
A Comprehensive Study of N-Butyl-1H-Benzimidazole
 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Result and Discussion
2.1. Structural Analysis of N-Butyl-1H-Benzimidazole
2.2. Topological Analysis
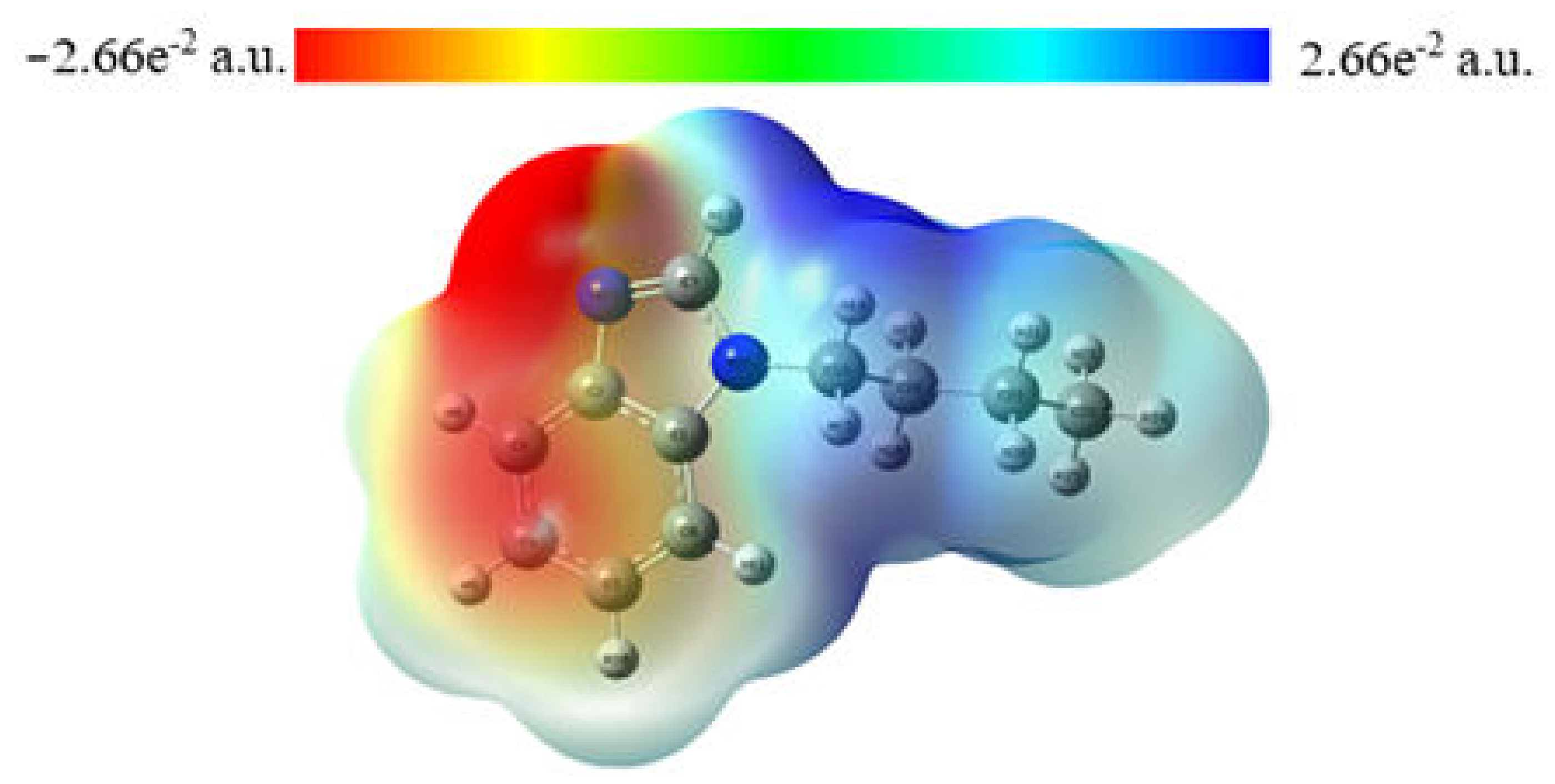
2.3. MEP Analysis
2.4. Frontier Molecular Orbital (FMO) Analysis
2.5. Fukui Functions
2.6. UV-Vis Analysis and NBO Analysis
2.7. FTIR Analysis
2.8. Mulliken Atomic Charges Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pathare, B.; Bansode, T. Review- biological active benzimidazole derivatives. Results Chem. 2021, 3, 100200. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorganic Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Cheng, J.; Xie, J.; Luo, X. Synthesis and antiviral activity against Coxsackie virus B3 of some novel benzimidazole derivatives. Bioorganic Med. Chem. Lett. 2005, 15, 267–269. [Google Scholar] [CrossRef]
- Charlson, A.J. The methanesulfonylation of 2-benzimidazolemethanol and α(2-benzimidazolyl)benzyl alcohol. Carbohydr. Res. 1973, 29, 89–98. [Google Scholar] [CrossRef]
- Walker, K.A.M.; Braemer, A.C.; Hitt, S.; Jones, R.E.; Matthews, T.R. 1-[4-(4-Chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-1H-imidazole nitrate, a new potent antifungal agent. J. Med. Chem. 1978, 21, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Nguyen, P.T.M.; Baldeck, J.D.; Olsson, J.; Marquis, R.E. Antimicrobial actions of benzimidazoles against the oral anaerobes Fusobacterium nucleatum and Prevotella intermedia. Arch. Oral Biol. 2006, 51, 1015–1023. [Google Scholar] [CrossRef]
- Infante-Castillo, R.; Rivera-Montalvo, L.A.; Hernández-Rivera, S.P. Theoretical DFT, vibrational and NMR studies of benzimidazole and alkyl derivatives. J. Mol. Struct. 2008, 877, 10–19. [Google Scholar] [CrossRef]
- Drolet, D.P.; Manuta, D.M.; Lees, A.J.; Katnani, A.D.; Coyle, G.J. FT-IR and XPS study of copper(II) complexes of imidazole and benzimidazole. Inorg. Chim. Acta 1988, 146, 173–180. [Google Scholar] [CrossRef]
- Shaharyar, M.; Mazumder, A. Benzimidazoles: A biologically active compounds. Arab. J. Chem. 2017, 10, S157–S173. [Google Scholar] [CrossRef] [Green Version]
- Shannon, M.S.; Hindman, M.S.; Danielsen, S.P.O.; Tedstone, J.M.; Gilmore, R.D.; Bara, J.E. Properties of alkylbenzimidazoles for CO2 and SO2 capture and comparisons to ionic liquids. Sci. China Chem. 2012, 55, 1638–1647. [Google Scholar] [CrossRef]
- Chen, S.-H.; Zhao, Q.; Xu, X.-W. Preparation and characterization of a novel benzimidazolium brønsted acidic ionic liquid and its application in esterifications. J. Chem. Sci. 2008, 120, 481–483. [Google Scholar] [CrossRef]
- Veerasamy, R.; Roy, A.; Karunakaran, R.; Rajak, H. Structure–Activity Relationship Analysis of Benzimidazoles as Emerging Anti-Inflammatory Agents: An Overview. Pharmaceuticals 2021, 14, 663. [Google Scholar] [CrossRef]
- Rutkowska, I.A.; Marszalek, M.; Orlowska, J.; Ozimek, W.; Zakeeruddin, S.M.; Kulesza, P.J.; Grätzel, M. Nanocomposite Semi-Solid Redox Ionic Liquid Electrolytes with Enhanced Charge-Transport Capabilities for Dye-Sensitized Solar Cells. ChemSusChem 2015, 8, 2560–2568. [Google Scholar] [CrossRef]
- Chen, J.-H.; Ahmed, W.; Li, M.-H.; Li, Z.-D.; Cui, Z.-N.; Tang, R.-Y. TEMPO-Mediated Synthesis of N-(Fluoroalkyl)imidazolones via Reaction of Imidazoles with Iodofluoroacetate. Adv. Synth. Catal. 2020, 362, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Hussaini, S.Y.; Haque, R.A.; Fatima, T.; Agha, T.M.; Abdul Majid, A.M.S.; Abdallah, H.H.; Razali, M.R. Nitrile functionalized silver(I) N-heterocyclic carbene complexes: DFT calculations and antitumor studies. Transit. Met. Chem. 2018, 43, 301–312. [Google Scholar] [CrossRef]
- Wu, H.; Jin, C.; Huang, G.; Wang, L.; Jiang, J.; Wang, L. Binaphthyl-bridged bis-imidazolinium salts as N-heterocyclic carbene ligand precursors in the palladium-catalyzed Heck reaction. Sci. China Chem. 2011, 54, 951–956. [Google Scholar] [CrossRef]
- Shukla, M.; Saha, S. Relationship between stabilization energy and thermophysical properties of different imidazolium ionic liquids: DFT studies. Comput. Theor. Chem. 2013, 1015, 27–33. [Google Scholar] [CrossRef]
- Dzyuba, S.V.; Bartsch, R.A. New room-temperature ionic liquids with -symmetrical imidazolium cations. Chem. Commun. 2001, 16, 1466–1467. [Google Scholar] [CrossRef]
- Aman, H.; Rashid, N.; Ashraf, Z.; Bibi, A.; Chen, H.-T.; Sathishkumar, N. Synthesis, density functional theory (DFT) studies and urease inhibition activity of chiral benzimidazoles. Heliyon 2020, 6, e05187. [Google Scholar] [CrossRef]
- Halls, M.D.; Velkovski, J.; Schlegel, H.B. Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theor. Chem. Acc. 2001, 105, 413–421. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Akman, F.; Malyar, Y.N.; Issaoui, N.; Vasilieva, N.Y.; Karacharov, A.A. Synthesis optimization, DFT and physicochemical study of chitosan sulfates. J. Mol. Struct. 2021, 1245, 131083. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Tomilin, F.N.; Pozdnyakova, A.A.; Vasilyeva, N.Y.; Malyar, Y.N.; Kuznetsova, S.A.; Avramov, P.V. Theoretical DFT interpretation of infrared spectra of biologically active arabinogalactan sulphated derivatives. Chem. Pap. 2020, 74, 4103–4113. [Google Scholar] [CrossRef]
- Brown, I.D. Topology and Chemistry. Struct. Chem. 2002, 13, 339–355. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. Atoms in molecules in external fields. J. Chem. Phys. 1989, 91, 6989–7001. [Google Scholar] [CrossRef]
- Akman, F.; Issaoui, N.; Kazachenko, A.S. Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J. Mol. Model. 2020, 26, 161. [Google Scholar] [CrossRef] [PubMed]
- Kazachenko, A.; Akman, F.; Medimagh, M.; Issaoui, N.; Vasilieva, N.; Malyar, Y.N.; Sudakova, I.G.; Karacharov, A.; Miroshnikova, A.; Al-Dossary, O.M. Sulfation of Diethylaminoethyl-Cellulose: QTAIM Topological Analysis and Experimental and DFT Studies of the Properties. ACS Omega 2021, 6, 22603–22615. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Akman, F.; Abdelmoulahi, H.; Issaoui, N.; Malyar, Y.N.; Al-Dossary, O.; Wojcik, M.J. Intermolecular hydrogen bonds interactions in water clusters of ammonium sulfamate: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NBO analysis. J. Mol. Liq. 2021, 342, 117475. [Google Scholar] [CrossRef]
- Medimagh, M.; Issaoui, N.; Gatfaoui, S.; Al-Dossary, O.S.; Kazachenko, A.; Marouani, H.; Wojcik, M.J. Molecular modeling and biological activity analysis of new organic-inorganic hybrid: 2-(3,4-dihydroxyphenyl) ethanaminium nitrate. J. King Saud Univ.-Sci. 2021, 33, 101616. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. A quantum chemical topological analysis of the C–C bond formation in organic reactions involving cationic species. Phys. Chem. Chem. Phys. 2014, 16, 14108–14115. [Google Scholar] [CrossRef]
- Fuster, F.; Sevin, A.; Silvi, B. Topological Analysis of the Electron Localization Function (ELF) Applied to the Electrophilic Aromatic Substitution. J. Phys. Chem. A 2000, 104, 852–858. [Google Scholar] [CrossRef]
- Hernández-Trujillo, J.; García-Cruz, I.; Martínez-Magadán, J.M. Topological analysis of the electron density and of the electron localization function of pyrene and its radicals. Chem. Phys. 2005, 308, 181–192. [Google Scholar] [CrossRef]
- Maulén, B.; Echeverri, A.; Gómez, T.; Fuentealba, P.; Cárdenas, C. Electron Localization Function in Excited States: The Case of the Ultrafast Proton Transfer of the Salicylidene Methylamine. J. Chem. Theory Comput. 2019, 15, 5532–5542. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Vincy, C.D.; Tarika, J.D.D.; Dexlin, X.D.D.; Rathika, A.; Beaula, T.J. Exploring the antibacterial activity of 1, 2 diaminoethane hexanedionic acid by spectroscopic, electronic, ELF, LOL, RDG analysis and molecular docking studies using DFT method. J. Mol. Struct. 2022, 1247, 131388. [Google Scholar] [CrossRef]
- Stewart, J.J.P. An examination of the nature of localized molecular orbitals and their value in understanding various phenomena that occur in organic chemistry. J. Mol. Model. 2018, 25, 7. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, H. Localized-orbital locator (LOL) profiles of chemical bonding. Can. J. Chem. 2008, 86, 695–702. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Non-covalent interaction via the reduced density gradient: Independent atom model vs experimental multipolar electron densities. Comput. Theor. Chem. 2012, 998, 148–163. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Gadre, S.R.; Suresh, C.H.; Mohan, N. Electrostatic Potential Topology for Probing Molecular Structure, Bonding and Reactivity. Molecules 2021, 26, 3289. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Sevvanthi, S.; Muthu, S.; Raja, M. Molecular docking, vibrational spectroscopy studies of (RS)-2-(tert-butylamino)-1-(3-chlorophenyl)propan-1-one: A potential adrenaline uptake inhibitor. J. Mol. Struct. 2018, 1173, 251–260. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Akman, F.; Sagaama, A.; Issaoui, N.; Malyar, Y.N.; Vasilieva, N.Y.; Borovkova, V.S. Theoretical and experimental study of guar gum sulfation. J. Mol. Model. 2021, 27, 5. [Google Scholar] [CrossRef] [PubMed]
- Barim, E.; Akman, F. Synthesis, characterization and spectroscopic investigation of N-(2-acetylbenzofuran-3-yl)acrylamide monomer: Molecular structure, HOMO–LUMO study, TD-DFT and MEP analysis. J. Mol. Struct. 2019, 1195, 506–513. [Google Scholar] [CrossRef]
- Akman, F.; Kazachenko, A.; Malyar, Y. A density functional theory study of sulfated monolignols: P-Coumaril and coniferyl alcohols. Cellul. Chem. Technol. 2021, 55, 41–54. [Google Scholar] [CrossRef]
- Noureddine, O.; Issaoui, N.; Medimagh, M.; Al-Dossary, O.; Marouani, H. Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. J. King Saud Univ.-Sci. 2021, 33, 101334. [Google Scholar] [CrossRef]
- Medimagh, M.; Issaoui, N.; Gatfaoui, S.; Antonia Brandán, S.; Al-Dossary, O.; Marouani, H.J.; Wojcik, M. Impact of non-covalent interactions on FT-IR spectrum and properties of 4-methylbenzylammonium nitrate. A DFT and molecular docking study. Heliyon 2021, 7, e08204. [Google Scholar] [CrossRef]
- Almeida, M.O.; Barros, D.A.S.; Araujo, S.C.; Faria, S.H.D.M.; Maltarollo, V.G.; Honorio, K.M. Study on molecular structure, spectroscopic properties (FTIR and UV–Vis), NBO, QTAIM, HOMO-LUMO energies and docking studies of 5-fluorouracil, a substance used to treat cancer. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 184, 169–176. [Google Scholar] [CrossRef]
- Akman, F.; Kazachenko, A.S.; Vasilyeva, N.Y.; Malyar, Y.N. Synthesis and characterization of starch sulfates obtained by the sulfamic acid-urea complex. J. Mol. Struct. 2020, 1208, 127899. [Google Scholar] [CrossRef]
- Buvaneswari, M.; Santhakumari, R.; Usha, C.; Jayasree, R.; Sagadevan, S. Synthesis, growth, structural, spectroscopic, optical, thermal, DFT, HOMO–LUMO, MEP, NBO analysis and thermodynamic properties of vanillin isonicotinic hydrazide single crystal. J. Mol. Struct. 2021, 1243, 130856. [Google Scholar] [CrossRef]
- Muthu, S.; Renuga, S. Vibrational spectra and normal coordinate analysis of 2-hydroxy-3-(2-methoxyphenoxy) propyl carbamate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 132, 313–325. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Alameen, A.A.; Abdalla, M.; Alshibl, H.M.; AlOthman, M.R.; Alkhulaifi, M.M.; Mirgany, T.O.; Elsayim, R. In-silico studies of glutathione peroxidase4 activators as candidate for multiple sclerosis management. J. Saudi Chem. Soc. 2022, 26, 101554. [Google Scholar] [CrossRef]
- Pineda, L.H.; Tecuapa-Flores, E.D.; Hernández, J.G.; Thangarasu, P.; Vázquez Ramos, J.M. Ruthenium complex of bis(benzimidazole-yl-ethyl)sulfide as chemo-sensor for selective recognition of chloride ion, and its application in real bacterial samples. Inorg. Chim. Acta 2021, 522, 120354. [Google Scholar] [CrossRef]
- Camacho-Mendoza, R.L.; Gutiérrez-Moreno, E.; Guzmán-Percástegui, E.; Aquino-Torres, E.; Cruz-Borbolla, J.; Rodríguez-Ávila, J.A.; Alvarado-Rodríguez, J.G.; Olvera-Neria, O.; Thangarasu, P.; Medina-Franco, J.L. Density Functional Theory and Electrochemical Studies: Structure–Efficiency Relationship on Corrosion Inhibition. J. Chem. Inf. Model. 2015, 55, 2391–2402. [Google Scholar] [CrossRef]
- Guadalupe, H.J.; Narayanan, J.; Pandiyan, T. Synthesis, molecular structure and spectral analysis: DFT–TDDFT computational study of ruthenium complex of tetradentate N,N′-bis(benzimidazole-2yl-ethyl)-ethylenediamine. J. Mol. Struct. 2011, 989, 70–79. [Google Scholar] [CrossRef]
- Najiya, A.; Panicker, C.Y.; Sapnakumari, M.; Narayana, B.; Sarojini, B.K.; Van Alsenoy, C. Molecular structure, FT-IR, first order hyperpolarizability, NBO analysis, HOMO and LUMO, MEP analysis of (E)-3-(4-chlorophenyl)-1-(4-fluorophenyl)prop-2-en-1-one by HF and density functional methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 133, 526–533. [Google Scholar] [CrossRef]
- Lutoshkin, M.A.; Petrov, A.I.; Kuznetsov, B.N.; Kazachenko, A.S. Aqueous Complexation of Morin and Its Sulfonate Derivative with Lanthanum(III) and Trivalent Lanthanides. J. Solut. Chem. 2019, 48, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Sarkar, U. Chapter 13 Chemical reactivity dynamics in ground and excited electronic states. In Theoretical and Computational Chemistry; Toro-Labbé, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 19, pp. 269–286. [Google Scholar]
- Kolandaivel, P.; Praveena, G.; Selvarengan, P. Study of atomic and condensed atomic indices for reactive sites of molecules. J. Chem. Sci. 2005, 117, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Raja, M.; Raj Muhamed, R.; Muthu, S.; Suresh, M.; Muthu, K. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV–Visible), Fukui function, antimicrobial and molecular docking study of (E)-1-(3-bromobenzylidene)semicarbazide by DFT method. J. Mol. Struct. 2017, 1130, 374–384. [Google Scholar] [CrossRef]
- Çankaya, N.; Tanış, E. Synthesis, characterization and in-silico estimation of the toxic potential of N-(4-nitrophenyl)methacrylamide. Mater. Res. Express 2018, 6, 025310. [Google Scholar] [CrossRef]
- Sheeba, B.Q.; Michael Mary, M.S.; Amalanathan, M.; Job, C.B. Structural and vibrational spectral investigation on the identification of Non-Linear Optical properties and wave function analyses (electrostatic potential, electron localisation function, localised orbital locator) of 3-Ethoxy Salicilaldehyde. Mol. Simul. 2021, 47, 1217–1233. [Google Scholar] [CrossRef]
- Lutoshkin, M.A.; Kazachenko, A.S. Assessment of various density functionals and solvation models to describe acid-base, spectral and complexing properties of thiobarbituric and barbituric acids in aqueous solution. J. Comput. Methods Sci. Eng. 2017, 17, 851–863. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Khalid, M.; Shaheen, M.A.; Tahir, M.N.; Khan, M.U.; Braga, A.A.C.; Shad, H.A. Synthesis and XRD, FT-IR vibrational, UV–vis, and nonlinear optical exploration of novel tetra substituted imidazole derivatives: A synergistic experimental-computational analysis. J. Phys. Chem. Solids 2018, 115, 265–276. [Google Scholar] [CrossRef]
- Tahir, M.N.; Khalid, M.; Islam, A.; Ali Mashhadi, S.M.; Braga, A.A.C. Facile synthesis, single crystal analysis, and computational studies of sulfanilamide derivatives. J. Mol. Struct. 2017, 1127, 766–776. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Sagaama, A.; Issaoui, N.; Al-Dossary, O.; Kazachenko, A.S.; Wojcik, M.J. Non covalent interactions and molecular docking studies on morphine compound. J. King Saud Univ.-Sci. 2021, 33, 101606. [Google Scholar] [CrossRef]
- Jayabharathi, J.; Thanikachalam, V.; Jayamoorthy, K. Optical properties of 1,2-diaryl benzimidazole derivatives – A combined experimental and theoretical studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 115, 74–78. [Google Scholar] [CrossRef]
- Lutoshkin, M.A.; Petrov, A.I.; Malyar, Y.N.; Kazachenko, A.S. Interaction of Rare-Earth Metals and Some Perfluorinated β-Diketones. Inorg. Chem. 2021, 60, 3291–3304. [Google Scholar] [CrossRef]
- Akman, F. A comparative study based on molecular structure, spectroscopic, electronic, thermodynamic and NBO analysis of some nitrogen-containing monomers. Polym. Bull. 2021, 78, 663–693. [Google Scholar] [CrossRef]
- Ouaket, A.; Chraka, A.; Raissouni, I.; Amrani, M.A.E.; Berrada, M.; Knouzi, N. Synthesis, spectroscopic (13C/1H-NMR, FT-IR) investigations, quantum chemical modelling (FMO, MEP, NBO analysis), and antioxidant activity of the bis-benzimidazole molecule. J. Mol. Struct. 2022, 1259, 132729. [Google Scholar] [CrossRef]
- Profant, V.; Johannessen, C.; Blanch, E.W.; Bouř, P.; Baumruk, V. Effects of sulfation and the environment on the structure of chondroitin sulfate studied via Raman optical activity. Phys. Chem. Chem. Phys. 2019, 21, 7367–7377. [Google Scholar] [CrossRef]
- Akman, F. Spectroscopic investigation, HOMO–LUMO energies, natural bond orbital (NBO) analysis and thermodynamic properties of two-armed macroinitiator containing coumarin with DFT quantum chemical calculations. Can. J. Phys. 2016, 94, 583–593. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Malyar, Y.N.; Ghatfaoui, S.; Issaoui, N.; Al-Dossary, O.; Wojcik, M.J.; Kazachenko, A.S.; Miroshnikova, A.V.; Berezhnaya, Y.D. A density functional theory calculations of infrared spectra of galactomannan butyl ether. J. Mol. Struct. 2022, 1251, 131998. [Google Scholar] [CrossRef]
- Majoube, M. Vibrational spectra of guanine. A normal coordinate analysis. J. Mol. Struct. 1984, 114, 403–406. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Guassian, Inc. GaussView; Carnergie Office Parck-Building 6; Guassian, Inc.: Pittsburgh, PA, USA; Semichem, Inc.: Shawnee, KS, USA, 2000–2003.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3LYP/6-311++G(d,p) | |||||
|---|---|---|---|---|---|
| Bond Lengths (A°) | Bond Angles (°) | ||||
| Atom Position | Theo. | Exp. | Atom Position | Theo. | Exp. |
| C1-C2 | 1.415 | 1.398(2) | C2-C1-C6 | 122.3 | 107.0 |
| C1-C6 | 1.396 | 1.390(3) | C2-C1-N26 | 105.1 | 107.6 |
| C1-N26 | 1.386 | 1.391(2) | C6-C1-N26 | 132.6 | 107.5 |
| C2-C3 | 1.399 | 1.371(3) | C1-C2-C3 | 119.9 | 107.6 |
| C2-N27 | 1.387 | 1.367(2) | C1-C3-N27 | 110.1 | 107.6 |
| C3-C4 | 1.389 | 1.393(3) | C3-C2-N27 | 130.0 | 118.9(2) |
| C3-H8 | 1.084 | 0.930 | C2-C3-C4 | 118.1 | 107.7(2) |
| C4-C5 | 1.408 | 1.368(3) | C2-C3-H8 | 120.2 | 124(1) |
| C4-H9 | 1.084 | 0.930 | C4-C3-H8 | 121.7 | 128(1) |
| C5-C6 | 1.391 | 1.371(3) | C3-C4-C5 | 121.4 | 119.8(2) |
| C5-H10 | 1.084 | 0.930 | C3-C4-H9 | 119.6 | 109.4(2) |
| C6-H11 | 1.084 | 0.930 | H9-C4-C9 | 119.0 | 130.8(2) |
| C7-H12 | 1.082 | 0.80(2) | C4-C5-C6 | 121.5 | 105.5(2) |
| C7-N26 | 1.377 | 1.365(3) | C4-C5-H10 | 119.2 | 132.3(2) |
| C7-N27 | 1.306 | 1.317(2) | C6-C5-H10 | 119.2 | 122.2(2) |
| C13-H14 | 1.094 | 0.970 | C1-C6-C5 | 116.9 | 121.0 |
| C13-H15 | 1.095 | 0.970 | C1-C6-H11 | 122.2 | 118.0(2) |
| C13-C16 | 1.534 | 1.449(4) | C5-C6-H12 | 120.9 | 121.0 |
| C13-N26 | 1.458 | - | H12-C7-N26 | 120.7 | 121.5 |
| C16-H17 | 1.096 | 0.969 | H12-C7-N27 | 125.0 | 116.9(2) |
| C16-H18 | 1.095 | 0.970 | N26-C7-N27 | 114.3 | 121.6 |
| C16-C19 | 1.533 | 1.510(4) | H14-C13-H15 | 106.7 | 119.2 |
| C19-H20 | 1.097 | 0.970 | H14-C13-C16 | 110.2 | 121.6(2) |
| C19-H21 | 1.097 | 0.971 | C14-C13-N26 | 107.4 | 119.2 |
| C19-C22 | 1.531 | 1.417(5) | H15-C13-C16 | 110.5 | 121.5(2) |
| C22-H23 | 1.094 | 0.960 | H15-C13-N26 | 108.2 | 119.2 |
| C22-H24 | 1.093 | 0.960 | C16-C13-N26 | 113.6 | 119.3 |
| C22-H25 | 1.094 | 0.959 | C13-C16-H17 | 108.8 | 123.1(2) |
| C13-C16-H18 | 109.3 | 124.8(2) | |||
| C13-C16-C19 | 112.4 | 112.1(2) | |||
| H17-C16-H18 | 106.4 | 108.3 | |||
| H17-C16-C19 | 109.9 | 108.3 | |||
| H18-C16-C19 | 109.8 | 115.7(2) | |||
| C16-C19-H20 | 109.4 | 107.5 | |||
| C16-C19-H21 | 109.4 | 108.4 | |||
| C16-C19-C22 | 112.9 | 108.3 | |||
| H20-C19-H21 | 106.1 | 107.8 | |||
| H20-C19-C22 | 109.4 | 107.9 | |||
| H21-C19-C22 | 109.4 | 117.8(3) | |||
| C19-C22-H23 | 111.2 | 107.3 | |||
| C19-C22-H24 | 111.3 | 107.9 | |||
| C19-C22-H25 | 111.2 | 107.8 | |||
| H23-C22-H24 | 107.7 | 109.5 | |||
| H23-C22-H25 | 107.6 | 109.5 | |||
| H24-C22-H25 | 107.7 | 109.5 | |||
| C1-N26-C7 | 105.9 | 109.5 | |||
| C1-N26-C13 | 127.4 | 109.4 | |||
| C7-N26-C13 | 126.7 | 109.4 | |||
| C2-N27-C7 | 104.6 | 105.4(2) | |||
| Interactions Types | ∇2ρ(r) (a.u.) | ρ (r) (a.u.) | G(r) (a.u.) | V(r) (a.u.) | H(r) (a.u.) | ε | Einteractions kJ/mol |
|---|---|---|---|---|---|---|---|
| RCP1 | 0.0140 | 0.0040 | 0.0029 | −0.0023 | 0.0006 | −2.0003 | −3.02 |
| RCP2 | −0.0176 | 0.3888 | 0.3888 | −0.6782 | −0.5589 | 0.0041 | −890.05 |
| RCP3 | 0.0071 | 0.0018 | 0.0013 | −0.0009 | 0.0005 | −1.9466 | −1.18 |
| RCP4 | 0.1673 | 0.0226 | 0.0342 | −0.0265 | 0.0077 | −1.2088 | −34.13 |
| NRCP1 | 0.3993 | 0.0560 | 0.0939 | −0.0879 | 0.0059 | −1.2860 | −112.89 |
| NRCP2 | 0.3980 | 0.0558 | 0.0936 | −0.0876 | 0.0059 | 1.2847 | −112.89 |
| NRCP3 | 0.1671 | 0.0225 | 0.0341 | −0.0265 | 0.0077 | −1.2076 | −34.13 |
| C43-H45…N26 | 0.0123 | 0.0033 | 0.0026 | −0.0022 | 0.0005 | 0.4900 | −28.88 |
| N31-H54…N26 | 0.0874 | 0.0211 | 0.0182 | −0.0145 | 0.0037 | 0.0298 | −18.90 |
| C7-H8…H16 | 0.0069 | 0.0019 | 0.0013 | −0.0009 | 0.0004 | 1.8492 | −1.05 |
| C36-H37…H2 | 0.0063 | 0.0017 | 0.0012 | −0.0008 | 0.0004 | 2.9057 | −1.05 |
| Quantum Parameters | DFT/B3LYP/6-31++G(d, p) |
|---|---|
| EHOMO(eV) | −6.26 |
| ELUMO(eV) | −0.82 |
| EHOMO+1(eV) | −6.37 |
| ELUMO-1 (eV) | −0.81 |
| │ΔEHOMO-LUMO│ (eV) | 5.44 |
| │ΔEHOMO+1-LUMO-1│ (eV) | 5.56 |
| I | 6.26 |
| A | 0.82 |
| χ | 3.39 |
| η | 2.72 |
| µ | −3.39 |
| ω | 2.11 |
| S | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazachenko, A.S.; Tanış, E.; Akman, F.; Medimagh, M.; Issaoui, N.; Al-Dossary, O.; Bousiakou, L.G.; Kazachenko, A.S.; Zimonin, D.; Skripnikov, A.M. A Comprehensive Study of N-Butyl-1H-Benzimidazole. Molecules 2022, 27, 7864. https://doi.org/10.3390/molecules27227864
Kazachenko AS, Tanış E, Akman F, Medimagh M, Issaoui N, Al-Dossary O, Bousiakou LG, Kazachenko AS, Zimonin D, Skripnikov AM. A Comprehensive Study of N-Butyl-1H-Benzimidazole. Molecules. 2022; 27(22):7864. https://doi.org/10.3390/molecules27227864
Chicago/Turabian StyleKazachenko, Aleksandr S., Emine Tanış, Feride Akman, Mouna Medimagh, Noureddine Issaoui, Omar Al-Dossary, Leda G. Bousiakou, Anna S. Kazachenko, Dmitry Zimonin, and Andrey M. Skripnikov. 2022. "A Comprehensive Study of N-Butyl-1H-Benzimidazole" Molecules 27, no. 22: 7864. https://doi.org/10.3390/molecules27227864