

Theoretical Study on the Aggregation and Adsorption Behaviors of Anticancer Drug Molecules on Graphene/Graphene Oxide Surface

Abstract
:1. Introduction
2. Computational Methods
2.1. Quantum Chemistry Calculations
2.2. Molecular Dynamics Simulation
3. Results and Discussion
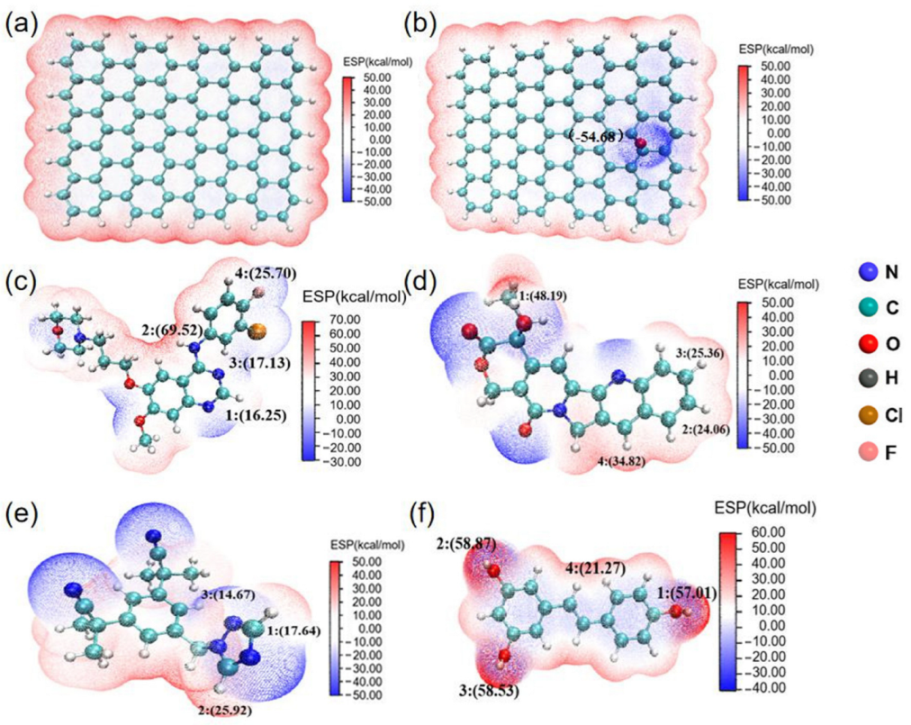
3.1. Electrostatic Potential (ESP) of Drug Molecules
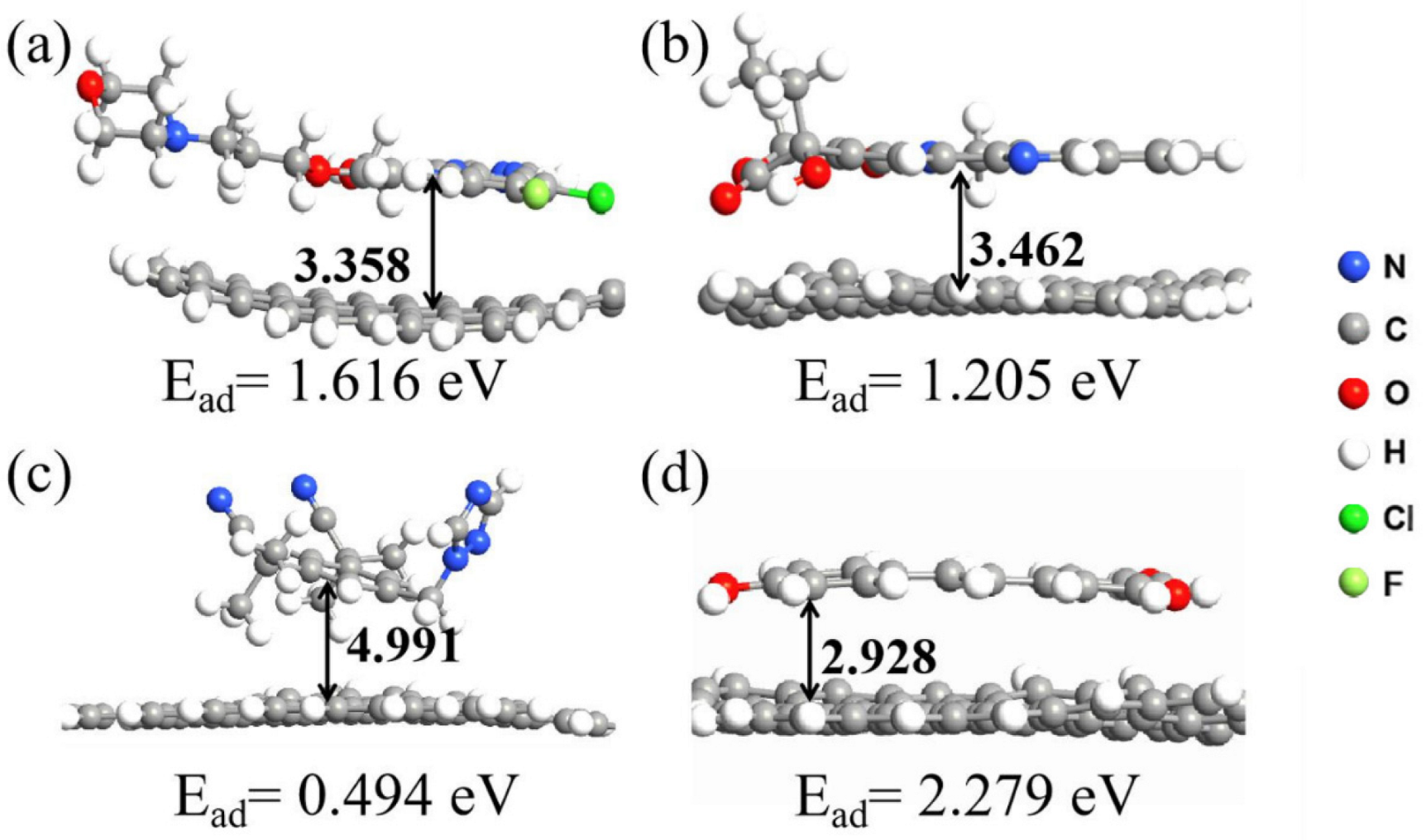
3.2. Simulation of Graphene Adsorption of Drug Molecules on Graphene
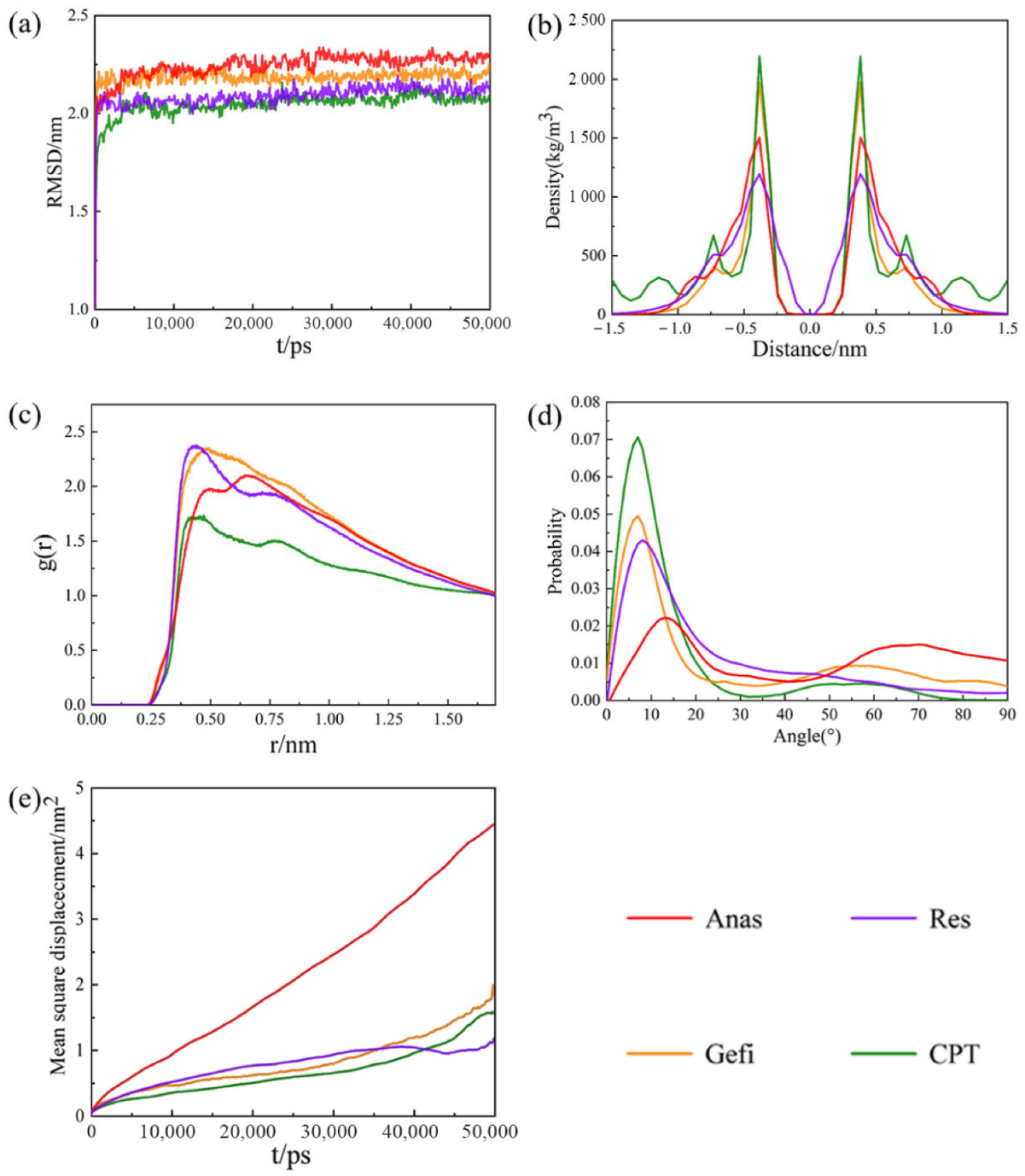
3.3. Simulation of the Adsorption of Drug Molecules on GO
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mostafavi, E.; Iravani, S. Mxene-graphene composites: A perspective on biomedical potentials. Nano-Micro Lett. 2022, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhu, R.; Shen, Y.; Tan, Q.; Chen, J. Comparison of loading and unloading of different small drugs on graphene and its oxide. J. Mol. Liq. 2021, 341, 117454. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, P.; Zhou, Z.; Wei, T. Interactions of graphene with mammalian cells: Molecular mechanisms and biomedical insights. Adv. Drug Deliv. Rev. 2016, 105, 145–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.M.L.; Machado, M.; Silva, G.A.; Bitoque, D.B.; Tavares Ferreira, J.; Pinto, L.A.; Ferreira, Q. Graphene oxide thin films with drug delivery function. Nanomaterials 2022, 12, 1149. [Google Scholar] [CrossRef]
- Fang, W.; Peng, L.; Liu, Y.; Wang, F.; Xu, Z.; Gao, C. A review on graphene oxide two-dimensional macromolecules: From single molecules to macro-assembly. Chin. J. Polym. Sci. 2021, 39, 267–308. [Google Scholar] [CrossRef]
- Alinejad, A.; Raissi, H.; Hashemzadeh, H. Understanding co-loading of doxorubicin and camptothecin on graphene and folic acid-conjugated graphene for targeting drug delivery: Classical md simulation and dft calculation. J. Biomol. Struct. Dyn. 2020, 38, 2737–2745. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C.; Cook, C.; Palmer, K.; Mcphail, A.T.; Sim, G. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata1,2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Thomas, C.J.; Rahier, N.J.; Hecht, S.M. Camptothecin: Current perspectives. Bioorg. Med. Chem. 2004, 12, 1585–1604. [Google Scholar] [CrossRef]
- Jena, N.R.; Mishra, P.C. A theoretical study of some new analogues of the anti-cancer drug camptothecin. J. Mol. Model. 2006, 13, 267–274. [Google Scholar] [CrossRef]
- Di Nunzio, M.R.; Cohen, B.; Douhal, A. Structural photodynamics of camptothecin, an anticancer drug in aqueous solutions. J. Phys. Chem. A 2011, 115, 5094–5104. [Google Scholar] [CrossRef]
- Liu, S.; Yuan, Y.; Okumura, Y.; Shinkai, N.; Yamauchi, H. Camptothecin disrupts androgen receptor signaling and suppresses prostate cancer cell growth. Biochem. Biophys. Res. Commun. 2010, 394, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.A.Q.; Queiroz, A.N.; Borges, R.S. A theoretical study of resveratrol oxidation. J. Comput. Theor. Nanosci. 2009, 6, 1637–1639. [Google Scholar] [CrossRef]
- Stivala, L.A.; Savio, M.; Carafoli, F.; Perucca, P.; Bianchi, L.; Maga, G.; Forti, L.; Pagnoni, U.M.; Albini, A.; Prosperi, E.; et al. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J. Biol. Chem. 2001, 276, 22586–22594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.H.; Cheng, H.Y.; Yu, Z.Q.; He, D.H.; Pan, Z.; Yang, D.T. Resveratrol induces apoptosis in pancreatic cancer cells. Chin. Med. J. 2007, 124, 1695–1699. [Google Scholar]
- Yokouchi, H.; Yamazaki, K.; Kinoshita, I.; Konishi, J.; Asahina, H.; Sukoh, N.; Harada, M.; Akie, K.; Ogura, S.; Ishida, T.; et al. Clinical benefit of readministration of gefitinib for initial gefitinib-responders with non-small cell lung cancer. BMC Cancer 2007, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kleeff, J.; Giese, N.; Buchler, M.W.; Korc, M.; Friess, H. Gefitinib (‘iressa’, zd1839), a selective epidermal growth factor receptor tyrosine kinase inhibitor, inhibits pancreatic cancer cell growth, invasion, and colony formation. Int. J. Oncol. 2004, 25, 203–210. [Google Scholar] [CrossRef]
- Geng, D.; Sun, D.; Zhang, L.; Zhang, W. Materials and methods the statement of ethics. Afr. Health Sci. 2015, 15, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Kalykaki, A.; Agelaki, S.; Kallergi, G.; Xyrafas, A.; Mavroudis, D.; Georgoulias, V. Elimination of egfr-expressing circulating tumor cells in patients with metastatic breast cancer treated with gefitinib. Cancer Chemother. Pharm. 2014, 73, 685–693. [Google Scholar] [CrossRef]
- Reese, D.; Nabholtz, J.M. Anastrozole in the management of breast cancer. Expert Opin. Pharmacother. 2002, 3, 1329–1339. [Google Scholar] [CrossRef]
- Cuzick, J.; Sestak, I.; Forbes, J.F.; Dowsett, M.; Cawthorn, S.; Mansel, R.E.; Loibl, S.; Bonanni, B.; Evans, D.G.; Howell, A. Use of anastrozole for breast cancer prevention (ibis-ii): Long-term results of a randomised controlled trial. Lancet 2020, 395, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Nabholtz, J. Role of anastrozole across the breast cancer continuum: From advanced to early disease and prevention. Oncology 2006, 70, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Graham, P.H. Anastrozole for malignant and benign conditions: Present applications and future therapeutic integrations. Expert Opin. Pharmacother. 2007, 8, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Ayissi, S.; Charpentier, P.A.; Farhangi, N.; Wood, J.A.; Palotas, K.; Hofer, W.A. Interaction of Titanium Oxide Nanostructures with Graphene and Functionalized Graphene Nanoribbons: A DFT Study. J. Phys. Chem. C 2013, 117, 25424–25432. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the dmol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Csonka, G.; Corminboeuf, C. Unified inter- and intramolecular dispersion correction formula for generalized gradient approximation density functional theory. J. Chem. Theory Comput. 2009, 11, 2950. [Google Scholar] [CrossRef]
- Janesko, B.G.; Barone, V.; Brothers, E.N. Accurate surface chemistry beyond the generalized gradient approximation: Illustrations for graphene adatoms. J. Chem. Theory Comput. 2013, 11, 4853–4859. [Google Scholar] [CrossRef]
- Hill, T.L. Statistical mechanics of multimolecular adsorption. I. J. Chem. Phys. 1946, 14, 263–267. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K. The nature of tt-tt interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Akman, F.; Sagaama, A.; Issaoui, N.; Malyar, Y.N.; Vasilieva, N.Y.; Borovkova, V.S. Theoretical and experimental study of guar gum sulfation. J. Mol. Model. 2021, 27, 5. [Google Scholar] [CrossRef] [PubMed]
- Pitoňák, M.; Neogrády, P.; Rezáč, J.; Jurečka, P.; Urban, M.; Hobza, P. Benzene dimer: High-level wave function and density functional theory calculations. J. Chem. Theory Comput. 2008, 4, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, Z. Dft study of no adsorption on pristine graphene. RSC Adv. 2017, 7, 13082–13091. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jia, Y. Udmh adsorption on graphene oxides: A first-principles study. Diam. Relat. Mater. 2021, 117, 108457. [Google Scholar] [CrossRef]
- Kozlov, S.M.; Viñes, F.; Görling, A. On the interaction of polycyclic aromatic compounds with graphene. Carbon 2012, 50, 2482–2492. [Google Scholar] [CrossRef]
- Zhao, D.; Li, L.; Zhou, J. Simulation insight into the cytochrome c adsorption on graphene and graphene oxide surfaces. Appl. Surf. Sci. 2018, 428, 825–834. [Google Scholar] [CrossRef]
- Maleki, R.; Khoshoei, A.; Ghasemy, E.; Rashidi, A. Molecular insight into the smart functionalized tmc-fullerene nanocarrier in the ph-responsive adsorption and release of anti-cancer drugs. J. Mol. Graph. Model. 2020, 100, 107660. [Google Scholar] [CrossRef]
- Kuisma, E.; Hansson, C.F.; Lindberg, T.B.; Gillberg, C.A.; Idh, S.; Schröder, E. Graphene oxide and adsorption of chloroform: A density functional study. J. Chem. Phys. 2016, 144, 184704. [Google Scholar] [CrossRef] [Green Version]
- Ai, Y.; Liu, Y.; Huo, Y.; Zhao, C.; Sun, L.; Han, B.; Cao, X.; Wang, X. Insights into the adsorption mechanism and dynamic behavior of tetracycline antibiotics on reduced graphene oxide (rgo) and graphene oxide (go) materials. Environ. Sci. Nano 2019, 6, 3336–3348. [Google Scholar] [CrossRef]
- Zhao, M.; Lai, Q.; Guo, J.; Guo, Y. Insights into the adsorption of resveratrol on graphene oxide: A first-principles study. ChemistrySelect 2017, 2, 6895–6900. [Google Scholar] [CrossRef]
- Kamel, M.; Raissi, H.; Hashemzadeh, H.; Mohammadifard, K. Theoretical elucidation of the amino acid interaction with graphene and functionalized graphene nanosheets: Insights from dft calculation and md simulation. Amino Acids 2020, 52, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, Y.; Shan, S.; Yang, X.; Liu, D.; Cui, F.; Xing, B. Theoretical insight into the adsorption of aromatic compounds on graphene oxide. Environ. Sci. Nano 2018, 5, 2357–2367. [Google Scholar] [CrossRef]
- Headen, T.F.; Howard, C.A.; Skipper, N.T.; Wilkinson, M.A.; Bowron, D.T.; Soper, A.K. Structure of π−π interactions in aromatic liquids. J. Am. Chem. Soc. 2010, 132, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Li, C.; Yu, J.; Xu, Q.; Wei, S.; Tian, Z.; Yang, Z.; Yang, W.; Shen, J. Insight into adsorption of combined antibiotic-heavy metal contaminants on graphene oxide in water. Sep. Purif. Technol. 2020, 236, 116278. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, S.; Huang, T.; Cui, F.; Xing, B. Ph-dependent adsorption of aromatic compounds on graphene oxide: An experimental, molecular dynamics simulation and density functional theory investigation. J. Hazard. Mater. 2020, 395, 122680. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Molecules | Gefi | CPT | Anas | Res |
|---|---|---|---|---|
| Diffusion coefficient (×10−5 cm2·s−1) | 0.004027 | 0.003376 | 0.0123 | 0.002793 |
| Drug Molecules | Gefi | CPT | Anas | Res |
|---|---|---|---|---|
| Diffusion coefficient (×10−5 cm2·s−1) | 0.001416 | 0.001032 | 0.007013 | 0.002307 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, P.; Zhou, Y.; Li, H.; Zhang, J.; Wu, Y.; Zheng, P.; Jiang, Y. Theoretical Study on the Aggregation and Adsorption Behaviors of Anticancer Drug Molecules on Graphene/Graphene Oxide Surface. Molecules 2022, 27, 6742. https://doi.org/10.3390/molecules27196742
Gong P, Zhou Y, Li H, Zhang J, Wu Y, Zheng P, Jiang Y. Theoretical Study on the Aggregation and Adsorption Behaviors of Anticancer Drug Molecules on Graphene/Graphene Oxide Surface. Molecules. 2022; 27(19):6742. https://doi.org/10.3390/molecules27196742
Chicago/Turabian StyleGong, Pengyu, Yi Zhou, Hui Li, Jie Zhang, Yuying Wu, Peiru Zheng, and Yanyan Jiang. 2022. "Theoretical Study on the Aggregation and Adsorption Behaviors of Anticancer Drug Molecules on Graphene/Graphene Oxide Surface" Molecules 27, no. 19: 6742. https://doi.org/10.3390/molecules27196742