, 2011, 473–476. [Google Scholar] [CrossRef]
- Yu, F.; Sun, X.; Jin, Z.; Wen, S.; Liang, X.; Ye, J. Enantioselective Michael addition of ketones to maleimides catalyzed by bifunctional monosulfonyl DPEN salt. Chem. Commun. 2010, 46, 4589–4591. [Google Scholar] [CrossRef]
- Muramulla, S.; Ma, J.-A.; Zhao, J.C.-G. Michael addition of ketones and aldehydes to maleimides catalyzed by modularly designed organocatalysts. Adv. Synth. Catal. 2013, 355, 1260–1264. [Google Scholar] [CrossRef]
- Nakashima, K.; Kawada, M.; Hirashima, S.-I.; Kato, M.; Koseki, Y.; Miura, T. Asymmetric conjugate addition of ketones to maleimides using diaminomethyleneindenedione organocatalyst. Synlett 2015, 26, 1248–1252. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Kawada, M.; Hirashima, S.-I.; Kosugi, A.; Kato, M.; Yoshida, A.; Koseki, Y.; Miura, T. Stereoselective conjugate addition of carbonyl compounds to maleimides using a diaminomethyleneindenedione organocatalyst. Tetrahedron Asymmetry 2016, 27, 888–895. [Google Scholar] [CrossRef]
- Jan, M.S.; Ahmad, S.; Hussain, F.; Ahmad, A.; Mahmood, F.; Rashid, U.; Abid, O.-U.-R.; Ullah, F.; Ayaz, M.; Sadiq, A. Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2,5-dione derivatives as multitarget anti-inflammatory agents. Eur. J. Med. Chem. 2020, 186, 111863. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Organocatalysts employed in the asymmetric conjugate addition of ketones to maleimides.

Figure 2.
Organocatalyst employed in this study.
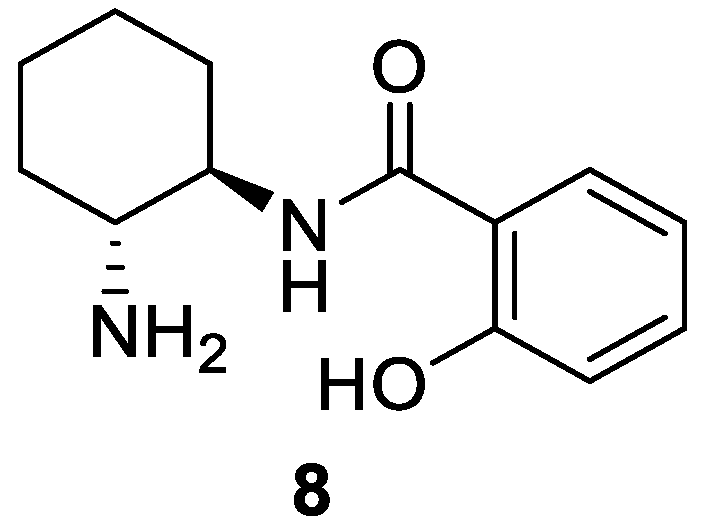
Figure 3.
Products obtained in the conjugate addition of ketones to maleimides using 8 as organocatalyst. Only the major stereoisomer is represented; 4-O2NC6H4CO2H (10 mol%) was used as co-catalyst for adducts 11ba, 11ca, 11da and 11di. Compounds 11ba-11ga, 11di and 11gi were obtained at room temperature.
Figure 3.
Products obtained in the conjugate addition of ketones to maleimides using 8 as organocatalyst. Only the major stereoisomer is represented; 4-O2NC6H4CO2H (10 mol%) was used as co-catalyst for adducts 11ba, 11ca, 11da and 11di. Compounds 11ba-11ga, 11di and 11gi were obtained at room temperature.
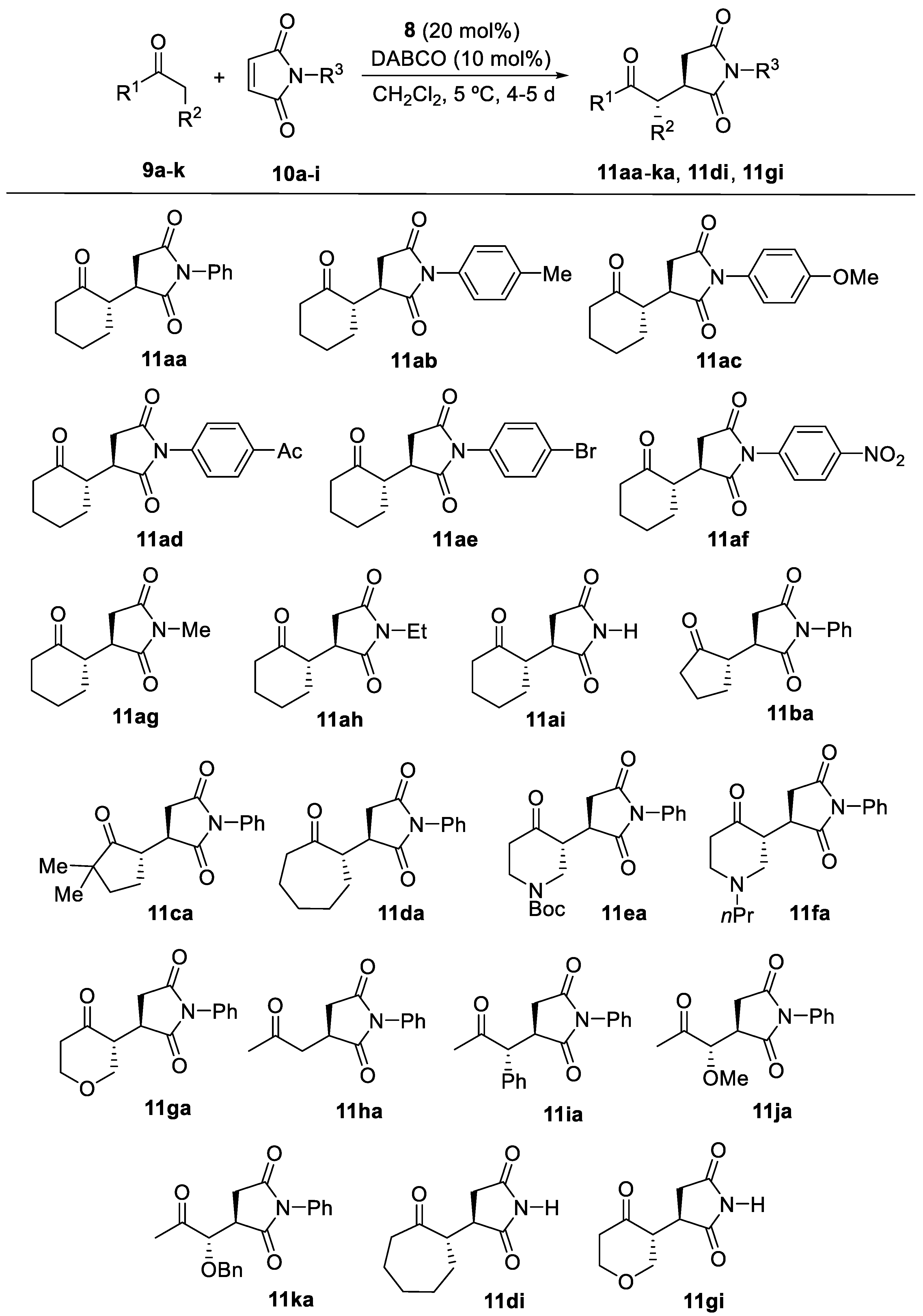
Figure 4.
Suggested transition state leading to the formation of 11aa.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Organocatalytic conjugate addition of cyclohexanone to N-phenylmaleimide. Optimization of the reaction conditions.
Table 1.
Organocatalytic conjugate addition of cyclohexanone to N-phenylmaleimide. Optimization of the reaction conditions.
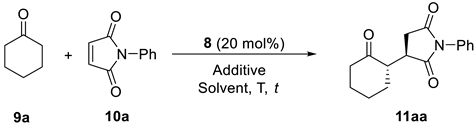 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Additive (mol%) a | Solvent a | T (°C) | t (d) | Conv. (%) b | drc | ee (%) d |
| 1 | - | Toluene | 25 | 3 | 93 | 76/24 | 94 (55) |
| 2 | - | Xylene | 25 | 3 | 76 | 79/21 | 89 (49) |
| 3 | - | Hexane | 25 | 3 | 91 | 63/37 | 31 (8) |
| 4 | - | CH2Cl2 | 25 | 3 | 100 | 80/20 | 99 (40) |
| 5 | - | CHCl3 | 25 | 3 | 100 | 80/20 | 92 (50) |
| 6 | - | DCE | 25 | 3 | 100 | 78/22 | 97 (30) |
| 7 | - | Et2O | 25 | 3 | 55 | 72/28 | 78 (25) |
| 8 | - | THF | 25 | 3 | 63 | 67/33 | 38 (32) |
| 9 | - | MeCN | 25 | 3 | 69 | 71/29 | 41 (5) |
| 10 | - | MeOH | 25 | 3 | n.r. | n.d. | n.d. |
| 11 | - | iPrOH | 25 | 3 | 49 | 71/29 | 50 (11) |
| 12 | - | H2O | 25 | 3 | 100 | 67/33 | 95 (6) |
| 13 | - | CH2Cl2 | 25 | 5 | 100 | 80/20 | 99 (41) |
| 14 | PhCO2H (10) | CH2Cl2 | 25 | 3 | 100 | 77/23 | 51 (40) |
| 15 | 4-O2NC6H4CO2H (10) | CH2Cl2 | 25 | 3 | 100 | 77/23 | 47 (40) |
| 16 | Salicylic acid (10) | CH2Cl2 | 25 | 3 | 100 | 74/26 | 33 (30) |
| 17 | HDA (10) | CH2Cl2 | 25 | 3 | 100 | 76/24 | 83 (58) |
| 18 | DMAP (10) | CH2Cl2 | 25 | 3 | 100 | 69/31 | 87 (22) |
| 19 | Imidazole (10) | CH2Cl2 | 25 | 3 | 100 | 83/17 | 83 (74) |
| 20 | DBU (10) | CH2Cl2 | 25 | 3 | n.r. | n.d. | n.d |
| 21 | 2,6-Lutidine (10) | CH2Cl2 | 25 | 3 | 100 | 81/19 | 84 (6) |
| 22 | DABCO (10) | CH2Cl2 | 25 | 3 | 100 | 85/15 | 99 (9) |
| 23 | DABCO (5) | CH2Cl2 | 25 | 3 | 100 | 85/15 | 97 (9) |
| 24 | DABCO (20) | CH2Cl2 | 25 | 3 | 100 | 85/15 | 64 (16) |
| 25 | DABCO (10) | CH2Cl2 | 5 | 4 | 100 | 93/7 | 99 (19) |
a Abbreviations: DABCO: 1,4-Diazabicyclo[2.2.2]octane; DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; DCE: 1,2-Dichloroethane; DMAP: 4-(Dimethylamino)pyridine; HDA: Hexanedioic acid. b Determined by 1H NMR based on the remaining 10a. c Diastereomeric ratio determined by 1H NMR on the reaction crude. d Determined by chiral HPLC on the reaction crude (see Section 3). Enantiomeric excess of the minor anti diastereomer in parentheses.
Table 2.
Conjugate addition of ketones to maleimides organocatalyzed by 8.
| Entry | t (d) | Product 11 | Yield (%) a | drb | ee (%) c,d |
|---|---|---|---|---|---|
| 1 | 4 | 11aa | 98 | 93/7 | 99 (19) |
| 2 | 4 | 11ab | 91 | 90/10 | 76 (53) |
| 3 | 4 | 11ac | 90 | 85/15 | 64 (53) |
| 4 | 4 | 11ad | 82 | 88/12 | 78 (46) |
| 5 | 4 | 11ae | 95 | 92/8 | 73 (18) |
| 6 | 4 | 11af | 80 | 84/16 | 62 (40) |
| 7 | 4 | 11ag | 94 | 91/9 | 76 (25) |
| 8 | 4 | 11ah | 93 | 83/17 | 77 (63) |
| 9 | 4 | 11ai | 92 | 76/24 | 99 (99) |
| 10 e,f | 5 | 11ba | 89 | 51/49 | 62 (22) |
| 11 e,f | 5 | 11ca | 65 | 64/36 | 93 (81) |
| 12 e,f | 5 | 11da | 92 | 66/34 | 85 (21) |
| 13 f | 4 | 11ea | 94 | 56/44 | 62 (54) |
| 14 f | 4 | 11fa | 91 | 65/35 | 76 (76) |
| 15 f | 4 | 11ga | 93 | 58/42 | 99 (99) |
| 16 | 4 | 11ha | 71 | - | 72 |
| 17 | 5 | 11ia | 56 | 52/48 | 96 (26) |
| 18 | 4 | 11ja | 94 | 62/38 | 36 (15) |
| 19 | 4 | 11ka | 96 | 75/25 | 75 (25) |
| 20 e,f | 4 | 11di | 85 | 60/40 | 95 (75) |
| 21 f | 4 | 11gi | 90 | 51/49 | 99 (99) |
| 22 g | 4 | 11aa | 92 | 91/9 | 97 (19) |
a Combined isolated yield of both diastereomers after flash chromatography. b Diastereomeric anti/syn ratio determined by 1H NMR on the reaction crude. c Determined by chiral HPLC on the reaction crude (see Materials and Methods). Enantiomeric excess of the minor diastereomer in parentheses. d The absolute stereochemistry was determined by comparing the elution order in chiral HPLC with the reported in literature, whereas the stereochemistry of unknown compounds was assigned by analogy (see Materials and Methods). e 4-O2NC6H4CO2H (10 mol%) was used as co-catalyst. f Reaction was carried out at 25 °C. g Scaled-up reaction (see Materials and Methods).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Torregrosa-Chinillach, A.; Chinchilla, R. Asymmetric Conjugate Addition of Ketones to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide. Molecules 2022, 27, 6668. https://doi.org/10.3390/molecules27196668
AMA Style
Torregrosa-Chinillach A, Chinchilla R. Asymmetric Conjugate Addition of Ketones to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide. Molecules. 2022; 27(19):6668. https://doi.org/10.3390/molecules27196668
Chicago/Turabian StyleTorregrosa-Chinillach, Alejandro, and Rafael Chinchilla. 2022. "Asymmetric Conjugate Addition of Ketones to Maleimides Organocatalyzed by a Chiral Primary Amine-Salicylamide" Molecules 27, no. 19: 6668. https://doi.org/10.3390/molecules27196668