
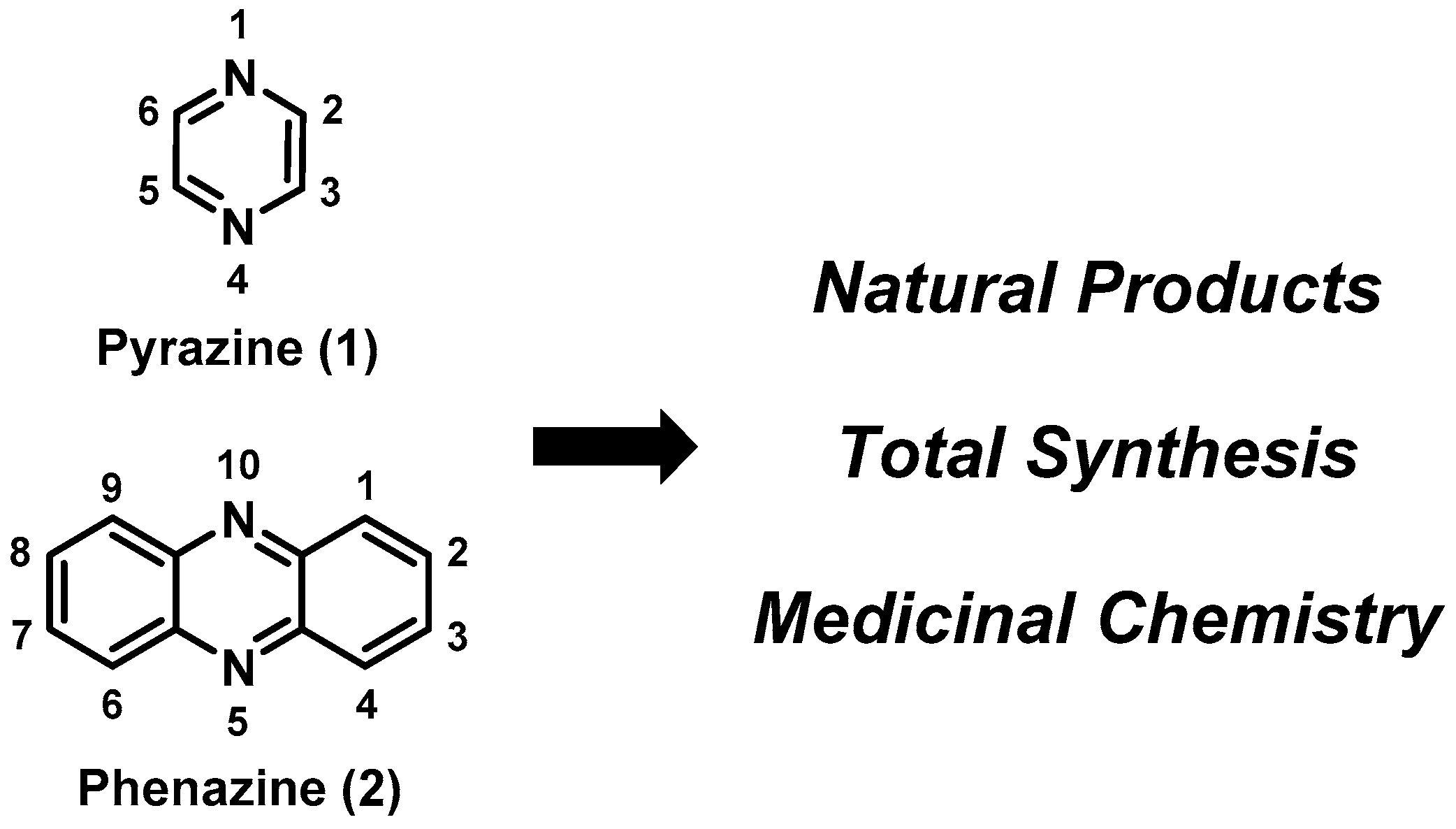
Pyrazine and Phenazine Heterocycles: Platforms for Total Synthesis and Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
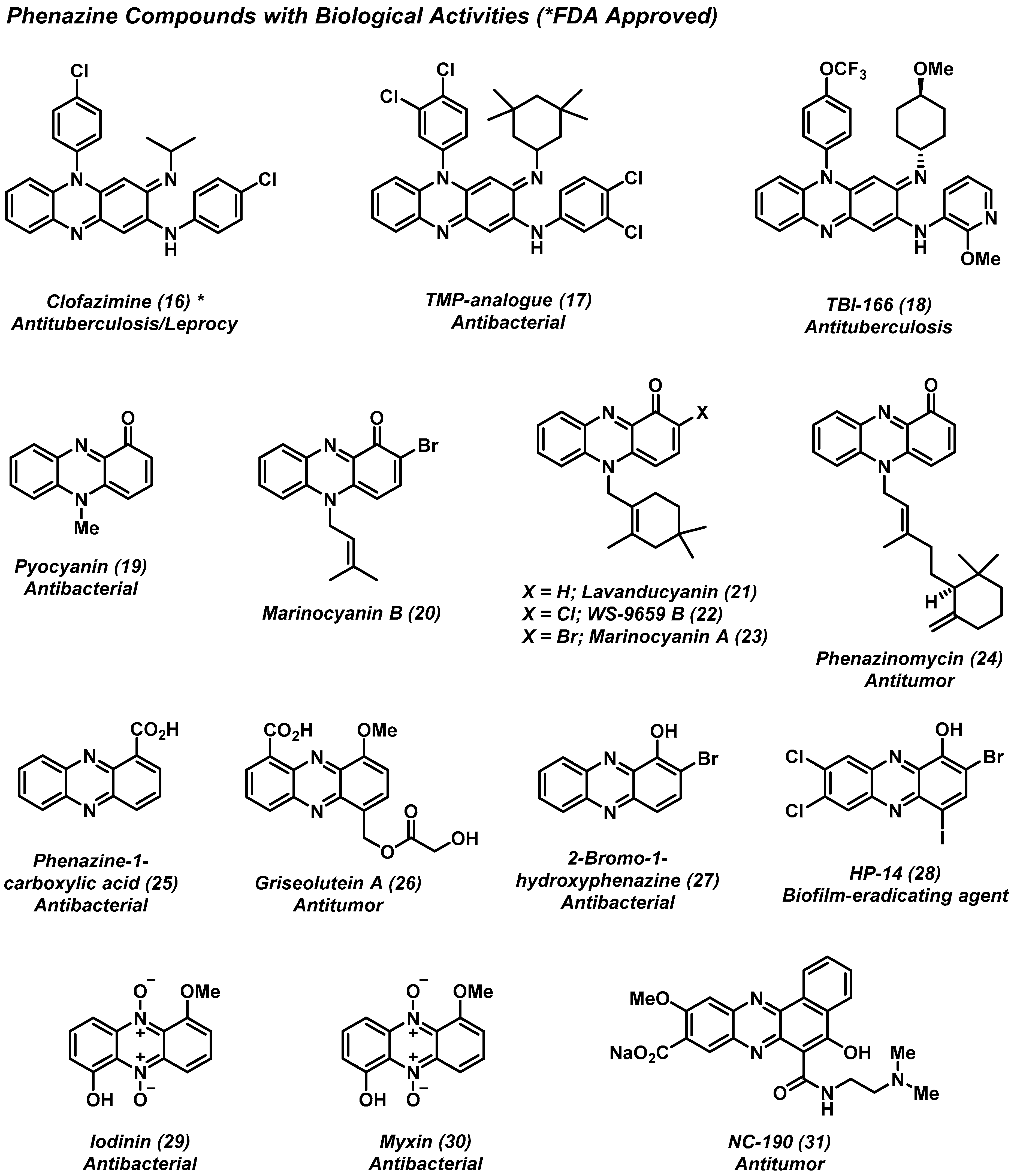
2. Pyrazines and Phenazines of Therapeutic Interest
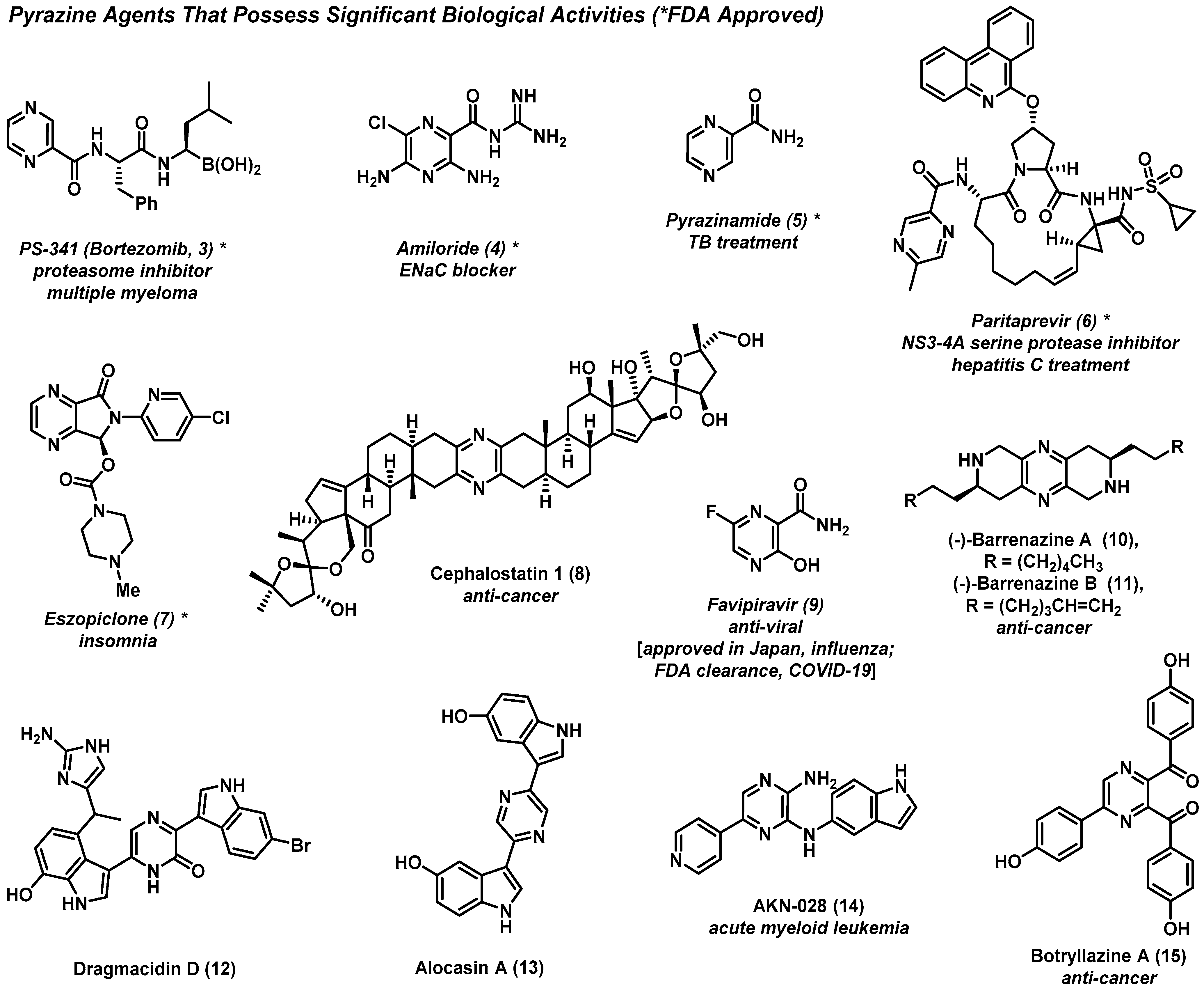
2.1. Pyrazines
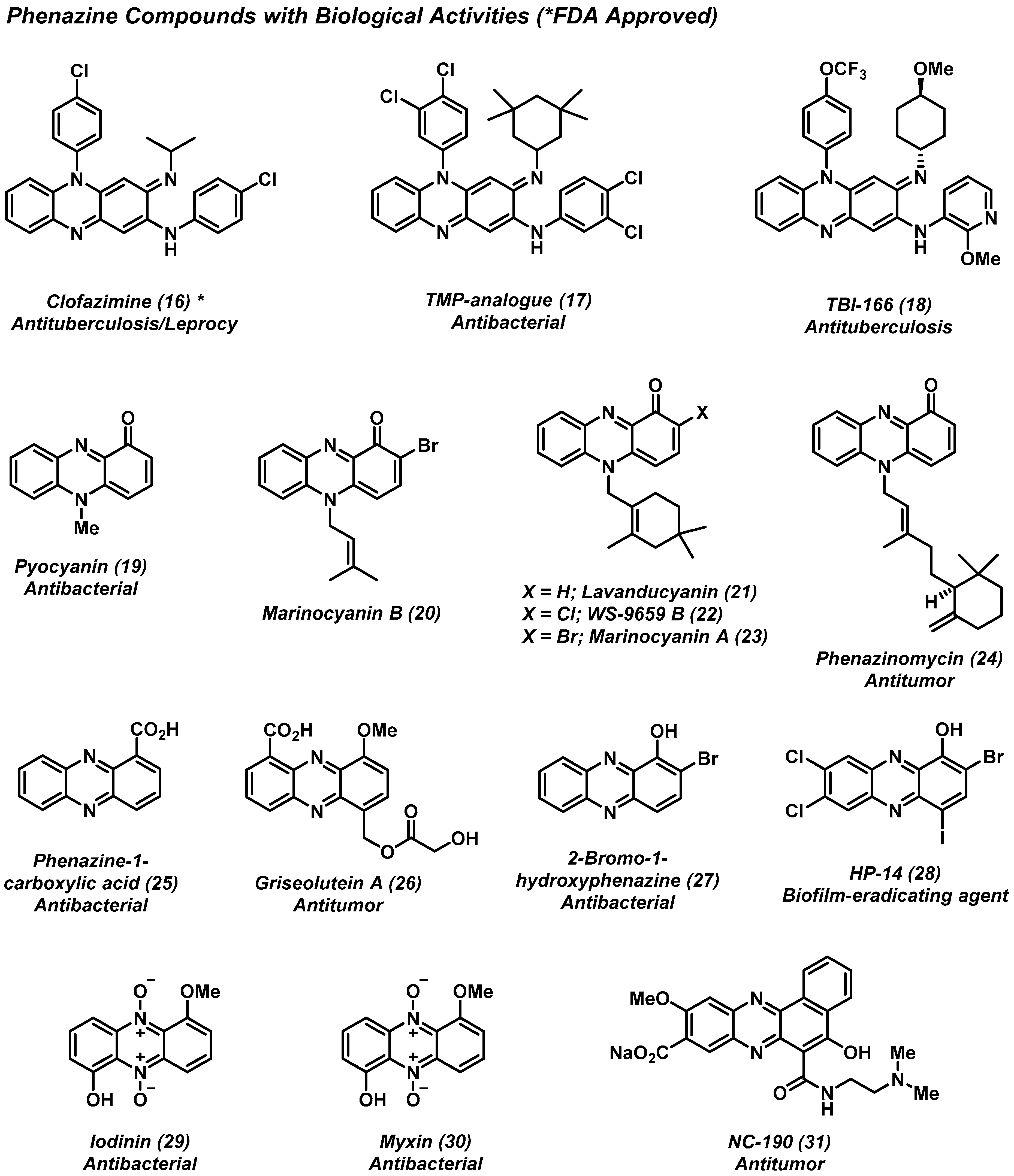
2.2. Phenazines
3. Select Total Syntheses of Natural Pyrazine Products
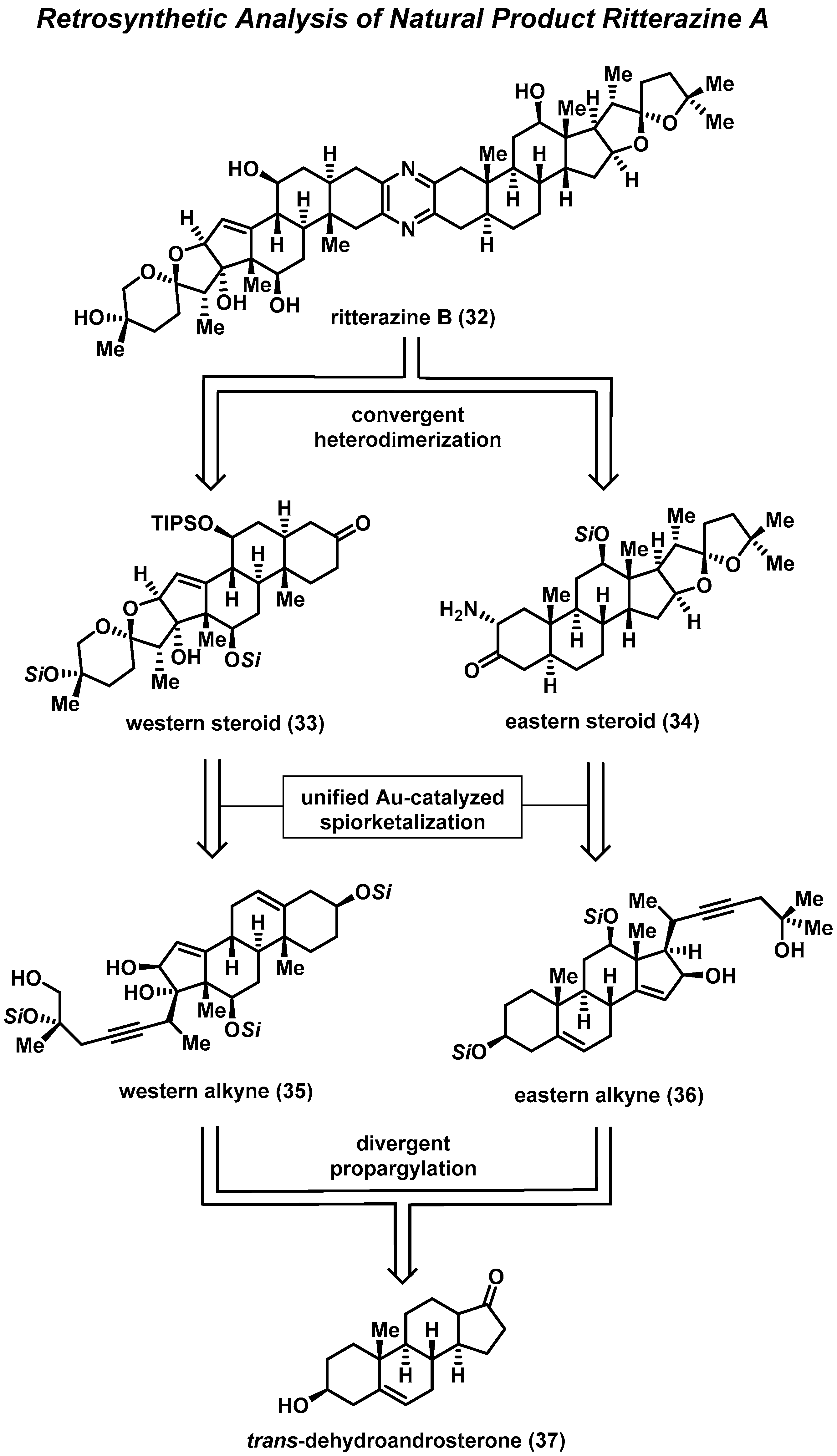
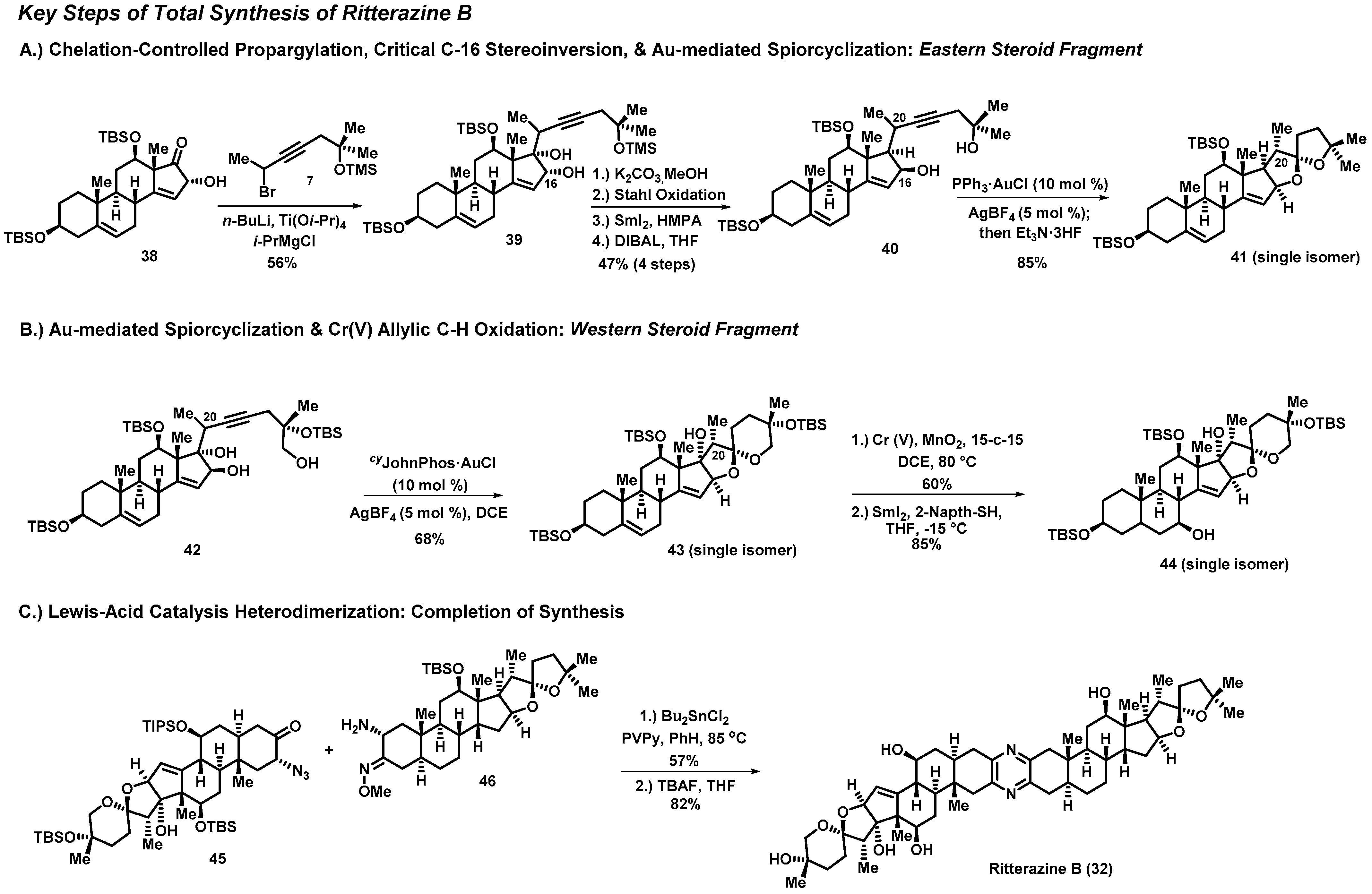
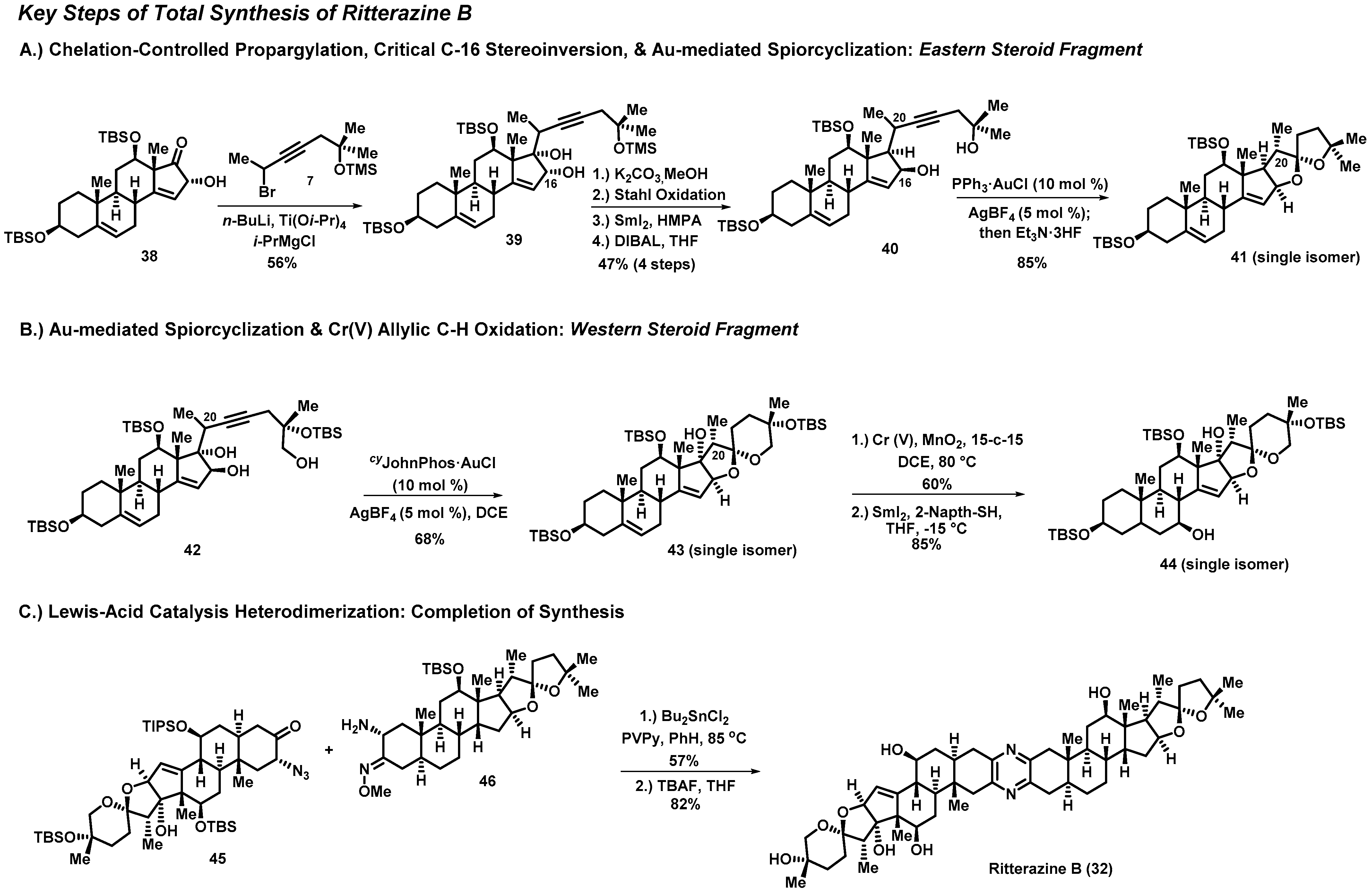
3.1. Total Synthesis of Ritterazine B
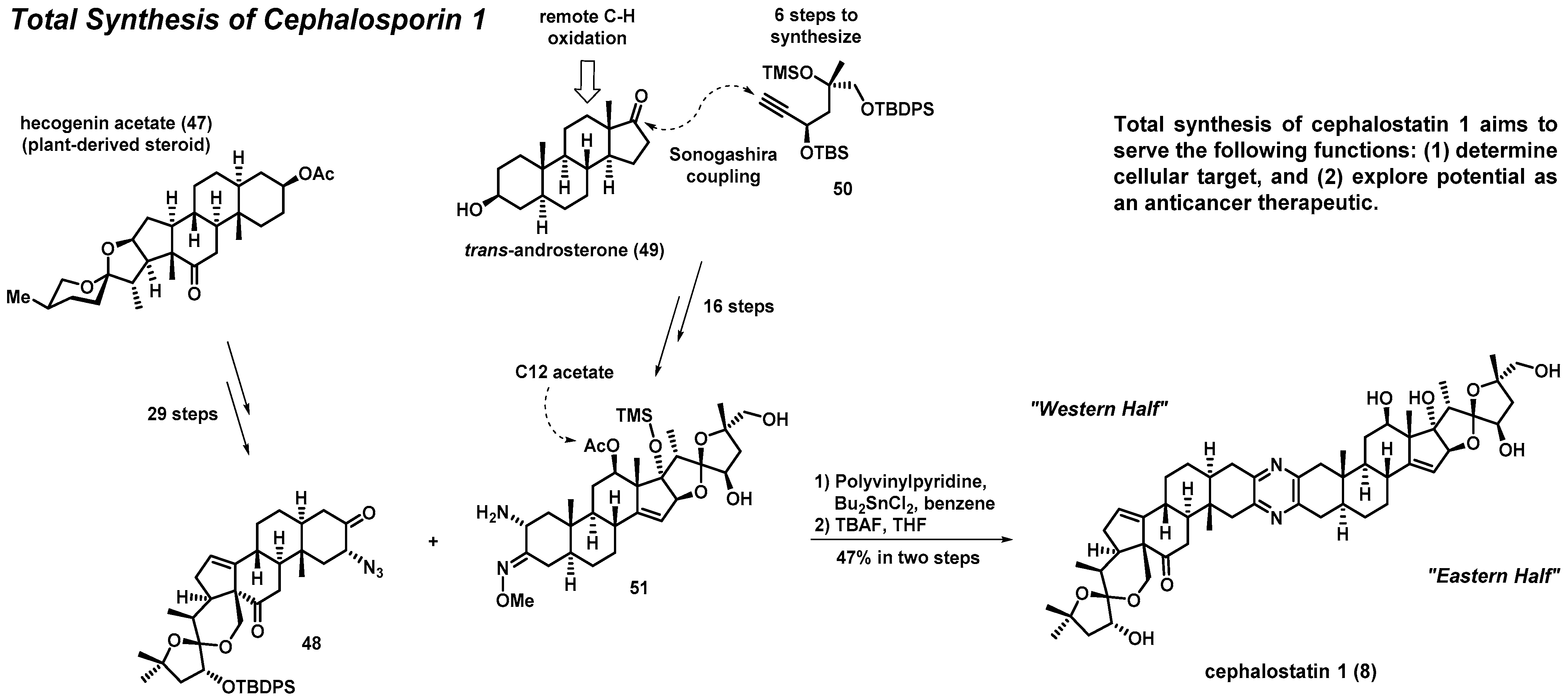
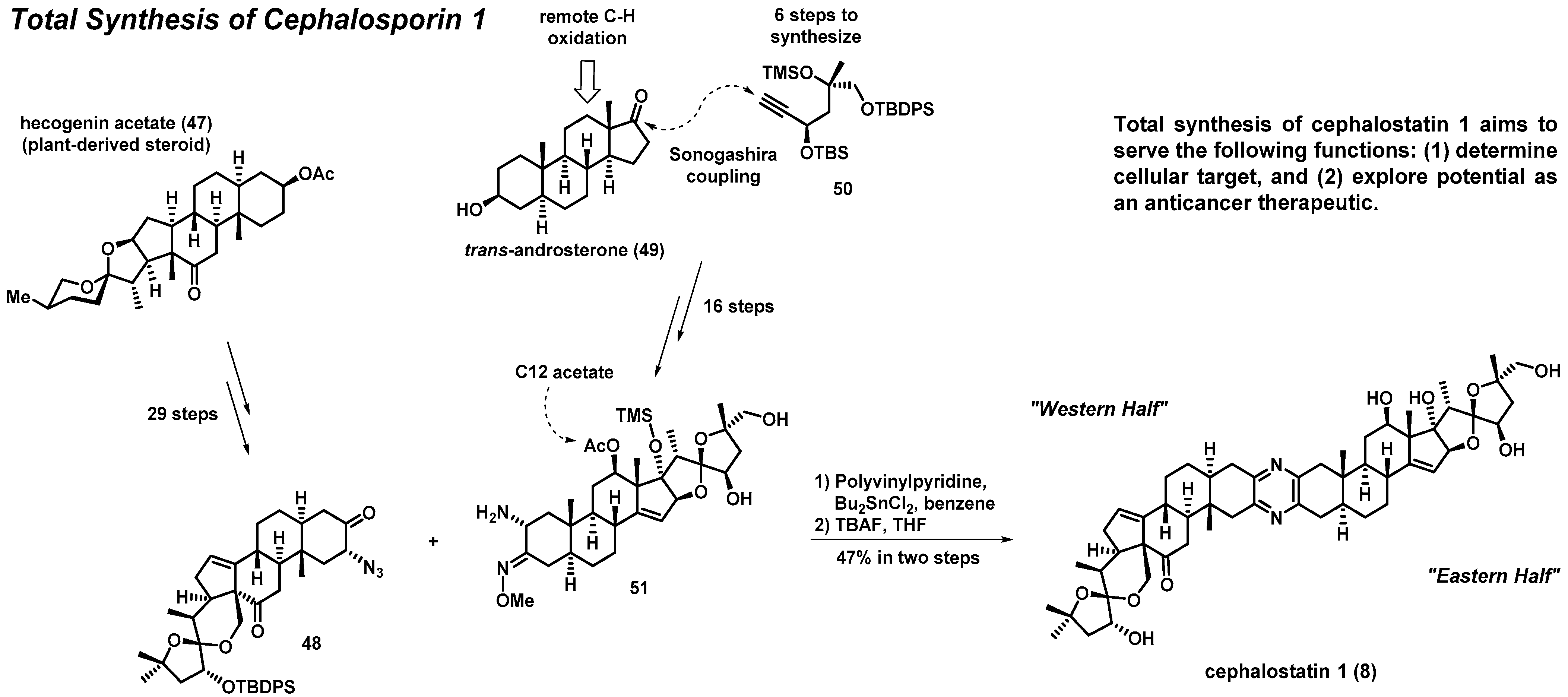
3.2. Total Synthesis of Cephalostatin 1
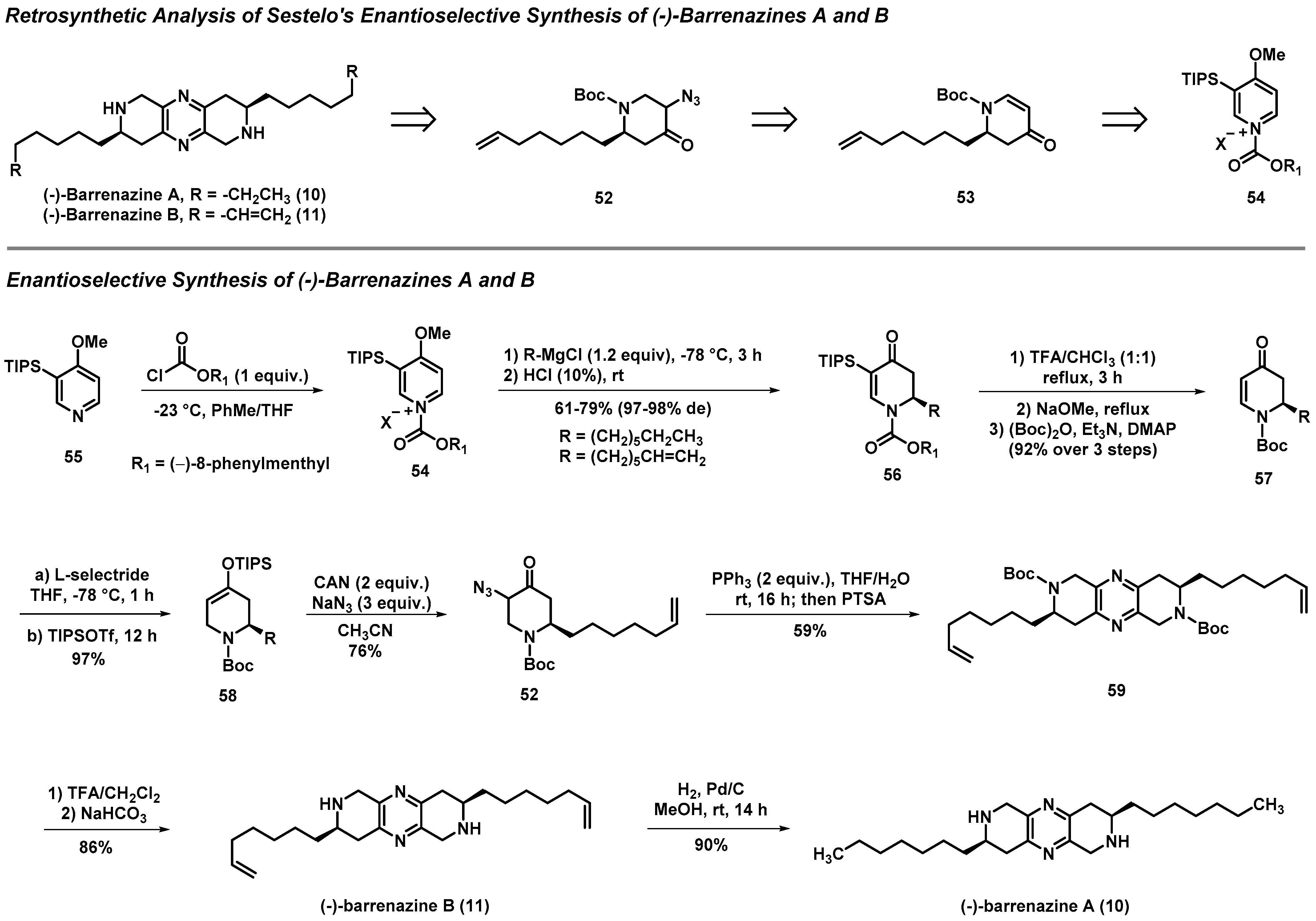
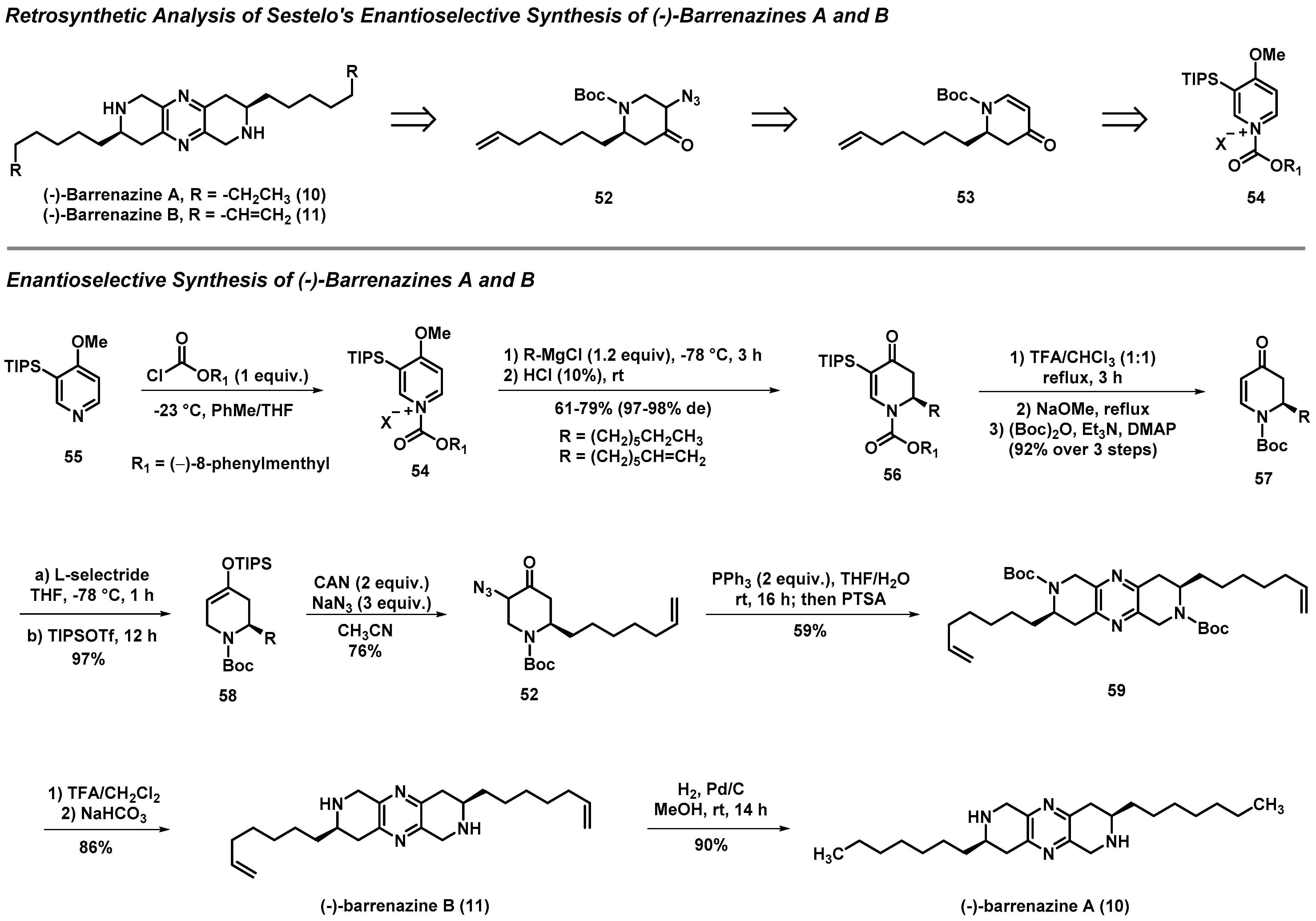
3.3. Total Syntheses of (-)-Barrenazines A and B
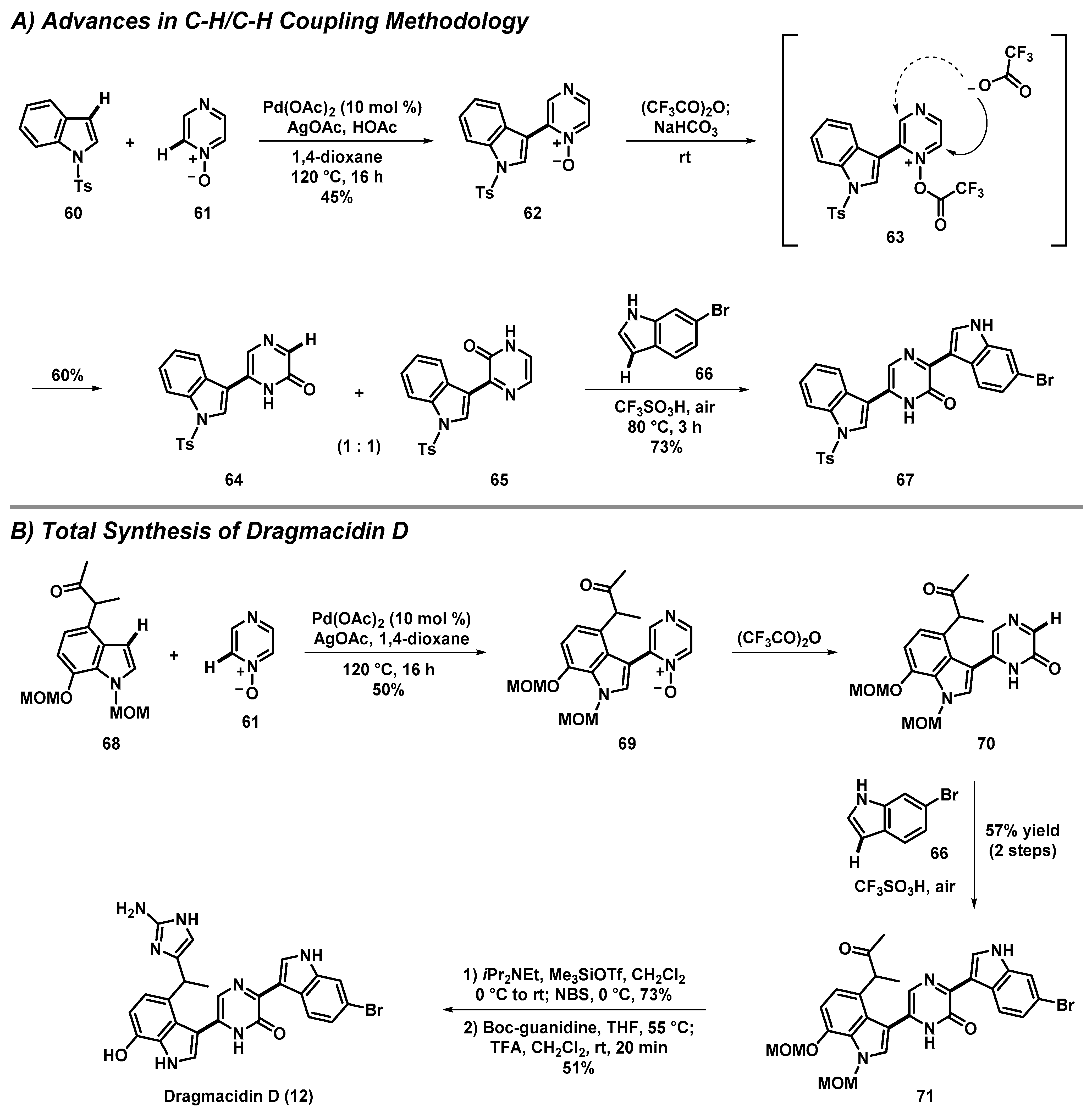
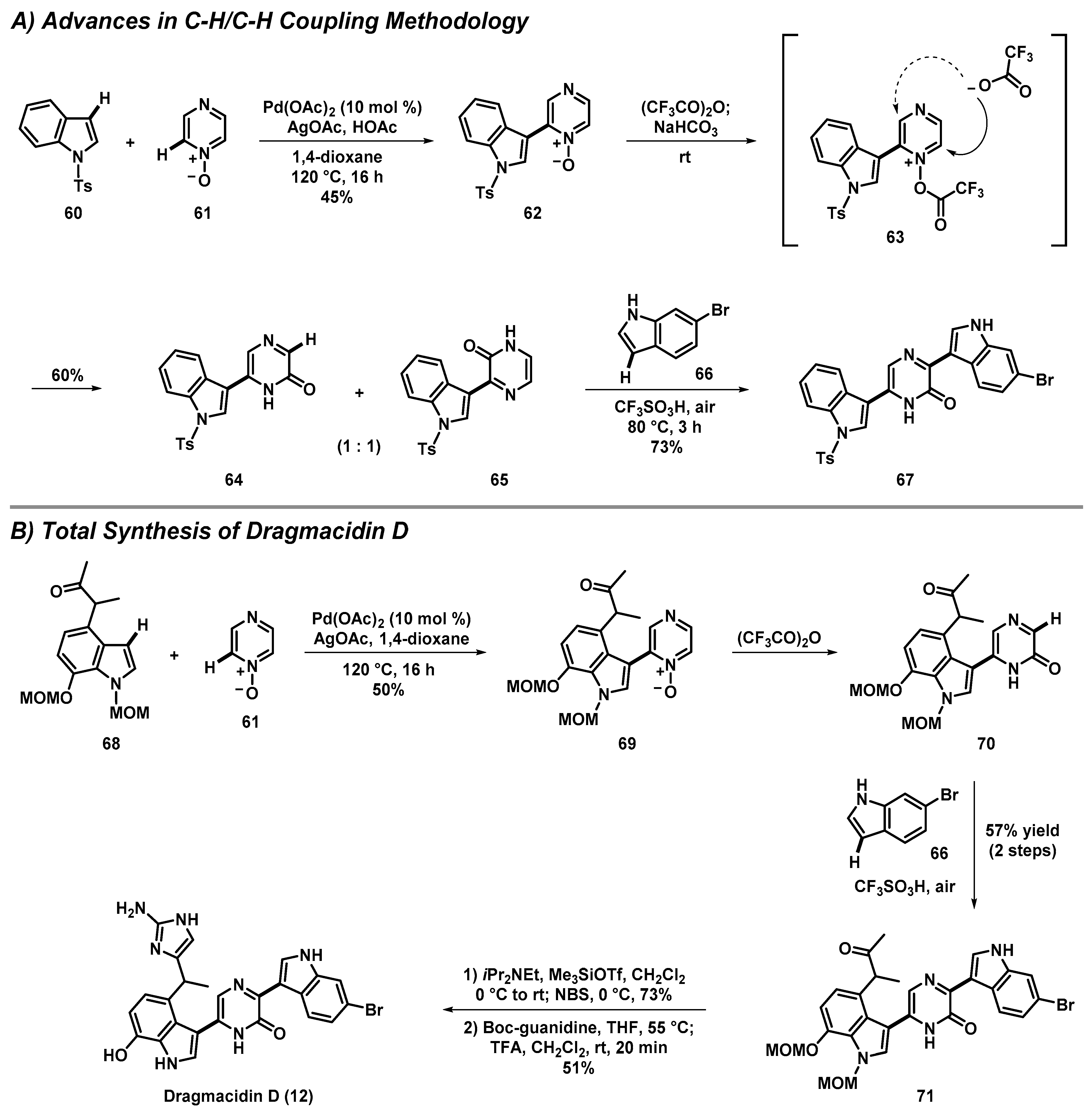
3.4. Total Synthesis of Dragmacidin D through Advances in C-H/C-H Cross-Coupling
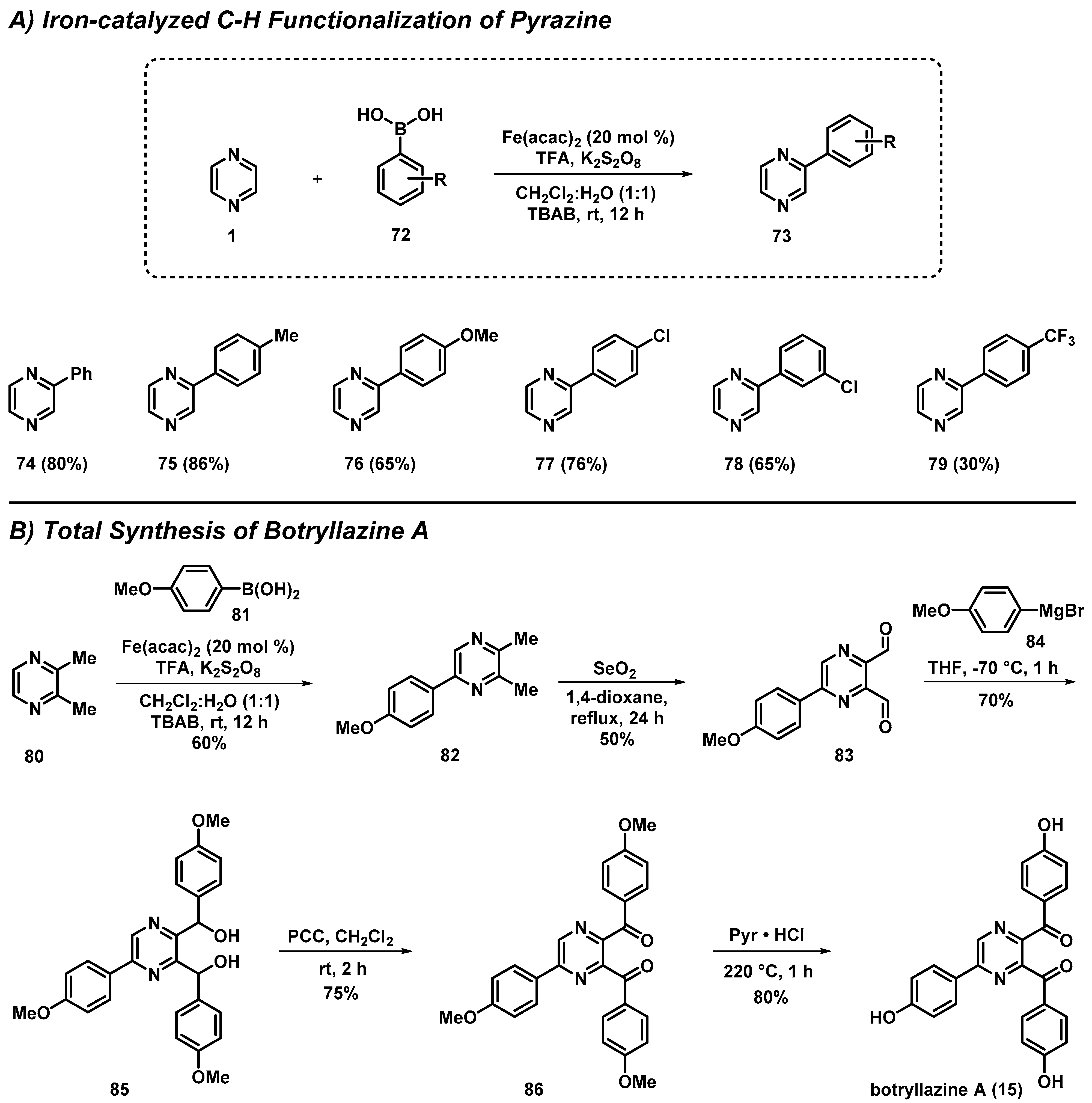
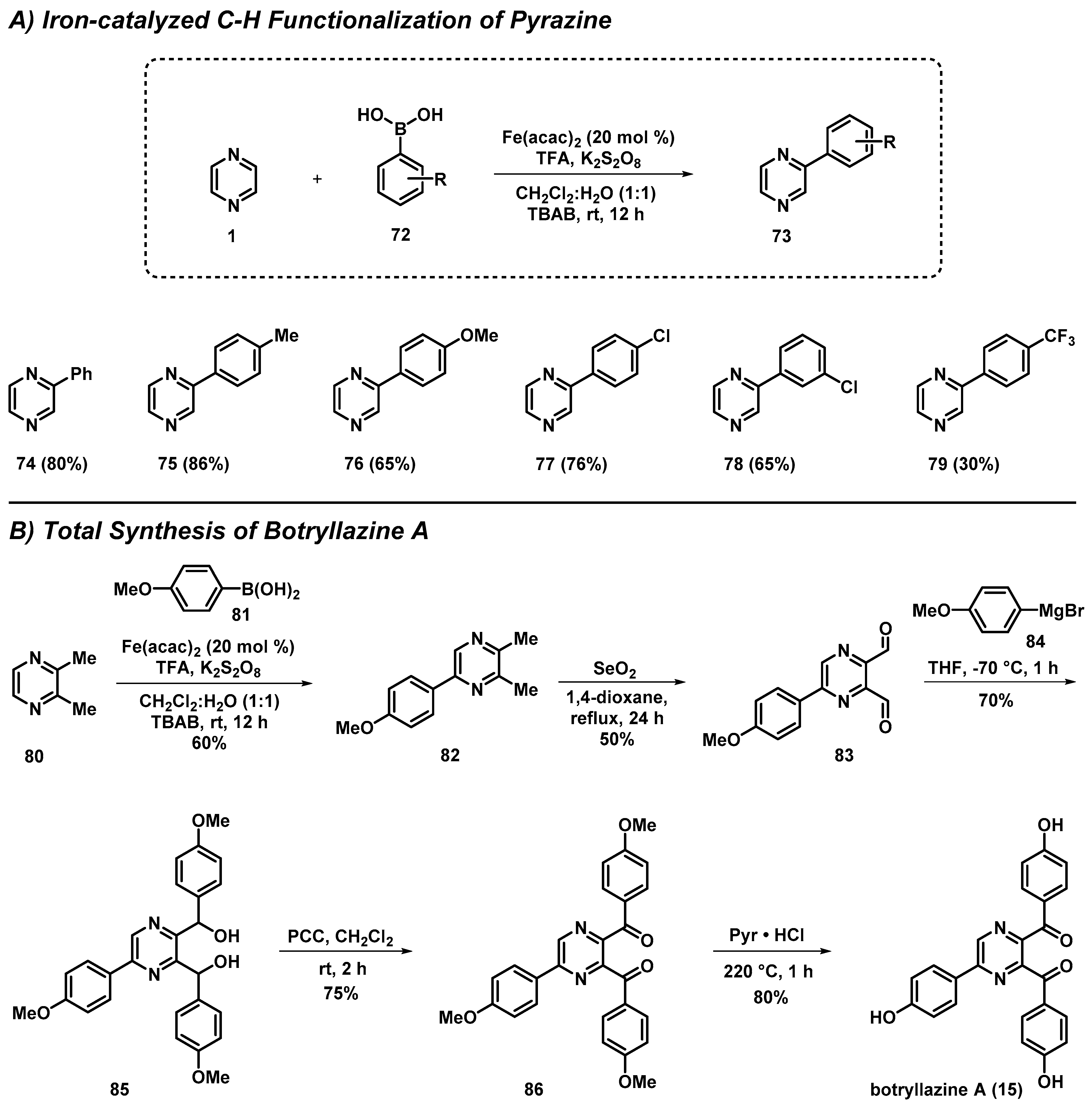
3.5. Total Synthesis of Botryllazine Involving a New C-H Functionalization of Pyrazines
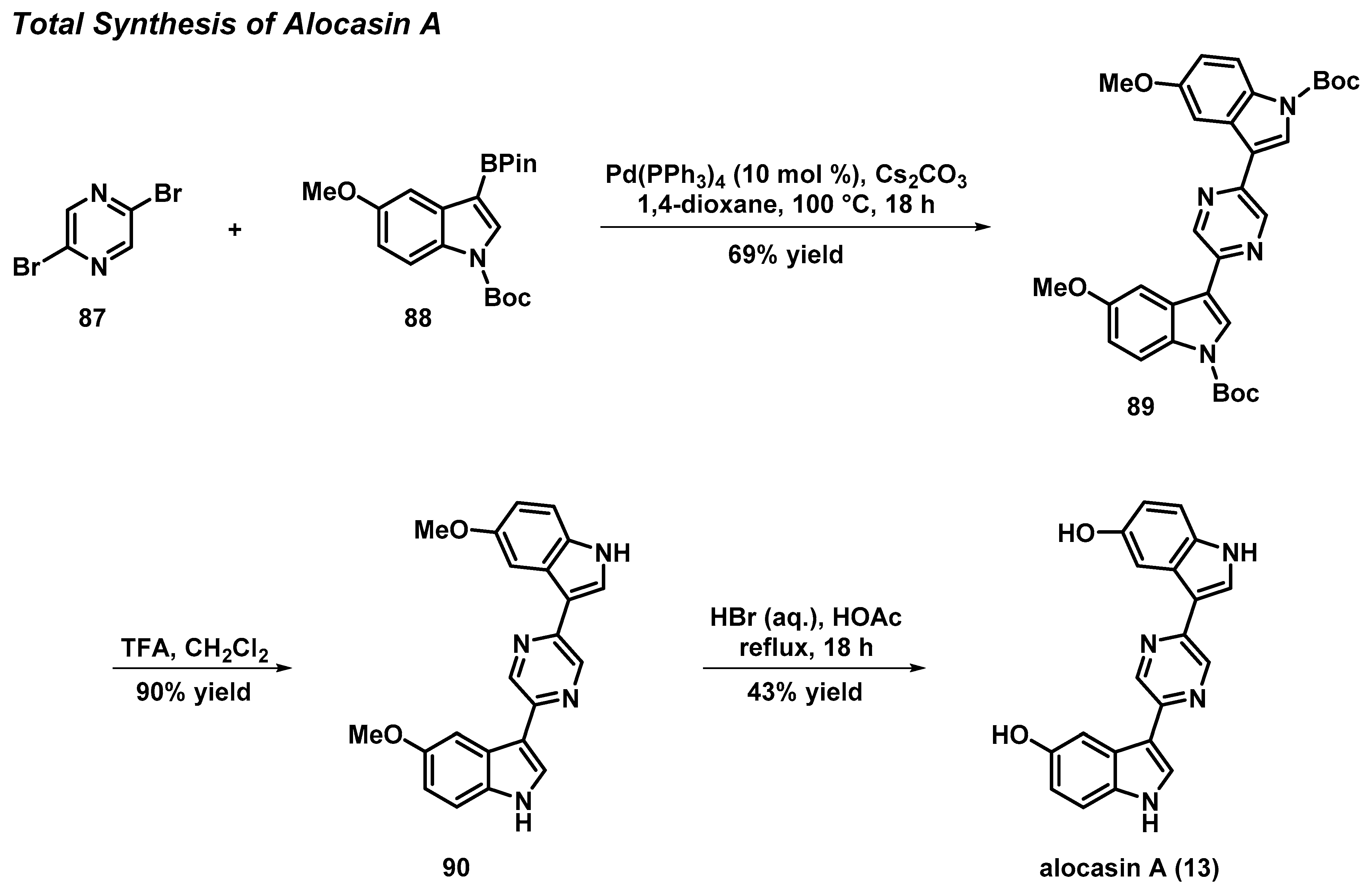
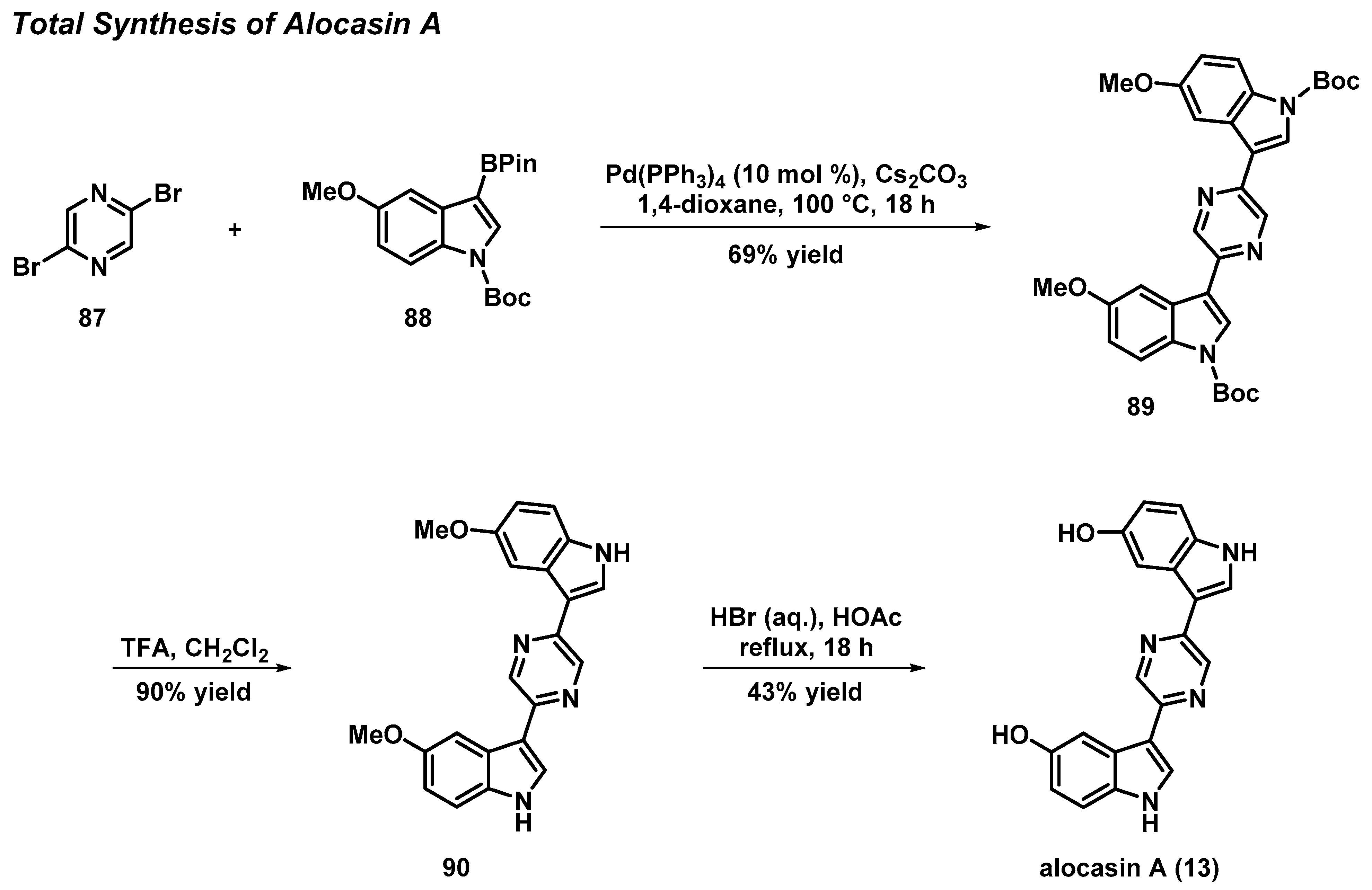
3.6. Total Synthesis of the Pyrazine Bisindole Alkaloid Alocasin A
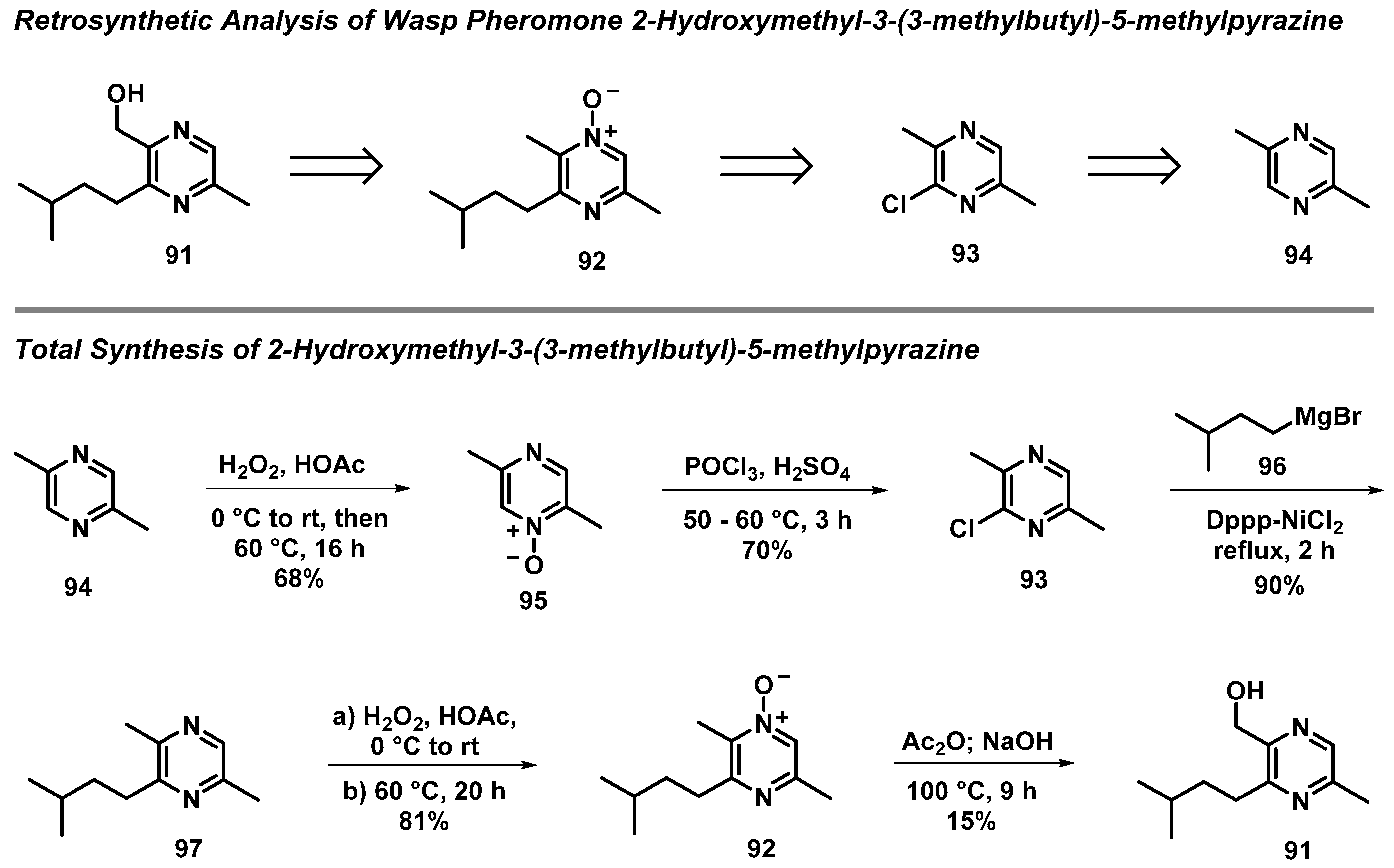
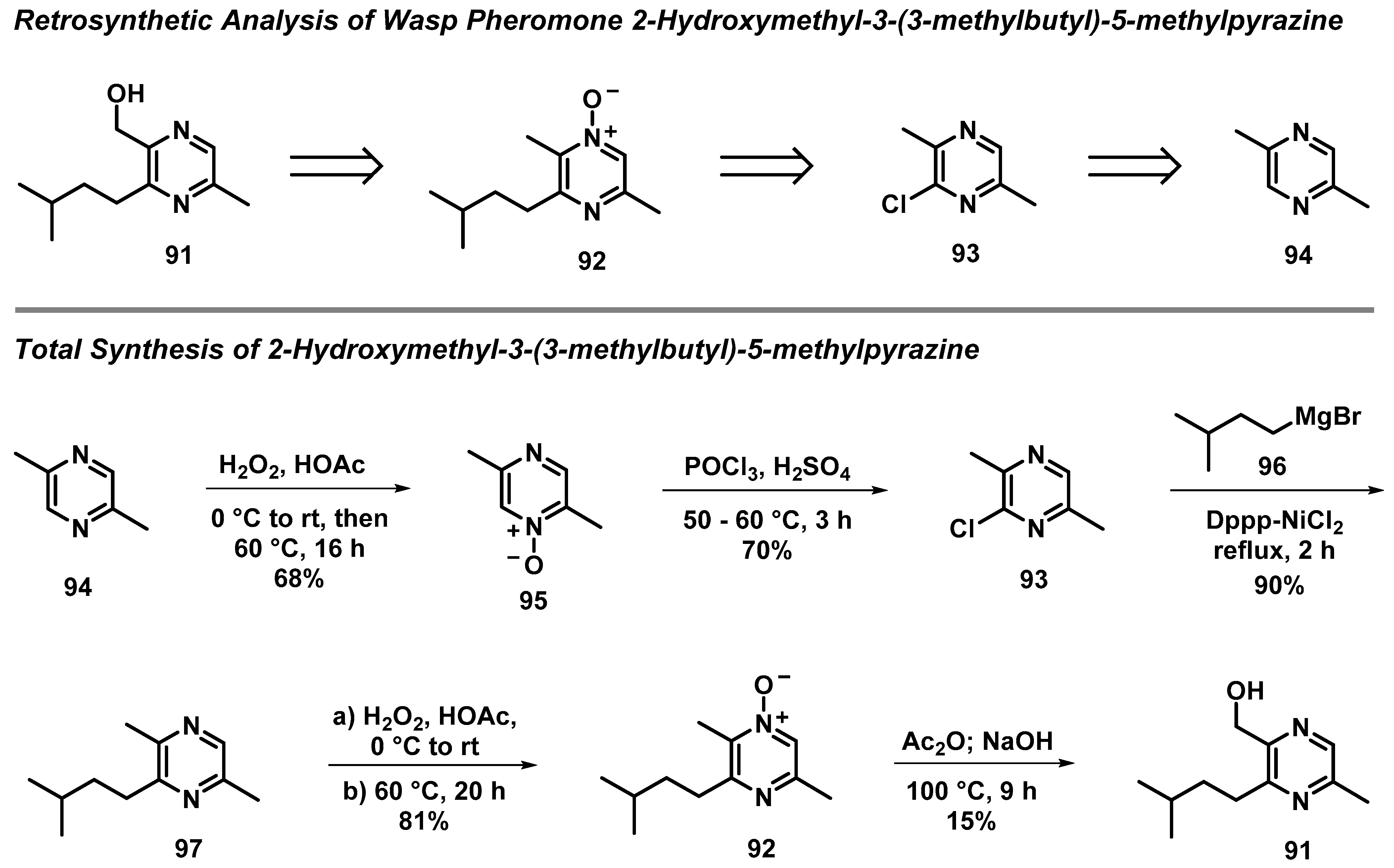
3.7. Total Synthesis of Wasp Pheromone 2-Hydroxymethyl-3-(3-methylbutyl)-5-methylpyrazine
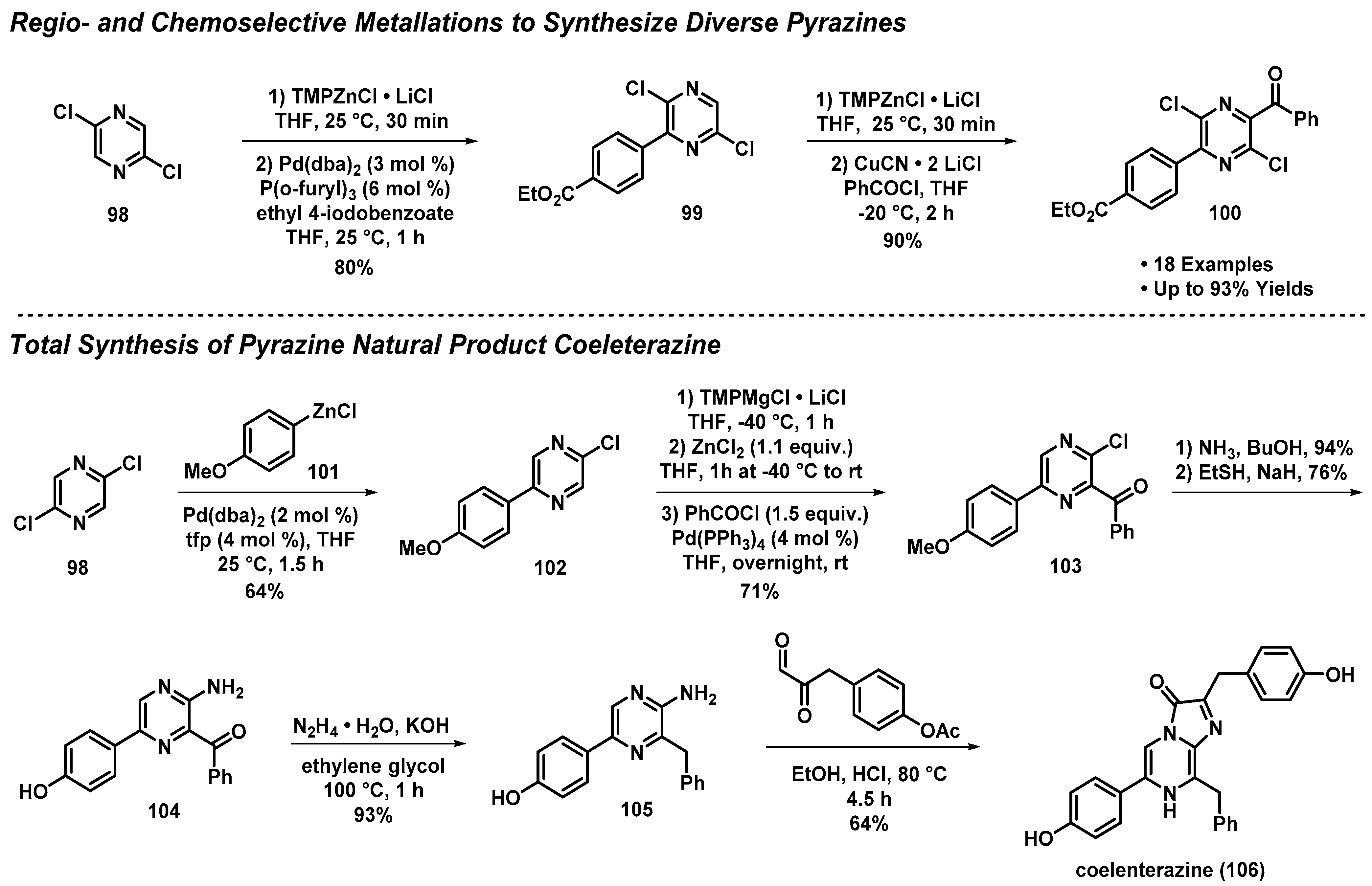
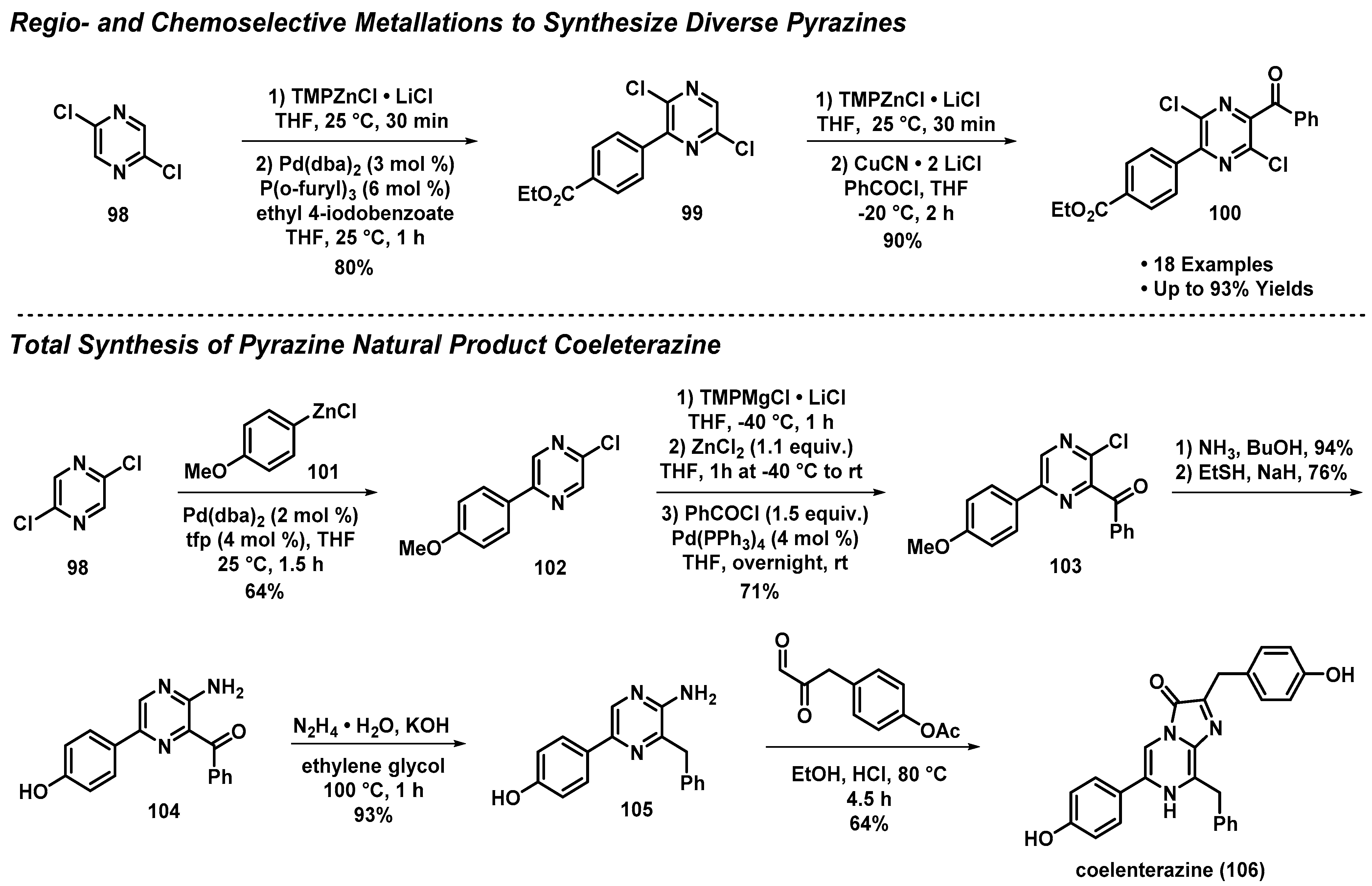
3.8. Regio- and Chemoselective Metallations to Access the Pyrazine Natural Product Coeleterazine
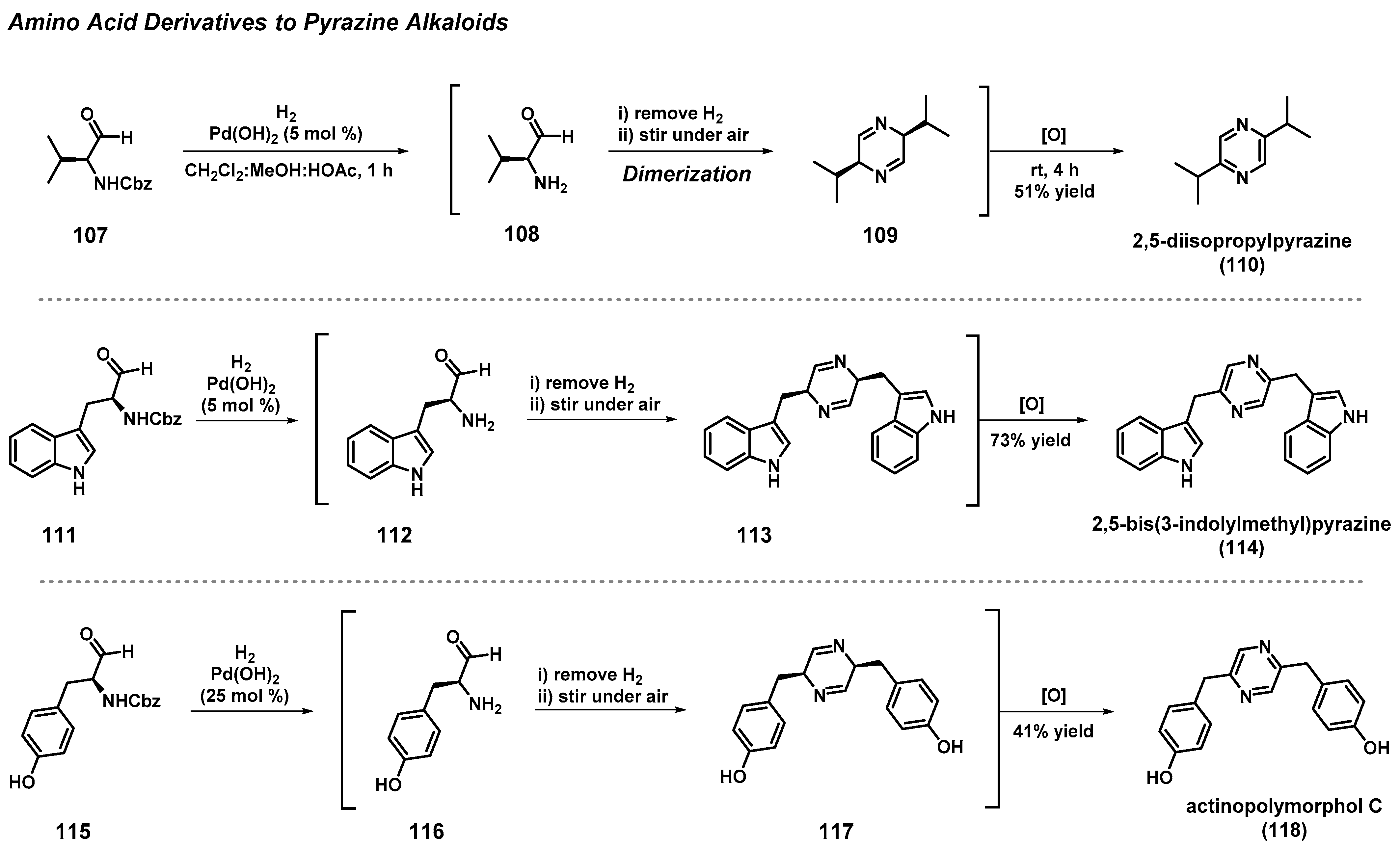
3.9. Total Synthesis of Pyrazine Alkaloids from Amino Acids
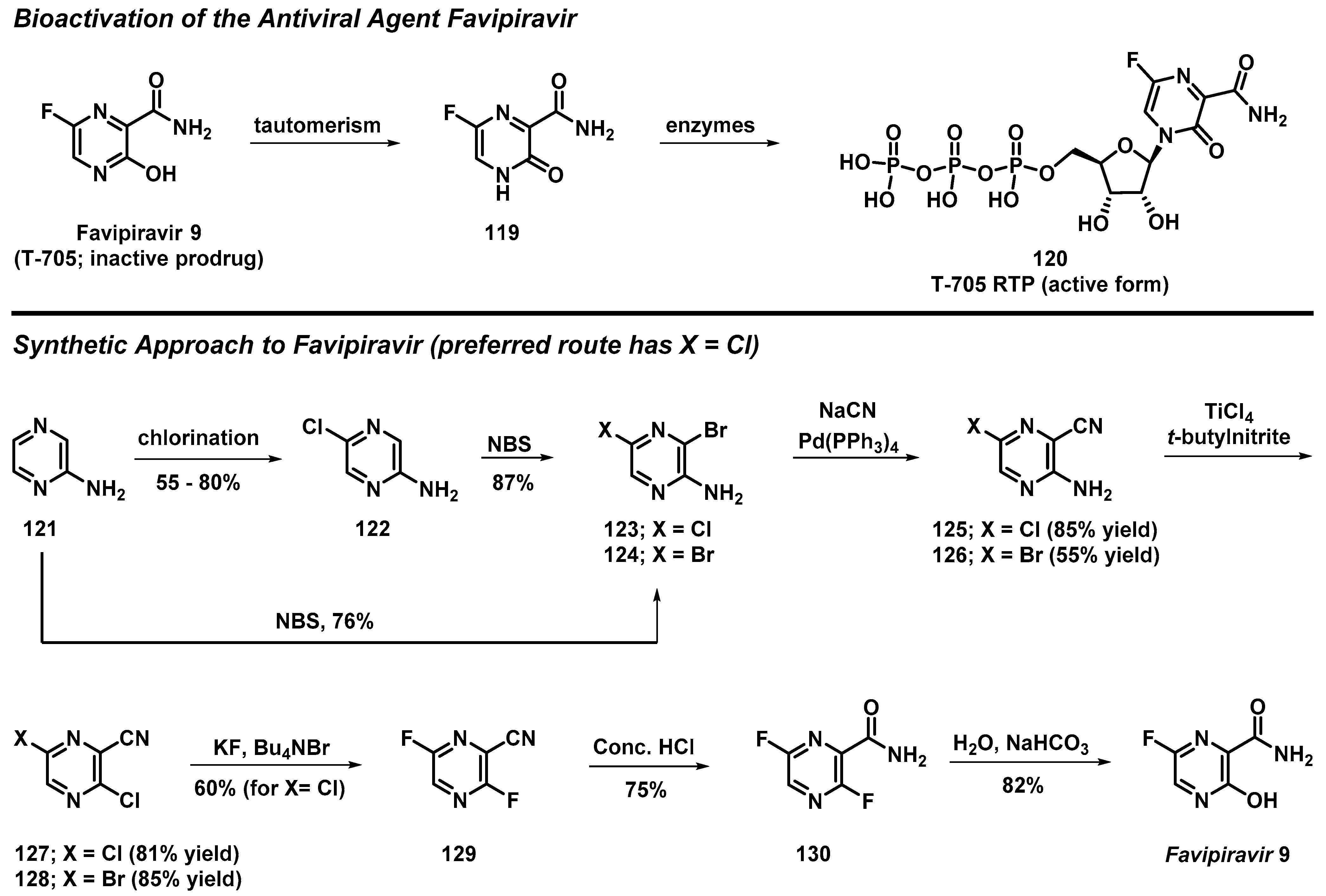
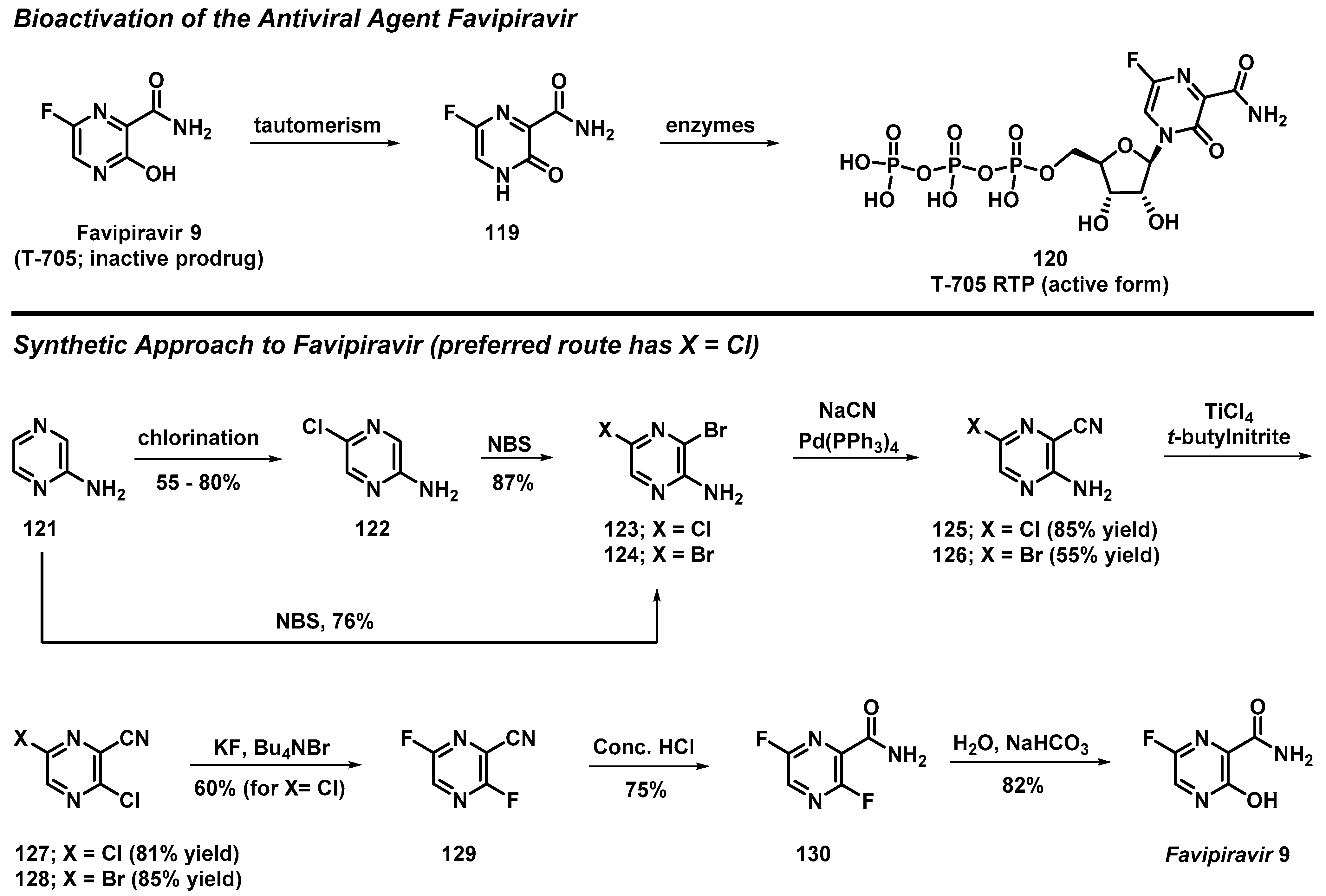
3.10. Total Synthesis of Favipiravir from 2-Aminopyrazine
4. Recent Total Synthesis of Phenazine-Containing Natural Products
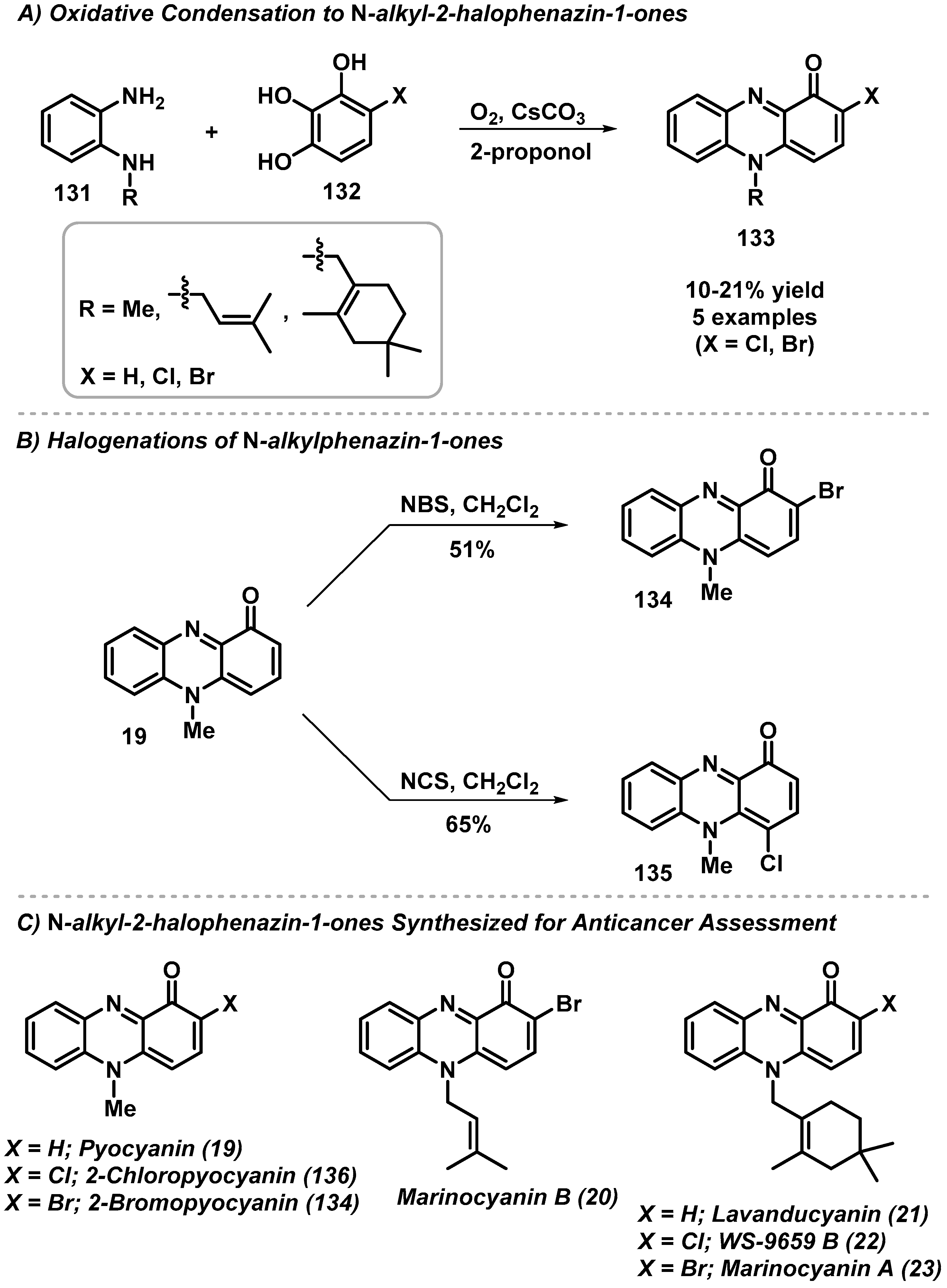
4.1. Total Synthesis of N-Alkyl-2-halophenazin-1-ones
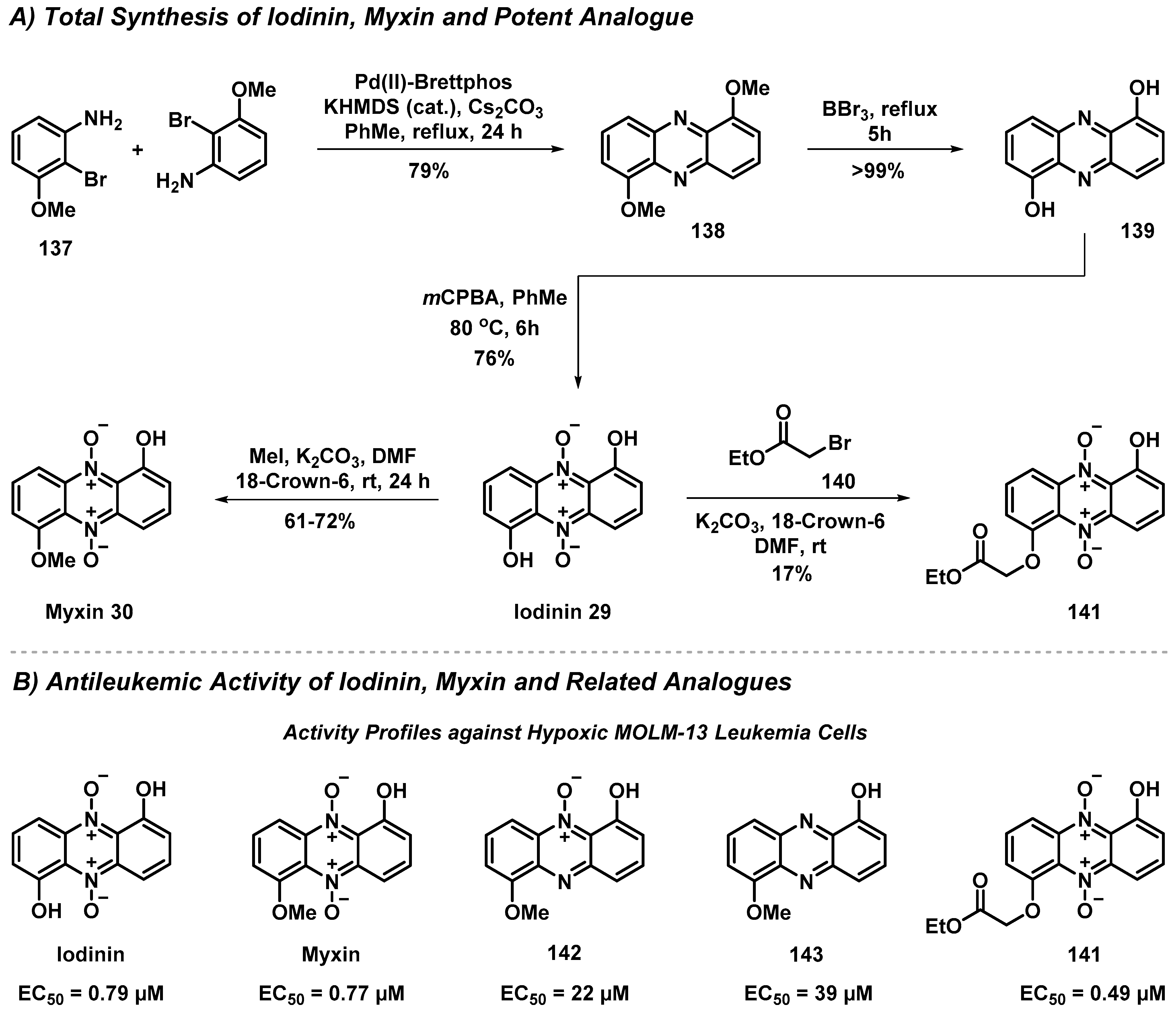
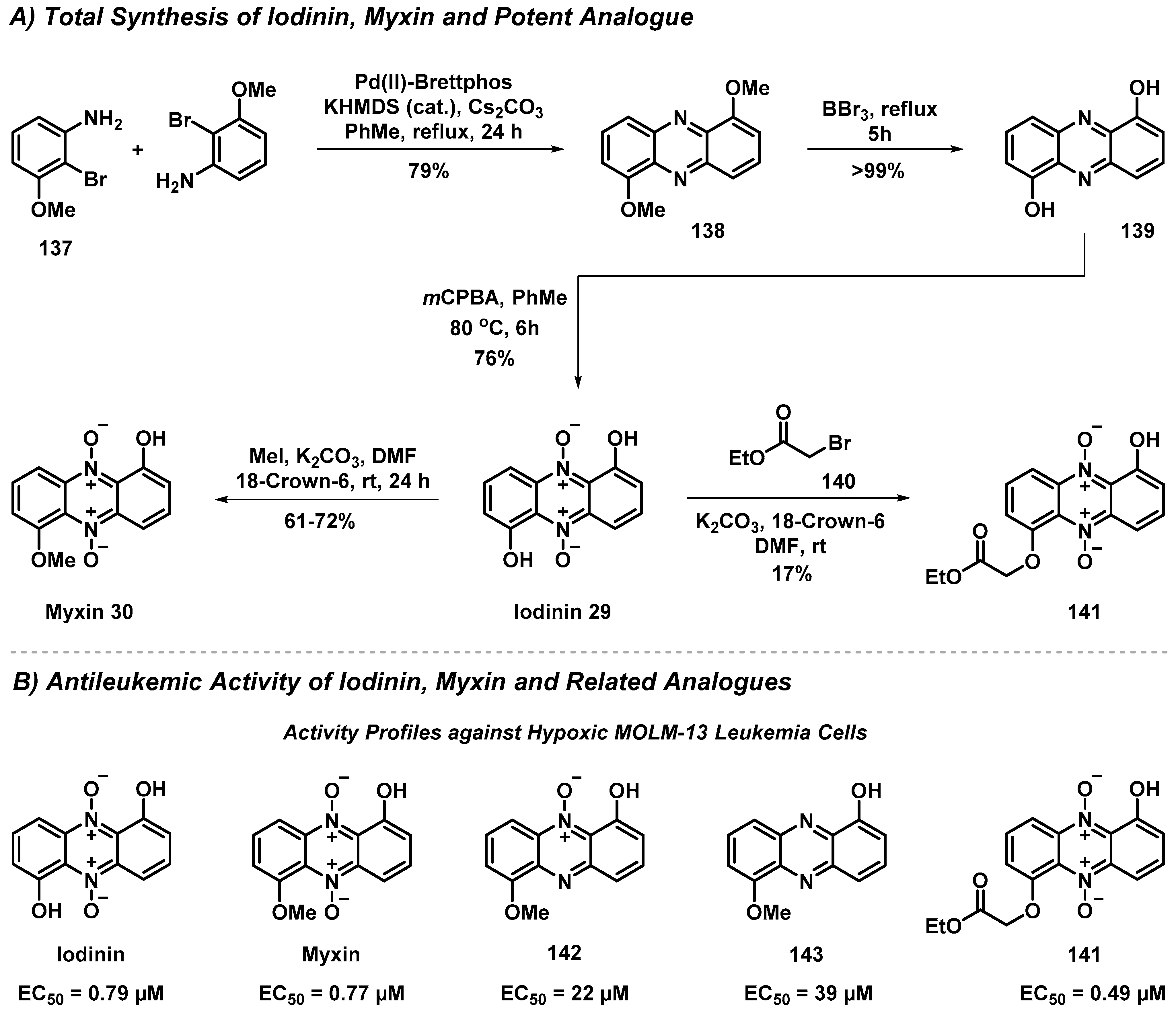
4.2. Total Synthesis and Antileukemic Evaluation of Iodinin, Myxin and Derivatives
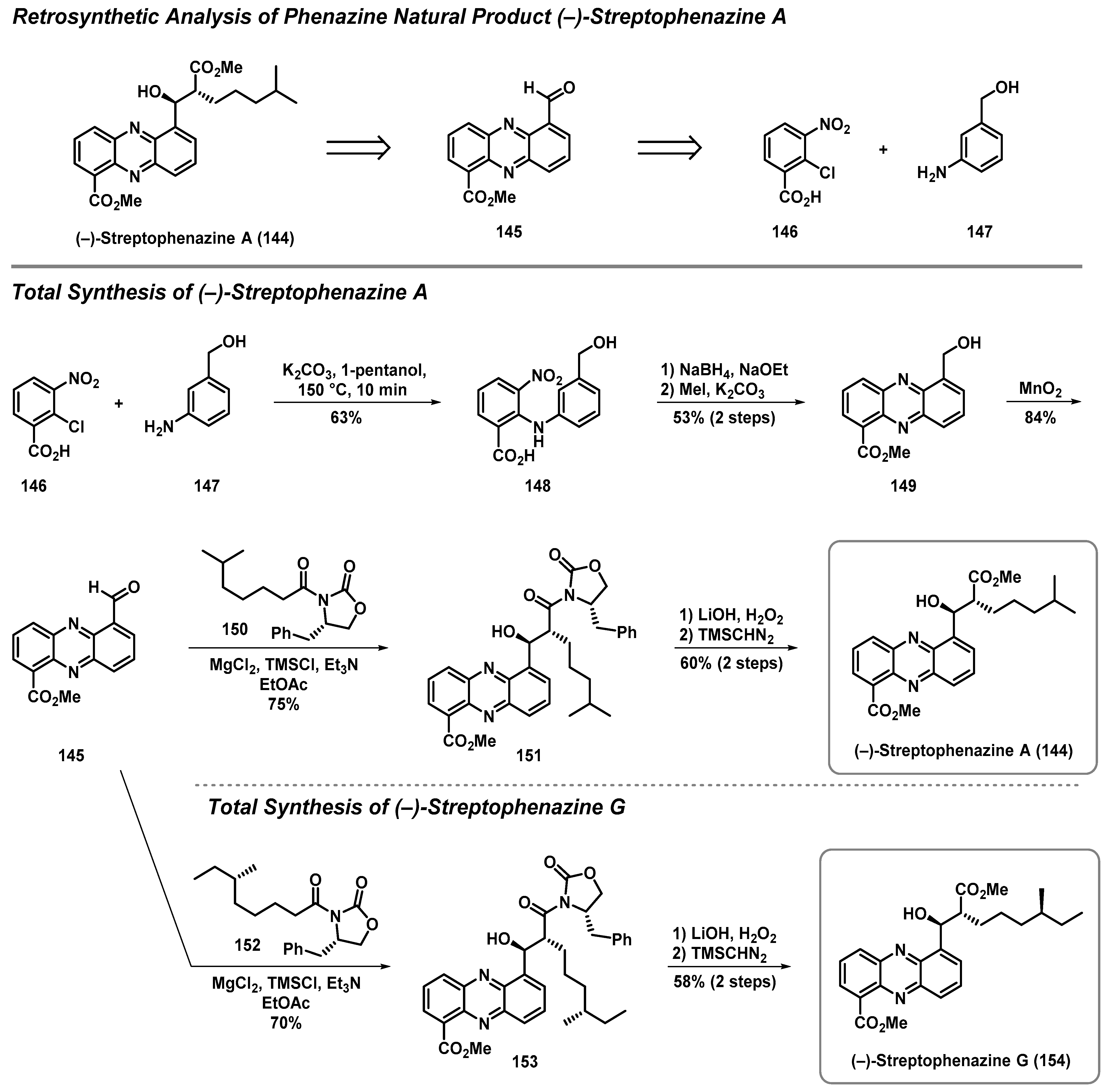
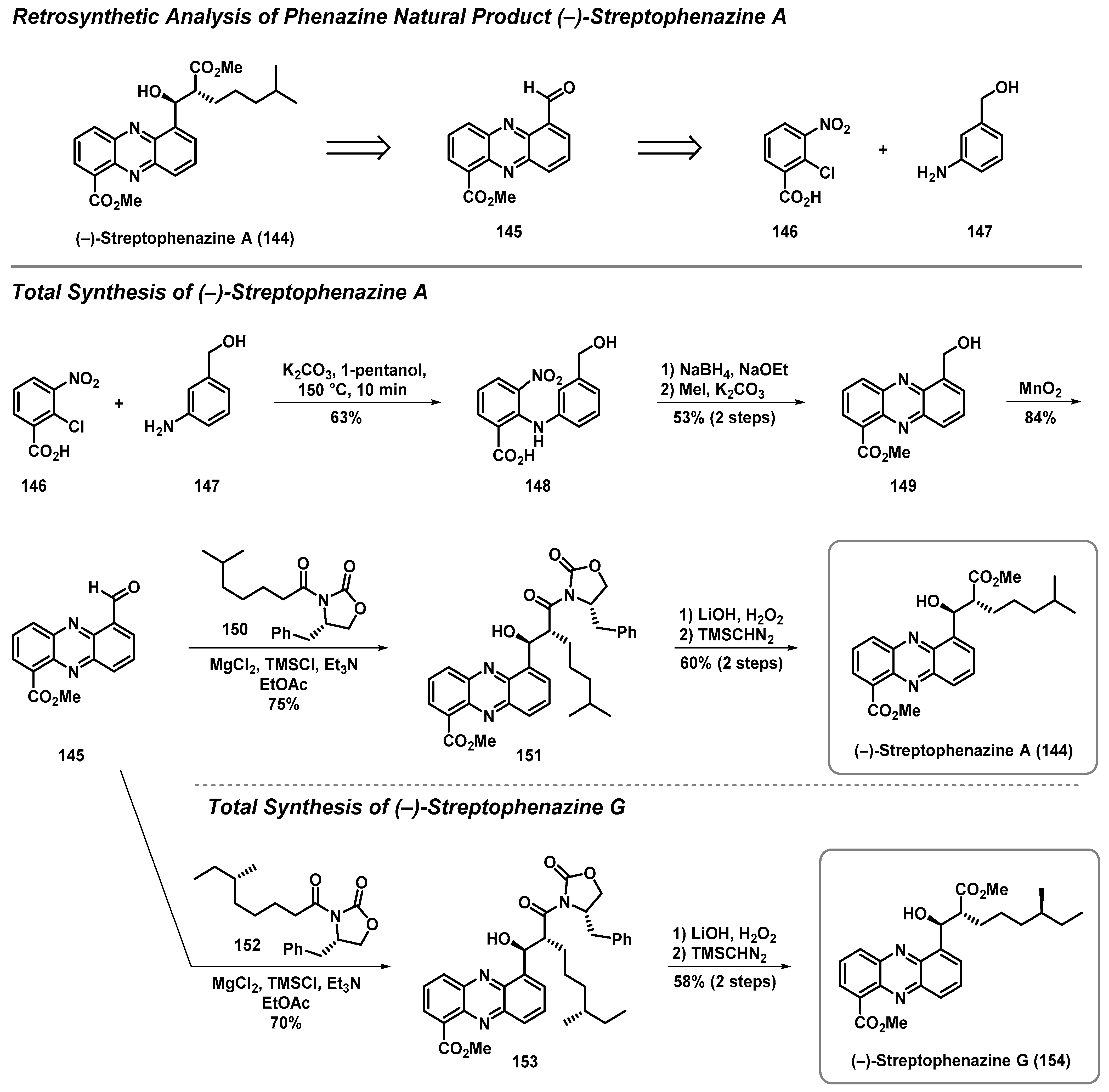
4.3. Total Synthesis of Streptophenazine A and G
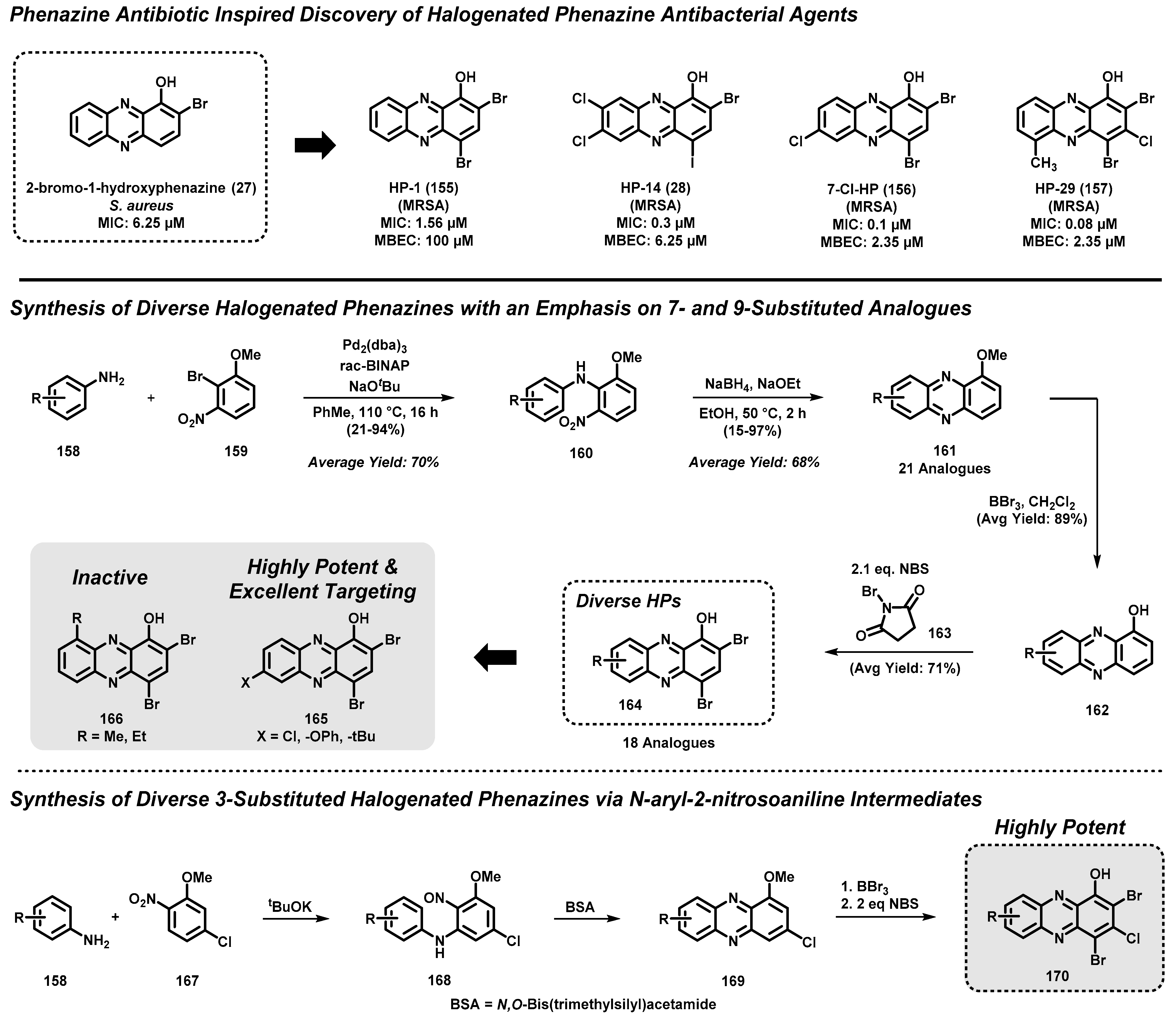
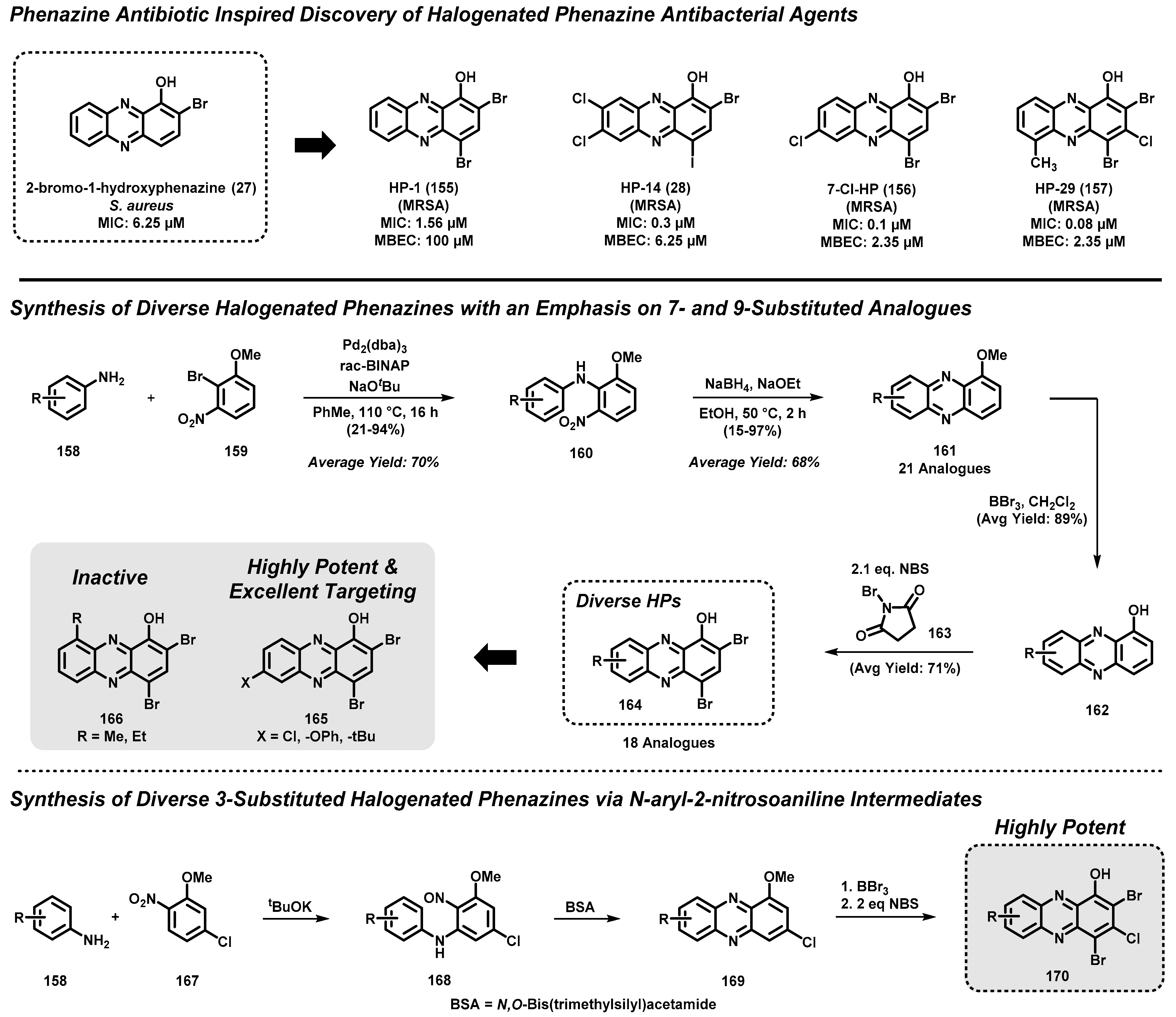
4.4. Marine Phenazine 2-Bromo-1-Hydroxyphenazine-Inspired Antibacterial Agents
5. Conclusions
Funding
Dedication
Conflicts of Interest
References
- Huigens, R.W., III; Tenneti, S.; Xiao, T.; Garrison, A.T. Pyrazines and Their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry IV; Stevens, C., Black, D., Cossy, J., Weinreb, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 8, pp. 229–282. [Google Scholar]
- Li, J.J. Heterocyclic Chemistry in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 536–567. [Google Scholar]
- Sato, N. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Pergamon: Oxford, UK, 2008; Volume 7, pp. 273–331. [Google Scholar]
- Laursen, J.B.; Nielsen, J. Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity. Chem. Rev. 2004, 104, 1663–1686. [Google Scholar] [CrossRef] [PubMed]
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent Developments in the Isolation, Biological Function, Biosynthesis, and Synthesis of Phenazine Natural Products. Bioorg. Med. Chem. 2017, 25, 6149–6166. [Google Scholar] [CrossRef] [PubMed]
- Nikishkin, N.I.; Huskens, J.; Verboom, W. Transition Metal-Catalyzed Functionalization of Pyrazines. Org. Biomol. Chem. 2013, 11, 3583–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, M.; Cargnin, S.; Massarotti, A.; Pirali, T.; Genazzani, A.A. Essential Medicinal Chemistry of Essential Medicines. J. Med. Chem. 2020, 63, 10170–10187. [Google Scholar] [CrossRef]
- Juhás, M.; Zitko, J. Molecular Interactions of Pyrazine-Based Compounds to Proteins. J. Med. Chem. 2020, 63, 8901–8916. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.P.; McKeage, K. Bortezomib: A Review of its Use in Patients with Multiple Myeloma. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef] [PubMed]
- Kouroukis, T.C.; Baldassarre, F.G.; Haynes, A.E.; Imrie, K.; Reece, D.E.; Cheung, M.C. Bortezomib in Multiple Myeloma: Systematic Review and Clinical Considerations. Curr. Oncol. 2014, 21, e573–e603. [Google Scholar] [CrossRef] [Green Version]
- Monti, J.M.; Pandi-Perumal, S.R. Eszopiclone: Its use in the treatment of insomnia. Neuropsychiatr. Dis. Treat. 2007, 3, 441–453. [Google Scholar]
- McCrae, C.S.; Ross, A.; Stripling, A.; Dautovich, N.D. Eszopiclone for Late-Life Insomnia. Clin. Interv. Aging 2007, 2, 313–326. [Google Scholar]
- Lee, S.; LaCour, T.G.; Fuchs, P.L. Chemistry of Trisdecacyclic Pyrazine Antineoplastics: The Cephalostatins and Ritterazines. Chem. Rev. 2009, 109, 2275–2314. [Google Scholar] [CrossRef] [Green Version]
- Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation and Structure Elucidation of Ritterazines B and C, Highly Cytotoxic Di-meric Steroidal Alkaloids, from the Tunicate Ritterella tokioka. J. Org. Chem. 1995, 60, 608–614. [Google Scholar] [CrossRef]
- Pereira, F. Have Marine Natural Product Drug Discovery Efforts Been Productive and How Can We Improve Their Efficiency? Exp. Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Xu, M.; Guo, S.; Zhu, F.; Xie, Y.; Shen, J. The Complete Synthesis of Favipiravir from 2-Aminopyrazine. Chem. Papers 2019, 73, 1043–1051. [Google Scholar] [CrossRef]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A New and Emerging Antiviral Option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Kambayshi, D.; Akatsu, H.; Kudo, K. Favipiravir for the Treatment of Patients with COVID-19: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2021, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Ericksson, A.; Hermanson, M.; Wickström, M.; Lindhagen, E.; Ekholm, C.; Janmalm Jensen, A.; Löhgren, A.; Lehmann, F.; Larsson, R.; Parrow, V.; et al. The Novel Tyrosine Kinase Inhibitor AKN-028 Has Significant Antileukemic Activity in Cell Lines and Primary Cultures of Acute Myeloid Leukemia. Blood Cancer J. 2012, 2, e81. [Google Scholar] [CrossRef] [Green Version]
- Bremberg, U.; Eriksson-Bajtner, J.; Lehmann, F.; Oltner, V.; Sölver, E.; Wennerberg, J. Development of a Synthesis of Kinase Inhibitor AKN028. Org. Process Res. Dev. 2018, 22, 1360–1364. [Google Scholar] [CrossRef]
- Durán, R.; Zubía, E.; Ortega, M.J.; Naranjo, S.; Salvá, J. Novel Alkaloids from the Red Ascidian Botryllus leachi. Tetrahedron 1999, 55, 13225–13232. [Google Scholar] [CrossRef]
- Saito, R.; Tokita, M.; Uda, K.; Ishikawa, C.; Satoh, M. Synthesis and In Vitro Evaluation of Botryllazine B Analogues as a New Class of Inhibitor against Human Aldose Reductase. Tetrahedron 2009, 65, 3019–3026. [Google Scholar] [CrossRef]
- Li, J.S.; Barber, C.C.; Zhang, W. Natural Products from Anerobes. J. Indust. Microbiol. Biotechnol. 2019, 46, 375–383. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Tan, H.; Kim, Y.S.; Jin, S.; Luesch, H.; Huigens, R.W., III. An Efficient Buch-wald-Hartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeing, Biofilm Eradication and Prodrug Exploration. J. Med. Chem. 2018, 61, 3962–3983. [Google Scholar] [CrossRef] [PubMed]
- Abouelhassan, Y.; Zhang, Y.; Jin, S.; Huigens, R.W., III. Transcript Profiling of MRSA Biofilms Treated with a Halogenated Phenazine Eradicating Agent: A Platform for Defining Cellular Targets and Pathways Critical to Biofilm Survival. Angew. Chem. Int. Ed. 2018, 57, 15523–15528. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.J.; Mistry, P.; Charlton, P.A.; Thomas, H.; Coley, H.M. Mode of Action of the Novel Phenazine Anticancer Agents XR11576 and XR5944. Anticancer Drugs 2007, 18, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Huigens, R.W., III; Abouelhassan, Y.; Yang, H. Phenazine Antibiotic-Inspired Discovery of Bacterial Biofilm-Eradicating Agents. ChemBioChem 2019, 20, 2885–2902. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Moschella, S.L. Clofazimine: A Review of its Medical Uses and Mechanisms of Action. J. Am. Acad. Dermatol. 1995, 32, 241–247. [Google Scholar] [CrossRef]
- Gopal, M.; Padayatchi, N.; Metcalfe, J.Z.; O’Donnell, M.R. Systematic Review of Clofazimine for the Treatment of Drug-Resistant Tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, R.R.; Krahenbuhl, J.L. Leprosy. Lancet 1999, 353, 655–660. [Google Scholar] [CrossRef]
- Ōmura, S.; Eda, S.; Funayama, S.; Komiyama, K.; Takahashi, Y.; Woodruff, H.B. Studies on a Novel Antitumor Antibiotic, Phenazinomycin: Taxonomy, Fermentation, Isolation, and Physicochemical and Biological Characteristics. J. Antibiot. 1989, 42, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Samata, K.; Yamagishi, T.; Ichihara, T.; Nanaumi, K.; Ikeda, T.; Ikeya, H.; Kuraishi, A.; Nakaike, S.; Kashiwagi, K.; Igarashi, K. Establishment and Characterization of a Mouse FM3A Cell Mutant Resistant to Topoisomerase II-Inhibitor NC-190. Cancer Chemother. Pharmacol. 2002, 50, 367–372. [Google Scholar] [CrossRef]
- Cimmino, A.; Andolfi, A.; Evidente, A. Microbial Phenazines: Biosynthesis, Agriculture and Health; Springer: Berlin/Heidelberg, Germany, 2013; pp. 217–243. [Google Scholar]
- Tarui, M.; Doi, M.; Ishida, T.; Inoue, M.; Nakaike, S.; Kitamura, K. DNA-Binding Characterization of a Novel Anti-tumour Benzo[a]phenazine Derivative NC-182: Spectroscopic and Viscometric Studies. Biochem. J. 1994, 304, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, T.; Nakaike, S.; Ikeda, T.; Ikeya, H.; Otomo, S. A Novel Antitumor Compound, NC-190, Induces Topoisomerase II-Dependent DNA Cleavage and DNA Fragmentation. Cancer Chemother. Pharmacol. 1996, 38, 29–34. [Google Scholar] [CrossRef]
- Komiya, T.; Fusetani, N.; Matsunaga, S.; Kubo, A.; Kaye, F.J.; Kelley, M.J.; Tamura, K.; Yoshida, M.; Fukuoka, M.; Nakagawa, K. Ritterazine B, a New Cytotoxic Natural Compound, Induces Apoptosis in Cancer Cells. Cancer Chemother. Pharmacol. 2003, 51, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.T.; Shair, M.D. Syntheses of the Eastern Halves of Ritterazines B, F, G, and H, Leading to Reassignment of the 5,5-Spiroketal Stereochemistry of Ritterazines B and F. J. Am. Chem. Soc. 2007, 129, 6589–6598. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation of 13 New Ritterazines from the Tunicate Ritterella tokioka and Chemical Transformation of Ritterazine B1. J. Org. Chem. 1997, 62, 4484–4491. [Google Scholar] [CrossRef] [PubMed]
- Budzikiewicz, H.; Flessner, T.; Jautelat, R.; Scholz, U.; Winterfeldt, E. Progress in the Chemistry of Organic Natural Products; Springer: Berlin/Heidelberg, Germany, 2004; pp. 1–80. [Google Scholar]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from Marine Invertebrates as Important Leads for Anticancer Drugs Discovery and Development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef]
- Burgett, A.W.G.; Poulsen, T.B.; Wangkanont, K.; Anderson, D.R.; Kikuchi, C.; Shimada, K.; Okubo, S.; Fortner, K.C.; Mimaki, Y.; Kuroda, M.; et al. Natural Products Reveal Cancer Cell Dependence on Oxysterol-Binding Proteins. Nat. Chem. Biol. 2011, 7, 639–647. [Google Scholar] [CrossRef] [Green Version]
- López-Antón, N.; Rudy, A.; Barth, N.; Schmitz, L.M.; Pettit, G.R.; Schulze-Osthoff, K.; Dirsch, V.M.; Vollmar, A.M. The Marine Product Cephalostatin 1 Activates an Endoplasmic Reticulum Stress Specific and Apoptosome-Independent Apoptotic Sig-naling Pathway. J. Biol. Chem. 2006, 281, 33078–33086. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, A.J.; Santos, E.A.; Jimenez, P.C.; Rocha, D.D.; Wilke, D.V.; Beuzer, P.; Axelrod, J.; Kumar Kanduluru, A.; Fuchs, P.L.; Cang, H.; et al. Ritterostatin GN1N, a Cephalostatin-Ritterazine Bis-Steroidal Pyrazine Hybrid, Selectively Targets GRP78. ChemBioChem 2017, 18, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Maser, M.R.; Okita, T.; Dubrovskiy, A.V.; Campbell, T.L.; Reisman, S.E. Total Synthesis of Ritterazine B. J. Am. Chem. Soc. 2021, 143, 4187–4192. [Google Scholar] [CrossRef]
- Fortner, K.C.; Kato, D.; Tanaka, Y.; Shair, M.D. Enantioselective Synthesis of (+)-Cephalostatin 1. J. Am. Chem. Soc. 2010, 132, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.M.; Sarandeses, L.A.; Sestelo, J.P. Enantioselective Synthesis of (-)-Barrenazines A and B. Tetrahedron Lett. 2007, 48, 8536–8539. [Google Scholar] [CrossRef]
- Mandal, D.; Yamaguchi, A.D.; Yamaguchi, J.; Itami, K. Synthesis of Dragmacidin D via Direct C-H Couplings. J. Am. Chem. Soc. 2011, 133, 19660–19663. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Aithagani, S.K.; Yadav, M.; Singh, V.P.; Vishwakarma, R.A. Iron-catalyzed Cross-Coupling of Electron-Deficient Heterocycles and Quinone with Organoboron Species via Innate C–H Functionalization: Application in Total Synthesis of Pyrazine Alkaloid Botryllazine A. J. Org. Chem. 2013, 78, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sperry, J. Synthesis of Alocasin A. J. Nat. Prod. 2015, 78, 3080–3082. [Google Scholar] [CrossRef] [PubMed]
- Bohman, B.; Jeffares, L.; Flematti, G.; Phillips, R.D.; Dixon, K.W.; Peakall, R.; Barrow, R.A. The Discovery of 2-Hydroxymethyl-3-(3-methylbutyl)-5-methylpyrazine: A Semiochemical in Orchid Pollination. Org. Lett. 2012, 14, 2576–2578. [Google Scholar] [CrossRef]
- Mosrin, M.; Bresser, T.; Knochel, P. Regio- and Chemoselective Multiple Functionalization of Chloropyrazine Derivatives. Application to the Synthesis of Coelenterazine. Org. Lett. 2009, 11, 3406–3409. [Google Scholar] [CrossRef]
- Badrinarayanan, S.; Sperry, J. Pyrazinealkaloids via Dimerization of Amino Acid-derived α-Amino Aldehydes: Biomimetic Synthesis of 2,5-Diisopropylpyrazine, 2,5-Bis(3-indolylmethyl)pyrazine and Actinopolymorphol C. Org. Biomol. Chem. 2012, 10, 2126–2132. [Google Scholar] [CrossRef]
- Kohatsu, H.; Kamo, S.; Furuta, M.; Tomoshige, S.; Kuramochi, K. Synthesis and Cytotoxic Evaluation of N-Alkyl-2-halophenazin-1-ones. ACS Omega 2020, 5, 27667. [Google Scholar] [CrossRef]
- Viktorsson, E.Ö.; Grøthe, B.M.; Aesoy, R.; Sabir, M.; Snellingen, S.; Prandina, A.; Åstrand, O.A.H.; Bonge-Hansen, T.; Døskeland, S.O.; Herfindal, L.; et al. Total Synthesis and Antileukemic Evaluations of the Pyrazine 5,10-Dioxide Natural Products Iodinin, Myxin and Their Derivatives. Bioorg. Med. Chem. 2017, 25, 2285. [Google Scholar] [CrossRef]
- Yang, Z.; Jin, X.; Guaciaro, M.; Molino, B.F.; Mocek, U.; Reategui, R.; Rhea, J.; Morley, T. The Revised Structure, Total Synthesis, and Absolute Configuration of Streptophenazine A. Org. Lett. 2011, 13, 5436–5439. [Google Scholar] [CrossRef]
- Yang, Z.; Jin, X.; Guaciaro, M.; Molino, B.F. Asymmetric Synthesis and Absolute Configuration of Streptophenazine G. J. Org. Chem. 2012, 77, 3191–3196. [Google Scholar] [CrossRef] [PubMed]
- Borrero, N.V.; Bai, F.; Perez, C.; Duong, B.Q.; Rocca, J.R.; Jin, S.; Huigens, R.W., III. Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 2014, 12, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Bai, F.; Ukhanova, M.; Mai, V.; Jin, S.; Luesch, H.; Huigens, R.W., III. Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-Biofilm Cultures and Mycobacterium tuberculosis. Angew. Chem. Int. Ed. 2015, 54, 14819–14823. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.T.; Abouelhassan, Y.; Norwood, V.M., IV; Kallifidas, D.; Bai, F.; Nguyen, M.T.; Rolfe, M.; Burch, G.M.; Jin, S.; Luesch, H.; et al. Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines that Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 3808–3825. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Abouelhassan, Y.; Burch, G.M.; Kallifdas, D.; Huang, G.; Yousaf, H.; Jin, S.; Luesch, H.; Huigens, R.W., III. A Highly Potent Class of Halogenated Phenazine Antibacterial and Biofilm-Eradicating Agents Accessed Through a Modular Wohl-Aue Synthesis. Sci. Rep. 2017, 7, 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Kundra, S.; Chojnacki, M.; Liu, K.; Fuse, M.A.; Abouelhassan, Y.; Kallifidas, D.; Zhang, P.; Huang, G.; Jin, S.; et al. A Modular Synthetic Route Involving N-Aryl-2-Nitrosoaniline Intermediates Leads to a New Series of 3-Substituted Halogenated Phenazine Antibacterial Agents. J. Med. Chem. 2021, 64, 7275–7295. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huigens, R.W., III; Brummel, B.R.; Tenneti, S.; Garrison, A.T.; Xiao, T. Pyrazine and Phenazine Heterocycles: Platforms for Total Synthesis and Drug Discovery. Molecules 2022, 27, 1112. https://doi.org/10.3390/molecules27031112
Huigens RW III, Brummel BR, Tenneti S, Garrison AT, Xiao T. Pyrazine and Phenazine Heterocycles: Platforms for Total Synthesis and Drug Discovery. Molecules. 2022; 27(3):1112. https://doi.org/10.3390/molecules27031112
Chicago/Turabian StyleHuigens, Robert W., III, Beau R. Brummel, Srinivasarao Tenneti, Aaron T. Garrison, and Tao Xiao. 2022. "Pyrazine and Phenazine Heterocycles: Platforms for Total Synthesis and Drug Discovery" Molecules 27, no. 3: 1112. https://doi.org/10.3390/molecules27031112