
Rational Design and Synthesis of HSF1-PROTACs for Anticancer Drug Development


College of Pharmacy, Keimyung University, Daegu 704-701, Korea
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(5), 1655; https://doi.org/10.3390/molecules27051655
Submission received: 4 February 2022
/
Revised: 24 February 2022
/
Accepted: 28 February 2022
/
Published: 2 March 2022
(This article belongs to the Section Medicinal Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:PROTACs employ the proteosome-mediated proteolysis via E3 ligase and recruit the natural protein degradation machinery to selectively degrade the cancerous proteins. Herein, we have designed and synthesized heterobifunctional small molecules that consist of different linkers tethering KRIBB11, a HSF1 inhibitor, with pomalidomide, a commonly used E3 ligase ligand for anticancer drug development.
1. Introduction
Cancer continues to be one of the leading causes of morbidity and mortality worldwide, accounting for 10 million deaths in 2020 [1]. It has an immense economic impact, which is escalating, in addition to the huge loss of human lives. According to the WHO report, the total annual economic cost of cancer in 2010 was estimated at approximately USD 1.16 trillion [2]. The existing treatment by traditional chemotherapy, targeted therapy, and immunotherapy is fraught with the disadvantages of severe adverse reactions and toxicity, resistance to the treatment therapy, and excessive uncontrollable activity of the immune system [3,4,5,6]. Thus, new strategies to combat cancer are an urgent medical need.
Targeting proteins that contribute to cancer development for therapeutic purposes is challenging, and most of the proteome is still considered beyond reach that is deemed undruggable. Sakamoto et al. reported a novel chemical strategy in 2001, which can be used to rationally manipulate the intracellular levels of many proteins that are regulated by the molecular mechanism of ubiquitin-dependent proteolysis [7]. This technology opened new avenues in anticancer drug discovery and was also commercialized as PROTAC® for targeted protein degradation [8]. Proteolysis-targeting chimeras are heterobifunctional molecules that can target any protein for ubiquitination and subsequent proteasomal degradation [9]. Further, lysosomal targeting with lysosomal-targeting chimeras (LYTACs), autophagy targeting with autophagy-targeting chimeras (AUTACs), and RNA targeting with ribonuclease-targeting chimeras (RIBOTACs) have been reported by recruiting alternative degradation pathways [10].
HSF1 is a master regulator of transcriptional responses to proteotoxic stress and an effective drug target for anticancer therapies including radiotherapy and thermotherapy [11]. HSF1 is known to be overactive or overexpressed in many cancer types including prostate cancer, pancreatic cancers, breast cancer, colorectal, lymphomas, melanoma, oral cancers, and so forth [12]. The activation of HSF1 essentially results in the over-expression of various heat shock proteins, which affects the outcome of the treatment.
The over-expression of heat shock proteins caused by HSF1 is reported as the main cause of resistance to chemotherapy, radiation therapy, and hypothermia used in cancer treatment [13]. Although HSF1 has been found to be a significant factor in the development and low prognosis of various chemotherapy cancer treatments, there are still very few effective inhibitors of HSF1 [14]. Most HSF1 inhibitors reported to date are indirect inhibitors through high signal disturbance in HSF1 rather than directly inhibiting HSF1. Continuing our effort of developing anticancer drug candidates [15,16], we report the rational design and synthesis of HSF1-PROTACs in an endeavor to create new chemical agents for the development of appropriate clinical candidates in anticancer drug discovery.
2. Results and Discussion
Our design rationale for making PROTAC involved tagging a known binder of HSF1 protein. A 3-nitropyridine derivative (N2-(1H-indazole-5-yl)-N6-methyl-3-nitropyridine-2,6-diamine) named KRIBB11 is a well-known HSF1 inhibitor used to treat tumor tissues without significant body weight loss [17]. The docking between the crystal structure HSF1 [18] and KRIBB11 revealed that the hydrogen bonding by the oxygen atoms of the nitro group of the inhibitor stabilizes the binding to HSF1 protein. The nitrogen atoms of the pyridine ring and the N-methyl group of the inhibitor form hydrogen bonding interactions with the amide group of Gln72 and the phenol group of Tyr76, respectively. The N-methyl moiety in KRIBB11 is projected away from the protein, as shown in Figure 1. Thus, we decided to link the E-3 ligase ligand to the N-methyl part of KRIBB11. We employed three different linkers. Phthalimide conjugation is the most effective strategy for proteasome-mediated proteolysis [19]. Accordingly, we designed the PROTAC molecules by connecting KRIBB11 and pomalidomide as a ligand for E3 ligase.
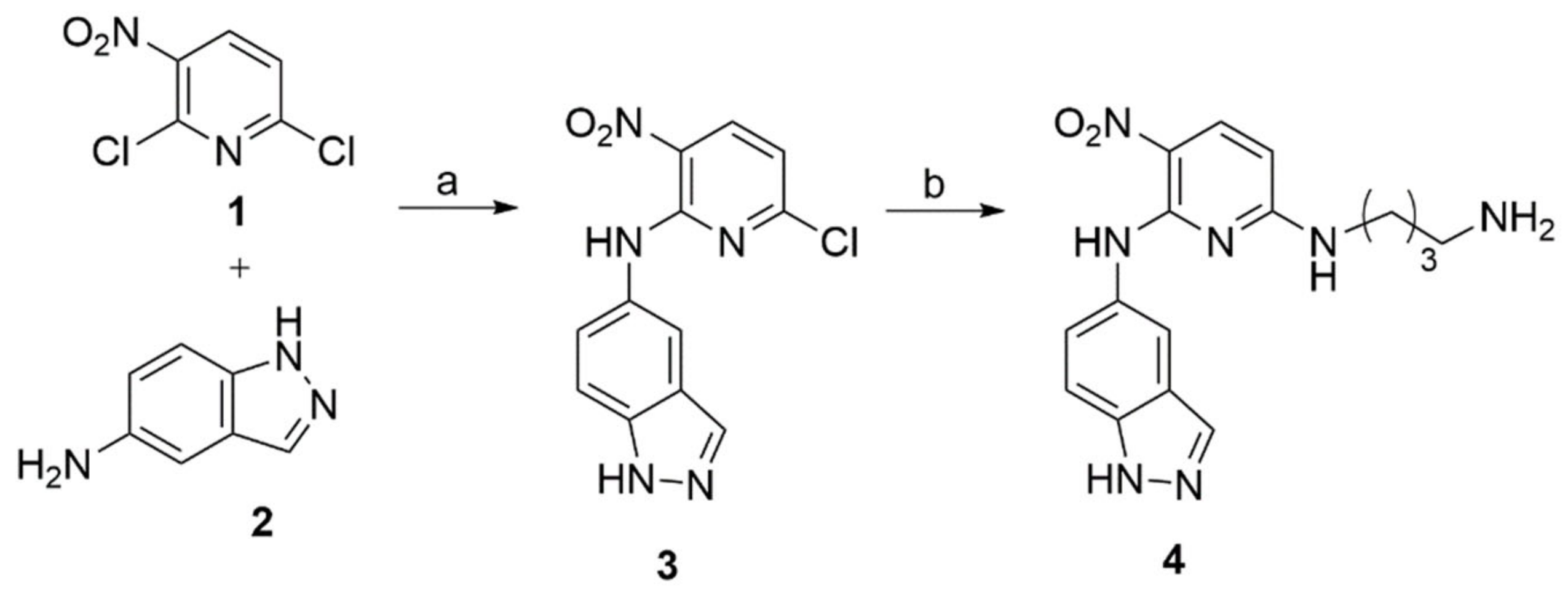
We began the synthesis for making the KRIBB11 part by treating 2,6-dichloro-3-nitropyridine with 5-aminoindazole in the presence of triethylamine in ethanol, resulting in the formation of 3 in good yield [20]. Subsequent reaction with 1,4-diaminobutane in acetonitrile at room temperature yielded 4, as shown in Scheme 1. The pomalidomide part 9c and a precursor 9a was synthesized using the previously reported procedure, as illustrated in Scheme 2 [21].
The linkers were connected to the pomalidomide, as shown in Scheme 3. Firstly, the 4,7,10-Trioxatridecane-1,13-diamine 10a was mono-Boc protected into 10b using Boc anhydride and triethylamine in anhydrous dichloromethane. It was then treated with 9a in the presence of diisopropylethylamine in DMF to form 12a, which was further reacted with TFA to give 12b in good yield. The mono Boc-protected 4,7,10-Trioxatridecane-1,13-diamine 10b was also reacted with chloroacetyl chloride in the presence of Cs2CO3 to give 11. It was then treated with 9c to form 13a, which, on subsequent TFA treatment, gave 13b. Finally, the HSF1-PROTACs were synthesized by nucleophilic aromatic substitution, as shown in Scheme 4. The PROTAC 14 was synthesized by heating 4 and 9a in DMF in the presence of diisopropylethylamine. The PROTACs 15 and 16 were synthesized by reacting 3 at room temperature with 12b and 13b, respectively, in the presence of triethylamine in acetonitrile. The copies of 1H and 13C spectra for all prepared compounds are given in Supplementary Figures S1–S19. PROTACs were purified by MPLC, and the purity was also determined by HPLC (Supplementary Figure S20).
With compound 14–16 in hand, we examined the dose-dependent effect of 14–16 on the cell viability of MDA-MB-231, which is a highly aggressive and invasive triple negative breast cancer (TNBC) cell line. The preliminary investigation suggested 14–16 exhibited a minimal in vitro inhibitory effect against MDA-MB-231 breast cancer cells, as shown by the cell viability data in Figure 2. None of compounds 14–16 could inhibit the 50% cell growth up to 50 μM treatment. Compound 15 provided a slightly better inhibitory effect than 14 and 16, in that compound 15 impaired 27% cell growth by the treatment of 30 and 50 μM concentration.
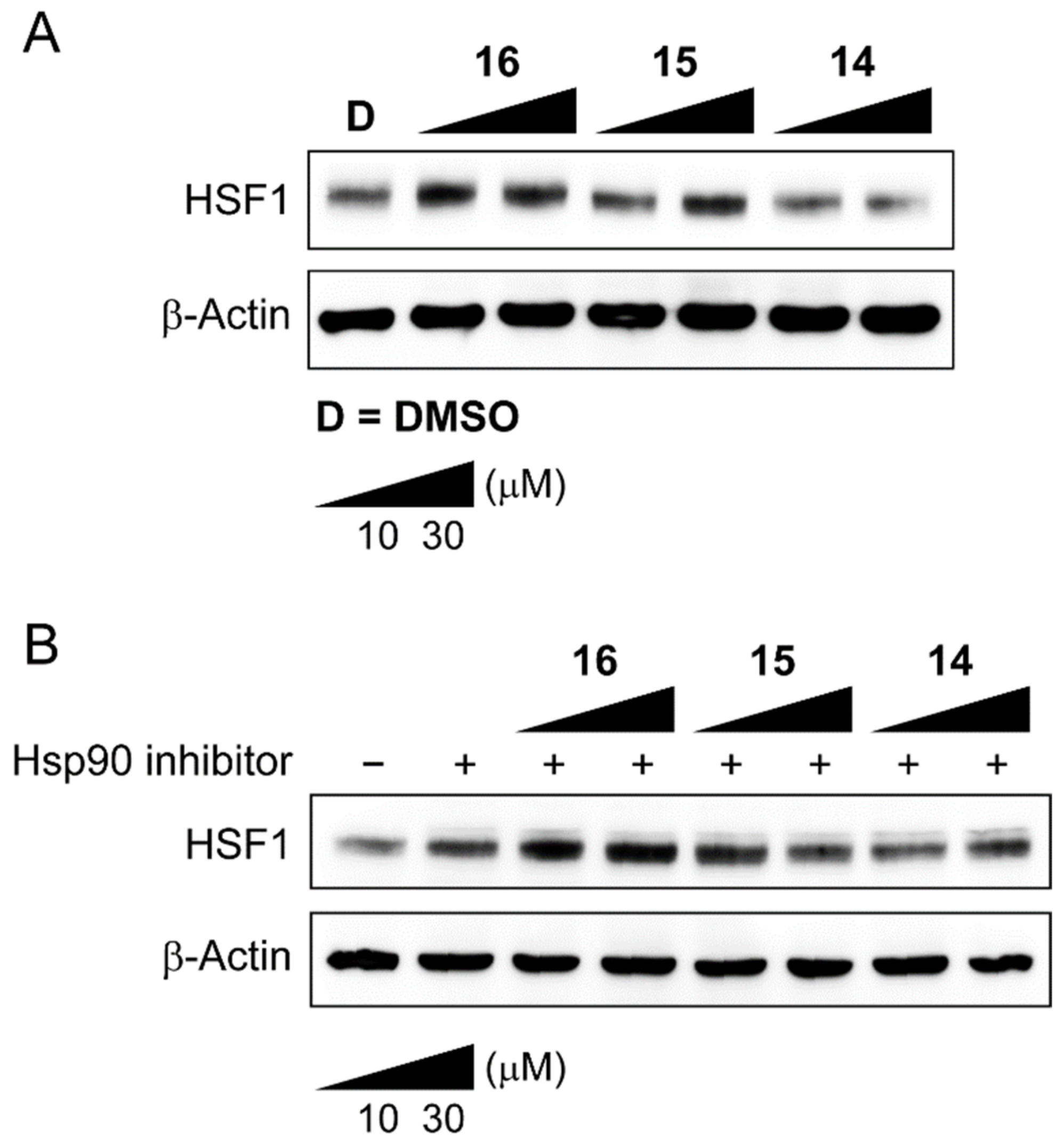
We further evaluated the proteolysis effect of compounds 14–16 on HSF1 (Figure 3). Our Western blotting data indicated that 14 showed a little degradation of HSF1. However, the results were not consistent despite several repetitions. We speculated that one reason for inconsistency could be due to the intrinsic formation of Hsp90-Hsp70-HSF1 multi-chaperone complex in the cancer cell, hampering compound 4 to interact with the binding site of HSF1. HSF1 is dynamically associated or dissociated from multi-chaperone complex under various stressed or non-stressed conditions. Hence, we decided to treat cancer cells with compounds 14–16, together with an Hsp90 inhibitor, which could induce a heat shock response (HSR) to dissociate HSF1 from the chaperone complex. Unfortunately, the results did not still show any meaningful degradation of HSF1 in MDA-MB-231 cells, shown in Figure 3B. To draw biological meaningful conclusions, our efforts are directed toward finding better PROTACs with new HSF1 ligands and E3 ligase ligands. The result will be reported in due course.
3. Materials and Methods
3.1. General Information
All reagents and solvents were purchased from commercial suppliers and used without further purification. All experiments dealing with moisture-sensitive compounds were carried out under argon atmosphere. Concentration or solvent removal under reduced pressure was carried out using rotary evaporator. Analytical thin layer chromatography was performed on precoated silica gel F254 TLC plates (E, Merck) with visualization under UV light or by staining using iodine. Column chromatography and medium-pressure liquid chromatography (MPLC) was conducted on silica (Merck Silica Gel 40–63 m) or performed by using a Biotage SP1 flash purification system with prepacked silica gel cartridges (Biotage, Uppsala, Sweden). NMR analyses were carried out using a JNM-ECZ500R (500 MHz) manufactured by Jeol resonance. Chemical shifts are reported in parts per million (δ). The deuterium lock signal of the sample solvent was used as a reference, and coupling constants (J) are given in hertz (Hz). The splitting pattern abbreviations are as follows: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublet; td, triplet of doublet; and m, multiplet. The purities of all final compounds were confirmed to be higher than 95% by analytical HPLC performed with a dual-pump Shimadzu LC-6AD system equipped with a VP-ODS C18 column (4.6 mm × 250 mm, 5 μm, Shimadzu, Kyoto, Japan). The LC-QTOF-MS analysis was performed using an Agilent 6530 Accurate-Mass Q-TOF LC/MS System with Agilent 1290 Infinity LC (Agilent Technologies, Palo Alto, CA, USA). The guard column and the analytical column were Zorbax SB-C8 (3.5 μm, 2.1 × 30 mm, Agilent Technologies) and Zorbax SB-Aq (1.8 μm, 2.1 × 100 mm, Agilent Technologies), respectively, and were maintained at 40 °C. The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient conditions were as follows: 0–30 min, 1–20% B; 30–40 min, 20–90% B; 40–45 min, 90% B; 45–47 min, 90–1% B; and 47–52 min 1% B at a flow rate of 400 μL/min. The MS system was operated using ESI in the positive and the negative ionization mode. The optimized conditions of the QTOF-MS system for both ionization modes were as follows: drying gas temperature, 300 °C; drying gas flow, 10 L/min; nebulization pressure, 45 psi; sheath gas temperature, 350 °C; sheath gas flow, 10 L/min; capillary voltage, 3500 V; nozzle voltage, 0 V; fragmentor voltage, 175 V; and skimmer voltage, 65 V. The mass range was 50–1700 m/z, and the scan rate was 2.00 spectra/sec for both MS and MS/MS analyses.
3.2. Synthetic Procedures
3.2.1. Synthesis of KRIBB11 Aanalogue 4
To 15 mL of ethanol were added 2,6-dichloronitropyridine (0.5 g, 2.59 mmol), 5-aminoindazole (0.36 g, 2.72 mmol), and triethylamine (0.4 mL, 2.85 mmol). The reaction mixture was refluxed for 90 °C for about 5 h. After the reaction was completed, the solid obtained was filtered, washed with methanol, and then dried under vacuum to afford compound 3 in 80.7% yield. 1H-NMR (500 MHz, DMSO-D6) δ 10.22 (s, 1H), 8.54 (d, J = 8.6 Hz, 1H), 8.09 (s, 1H), 7.92 (d, J = 1.7 Hz, 1H), 7.56 (d, J = 9.2 Hz, 1H), 7.44 (dd, J = 8.6, 1.7 Hz, 1H), and 6.95 (d, J = 8.6 Hz, 1H).
Compound 3 (0.289 g, 1 mmol) was dissolved in 25 mL acetonitrile, and 1,4-diaminobutane (0.44 mL, 5 mmol) was added. The reaction mixture was stirred at RT for 14 h. After completion of the reaction, acetonitrile was evaporated, and the residue was partitioned between 1N NaOH and ethyl acetate. The organic layer was dried with sodium sulfate and evaporated in vacuo to obtain compound 4 in 70.3% yield. 1H-NMR (500 MHz, DMSO-D6) δ 10.94 (s, 1H), 8.20 (s, 1H), 8.07 (d, J = 9.2 Hz, 1H), 8.04 (s, 1H), 7.53 (s, 2H), 6.09 (d, J = 9.7 Hz, 1H), 3.30 (t, J = 7.4 Hz, 2H), 2.48 (d, J = 6.9 Hz, 2H), 1.53 (t, J = 7.4 Hz, 2H), and 1.33 (t, J = 7.4 Hz, 2H).
3.2.2. Synthesis of E-3 Ligase Lligands (9a and 9c)
Synthesis of 9a. A mixture of N-Boc-L-asparagine (2 g, 8.61 mmol), CDI (1.39 g, 8.61 mmol), and catalytic amounts 4-DMAP in anhydrous THF (21.5 mL) was stirred and heated to reflux at 76 °C for 48 h. After the end of the reaction, the mixture was filtered, and the solid was washed with THF to give 6 in 73% yield as a colorless solid. 1H-NMR (500 MHz, DMSO-D6) δ 10.77 (s, 1H), 7.15 (d, J = 8.6 Hz, 1H), 4.20–4.25 (m, 1H), 2.71 (dt, J = 17.8, 6.3 Hz, 1H), 2.46 (t, J = 3.7 Hz, 1H), 1.87–1.94 (m, 2H), and 1.40 (s, 9H).
Compound 6 (1.54 g, 6.75 mmol) was dissolved in TFA (7.8 mL, 101.25 mmol) and stirred for 30 min. The excess of the acid was removed in vacuo, and the resulting product was dried under vacuum to give 7 in quantitative yield as an off-white solid. 1H-NMR (500 MHz, DMSO-D6) δ 11.25 (s, 1H), 8.58 (s, 3H), 4.21 (d, J = 8.6 Hz, 1H), 2.67–2.75 (m, 1H), 2.59 (dt, J = 15.3, 2.3 Hz, 1H), 2.14–2.19 (m, 1H), and 2.02 (td, J = 13.0, 4.8 Hz, 1H).
Briefly, a mixture of 3-fluorophthalic anhydride (1.25 g, 7.5 mmol), 7 (1.14 g, 5 mmol), and a solution of sodium acetate (0.50 g, 6.0 mmol) in glacial acetic acid (20 mL) was refluxed for 140 °C for 18 h. After cooling, it was poured onto H2O (100 mL), and the solid formed was collected by filtration and washed with H2O and petroleum ether. The purple solid was further dried in vacuo, resulting in 9a in 91% yield. 1H-NMR (500 MHz, DMSO-D6) δ 11.17 (s, 1H), 7.95 (td, J = 7.9, 4.0 Hz, 1H), 7.79 (d, J = 7.4 Hz, 1H), 7.74 (t, J = 8.9 Hz, 1H), 5.16 (dd, J = 12.9, 5.4 Hz, 1H), 2.85–2.91 (m, 1H), 2.58–2.63 (m, 1H), 2.52 (t, J = 2.0 Hz, 1H), and 2.04–2.07 (m, 1H).
3.2.3. Synthesis of 9c
A mixture of 36 mL glacial acetic acid and (0.308 g, 3.75 mmol) anhydrous sodium acetate was added to (0.58 g, 3 mmol) of 3-nitrophthalic anhydride (0.57 g, 2.5 mmol) of compound 7. The reaction mixture was heated at 140 °C for 18 h. After completion of the reaction, the reaction mixture was cooled to rt, water was added to the reaction mixture, and it was stirred for another 30 min. The solid was filtered and washed with water. The product was dried under vacuum to obtain 9b in 44.5% yield. 1H-NMR (500 MHz, DMSO-D6) δ 11.18 (s, 1H), 8.35 (d, J = 7.4 Hz, 1H), 8.24 (d, J = 6.9 Hz, 1H), 8.12 (t, J = 7.7 Hz, 1H), 5.20 (dd, J = 12.9, 5.4 Hz, 1H), 2.85–2.92 (m, 1H), 2.54–2.63 (m, 1H), 2.52–2.54 (m, 1H), and 2.05–2.10 (m, 1H).
To the mixture of compound 9b (0.61 g, 2.02 mmol) in acetone was added a catalytic amount of 10% Pd/C (0.24 g, 0.66 mmol). The mixture was stirred at room temperature for 72 h under H2 gas. Then, the mixture was filtered to remove the palladium and acetone was evaporated to afford 9c in 90.8% yield. 1H-NMR (500 MHz, DMSO-D6) δ 11.10 (s, 1H), 7.47 (dd, J = 8.3, 7.2 Hz, 1H), 7.01 (t, J = 7.4 Hz, 2H), 6.53 (s, 2H), 5.05 (dd, J = 12.9, 5.4 Hz, 1H), 2.84–2.89 (m, 1H), 2.52–2.60 (m, 2H), and 2.02 (td, J = 6.4, 2.1 Hz, 1H).
3.2.4. Synthesis of Linkers (12b and 13b)
Synthesis of 12b. A solution of 4,7,10-trioxa-1,13-tridecanediamine (1.0 g, 4.5 mmol) in anhydrous DCM (15 mL) was treated with Boc2O (0.164 g, 0.75 mmol) and TEA (0.627 mL, 4.5 mmol) in DCM (22.5 mL). The reaction was stirred at room temperature for 4 h. The resulting yellow oil was purified by column chromatography, resulting in 10b in 51.5% yield. 1H-NMR (500 MHz, CDCl3) δ 5.26 (s, 1H), 3.51–3.53 (m, 4H), 3.46–3.49 (m, 4H), 3.41–3.45 (m, 4H), 3.09 (q, J = 6.1 Hz, 2H), 2.69 (t, J = 6.6 Hz, 2H), 2.47 (s, 2H), 1.63 (td, J = 13.0, 6.7 Hz, 4H), and 1.31 (s, 9H).
A solution of 10b (0.2 g, 0.7 mmol) and 9a (0.14 g, 0.8 mmol) in DMF (2 mL) was treated with DIPEA (0.18 g, 1.4 mmol) under 90 °C for 12 h. The reaction mixture was poured into water and extracted with EtOAc. The combined organic phases were dried and concentrated under the reduced pressure. The crude product was purified to give compound 12a in 40% yield. 1H-NMR (500 MHz, CDCl3) δ 8.41 (s, 1H), 7.48 (dd, J = 8.6, 7.4 Hz, 1H), 7.07 (d, J = 6.9 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H), 6.43 (s, 1H), 4.99 (s, 1H), 4.90 (q, J = 5.7 Hz, 1H), 3.57–3.69 (m, 11H), 3.52 (t, J = 6.0 Hz, 2H), 3.39 (q, J = 6.1 Hz, 2H), 3.21 (q, J = 5.9 Hz, 2H), 2.69–2.89 (m, 4H), 2.09–2.14 (m, 1H), 1.89–1.94 (m, 2H), 1.71–1.76 (m, 2H), and 1.42 (s, 9H).
Compound 12a (0.154 g, 0.267 mmol) was dissolved in TFA (0.78 mL, 10.13 mmol) and stirred for 30 min. The excess of the acid was removed in vacuo, and the resulting product was dried under vacuum to give 12b in quantitative yield. 1H-NMR (500 MHz, CDCl3) δ 9.45 (s, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 7.4 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 7.01 (s, 2H), 5.29 (s, 1H), 5.01 (dd, J = 12.6, 5.2 Hz, 1H), 3.63–3.72 (m, 13H), 3.45 (t, J = 6.0 Hz, 2H), 3.24 (q, J = 5.2 Hz, 2H), 3.10 (s, 1H), 2.98 (s, 1H), 2.89 (s, 1H), 2.72–2.83 (m, 2H), 2.17–2.19 (m, 1H), and 1.94 (dt, J = 26.7, 5.3 Hz, 4H).
Synthesis of 13b. A solution of 10b (0.2 g, 0.7 mmol) dissolved in THF (2 mL) was treated with DIPEA (0.18 g, 1.4 mmol) and stirred for 6 h, resulting in 11 in 44.5% crude yield, which was used without further purification. 1H-NMR (500 MHz, CDCl3) δ 5.22–4.71 (1H), 4.02 (d, J = 1.7 Hz, 2H), 3.57–3.67 (m, 10H), 3.53 (td, J = 6.0, 1.7 CDCl3 Hz, 2H), 3.43 (q, J = 5.9 Hz, 2H), 3.21 (d, J = 4.0 Hz, 2H), 1.86 (s, 1H), 1.83 (q, J = 5.9 Hz, 3H), 1.75 (t, J = 6.3 Hz, 2H), and 1.43 (d, J = 2.3 Hz, 9H).
The compound 11 (0.14 g, 0.8 mmol) and (0.108 g, 0.96 mmol) chloroacetyl chloride in Acetonitrile (2 mL) was treated with cesium carbonate (0.18 g, 1.4 mmol) at rt for 3 h. The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by column to give compound 13a in 33% yield. 1H-NMR (500 MHz, CDCl3) δ 7.38 (dd, J = 8.6, 7.4 Hz, 1H), 7.09 (d, J = 6.9 Hz, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.73 (s, 1H), 5.03 (q, J = 5.9 Hz, 1H), 4.46 (dd, J = 22.6, 15.8 Hz, 2H), 3.60–3.62 (m, 5H), 3.55–3.57 (m, 5H), 3.49–3.53 (m, 5H), 3.35 (dd, J = 10.0, 6.0 Hz, 2H), 3.17 (t, J = 6.6 Hz, 2H), 2.92–2.97 (m, 1H), 2.83–2.86 (m, 1H), 2.69 (dd, J = 12.3, 4.3 Hz, 1H), 2.15 (t, J = 1.7 Hz, 1H), 1.71–1.78 (m, 4H), and 1.41 (d, J = 5.2 Hz, 10H).
Compound 13a (0.2 g, 0.31 mmol) was dissolved in TFA (0.78 mL, 10.13 mmol) and stirred for 30 min. The excess of the acid was removed in vacuo, and the resulting product was dried under vacuum to give 13b in quantitative yield. 1H-NMR (500 MHz, CD3OD) δ 7.45 (dd, J = 8.3, 7.2 Hz, 1H), 7.04 (d, J = 6.9 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 5.18 (dd, J = 12.9, 5.4 Hz, 1H), 4.44 (d, J = 7.4 Hz, 2H), 3.60–3.66 (m, 10H), 3.56–3.58 (m, 2H), 3.49 (t, J = 6.0 Hz, 2H), 3.28–3.29 (m, 2H), 3.09 (t, J = 6.3 Hz, 3H), 2.86–2.99 (m, 4H), 1.91 (dd, J = 6.3, 5.2 Hz, 2H), and 1.76 (t, J = 6.3 Hz, 2H).
3.2.5. Synthesis of PROTACS (14–16)
Synthesis of 14. The mixture of compound 4 (0.02 g, 0.07 mmol) and 9a (0.024 g, 0.07 mmol) dissolved in 1.5 mL DMF was added with DIPEA (0.03 mL, 0.14 mmol). The reaction mixture was refluxed at 90 °C for 48 h. The DMF was removed by evaporation, and the crude was purified by MPLC to obtain compound 13 in 58% yield. 1H-NMR (500 MHz, ACETONE-D6) δ 12.24 (s, 1H), 10.96 (s, 1H), 9.98 (s, 1H), 8.26 (s, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.02 (s, 1H), 7.58 (dd, J = 11.7, 8.9 Hz, 2H), 7.48 (q, J = 7.4 Hz, 2H), 6.96 (dd, J = 22.6, 7.7 Hz, 2H), 6.36 (s, 1H), 6.10 (d, J = 9.2 Hz, 1H), 5.06 (q, J = 6.1 Hz, 1H), 3.53 (d, J = 5.7 Hz, 2H), 3.30 (d, J = 6.3 Hz, 2H), 2.91–2.98 (m, 4H), 2.71–2.79 (m, 2H), 2.16–2.21 (m, 1H), 1.73 (s, 4H). 13C-NMR (126 MHz, ACETONE-D6) δ 172.74, 170.36, 170.26, 168.22, 161.61, 153.33, 147.61, 138.72, 136.86, 135.95, 134.63, 133.51, 132.57, 124.43, 124.29, 119.40, 117.45, 114.27, 111.27, 110.75, 103.27, 49.80, 42.64, 41.92, 31.96, 27.30 23.38 ESI MS (m/z); 598.2148 [M + H]+.
Synthesis of 15. The mixture of compound 3 (0.039 g, 0.12 mmol) and 12b (0.09 g, 0.18 mmol) dissolved in 20 mL CH3CN was added with TEA (0.05 mL, 0.36 mmol). The reaction mixture was stirred at RT for 24 h. The CH3CN was removed by evaporation, and the crude was purified by MPLC to obtain compound 15 in 47% yield. 1H-NMR (500 MHz, ACETONE-D6) δ 12.23 (s, 1H), 10.99 (s, 1H), 9.97 (s, 1H), 8.30 (s, 1H), 8.06–8.10 (m, 2H), 7.49–7.60 (m, 3H), 7.37 (s, 1H), 6.99 (dd, J = 26.1, 7.7 Hz, 2H), 6.56 (t, J = 5.4 Hz, 1H), 6.10 (d, J = 9.2 Hz, 1H), 5.05 (q, J = 6.1 Hz, 1H), 3.37–3.63 (m, 16H), 2.90–2.95 (m, 3H), 2.72–2.79 (m, 2H), 2.16–2.20 (m, 1H), 1.83–1.88 (m, 4H). 13C-NMR (126 MHz, ACETONE-D6) δ 172.75, 170.37, 170.14, 168.25, 161.54, 153.21, 147.73, 136.85, 135.85, 134.69, 124.22, 117.45, 113.99, 111.12, 103.39, 71.13, 71.09, 71.05, 70.86, 69.46, 49.78, 40.88, 23.39 ESI MS (m/z); 730.29 [M + H]+.
Synthesis of 16. The mixture of compound 3 (0.056 g, 0.19 mmol) and 13b (0.118 g, 0.22 mmol) dissolved in 20 mL CH3CN was added with TEA (0.03 mL, 0.23 mmol). The reaction mixture was stirred at RT for 18 h. The CH3CN was removed by evaporation, and the crude was purified by MPLC to obtain compound 16 in 38% yield. 1H-NMR (500 MHz, ACETONE-D6) δ 12.33 (s, 0H), 11.00 (s, 1H), 8.28 (s, 1H), 8.11 (d, J = 9.2 Hz, 1H), 7.45–7.63 (m, 4H), 7.26 (d, J = 14.9 Hz, 1H), 7.05 (dd, J = 13.7, 8.0 Hz, 2H), 6.20 (d, J = 7.4 Hz, 2H), 6.12 (d, J = 9.2 Hz, 1H), 5.15 (dd, J = 12.9, 5.4 Hz, 1H), 4.40 (s, 2H), 3.42–3.61 (m, 14H), 3.25 (tt, J = 20.4, 6.7 Hz, 2H), 3.05 (ddd, J = 18.3, 13.2, 4.6 Hz, 1H), 2.77–2.90 (m, 2H), 2.19–2.24 (m, 1H), 1.66–1.72 (m, 2H). 13C-NMR (126 MHz, ACETONE-D6) δ 171.92, 170.27, 169.85, 168.26, 167.23, 161.63, 153.31, 147.75, 136.28, 135.83, 134.61, 133.35, 132.60, 124.29, 122.34, 122.28, 119.31, 114.08, 112.25, 110.83, 110.59, 103.49, 71.06, 71.03, 70.82, 70.70, 69.46, 69.20, 50.32, 41.26, 37.38, 22.54. ESI MS (m/z); 787.31 [M + H]+.
3.3. Docking Studies
In silico docking of KRIBB11 with the 3D coordinates of the X-ray crystal structure of HSF1 (PDB code: 5d5U) was accomplished using the AutoDock 4.2 program downloaded from the Molecular Graphics Laboratory of the Scripps Research Institute. The AutoDock [22] program was chosen because it uses a genetic algorithm to generate the poses of the ligand inside a known or predicted binding site utilizing the Lamarckian version of the genetic algorithm, where the changes in conformations adopted by molecules after in situ optimization are used as subsequent poses for the offspring. In the docking experiments carried out, Gasteiger charges were placed on the X-ray structure of HSF1 along with KRIBB11, using tools from the AutoDock suite [23]. A grid box centered on the substrate binding pocket of HDACs enzyme with definitions of 50 × 50 × 50 points and 0.375 Å spacing was chosen for ligand docking experiments. The docking parameters consisted of setting the population size to 150, the number of generations to 27,000, and the number of evaluations to 2,500,000;the number of docking runs was set to 100, with a cutoff of 1 Å for the root-mean-square tolerance for the grouping of each docking run. The docking pose of HSF1 with compound KRIBB11 was depicted in Figure 1, and rendering of the picture was generated using PyMol (DeLanoScientific, South San Francisco, CA, USA) [24].
3.4. Cell Culture and Materials
Triple-negative breast cancer cells MDA-MB-231 were grown in DMEM high glucose and supplemented with streptomycin (500 mg/mL), penicillin (100 units/mL), and 10% fetal bovine serum (FBS). Cells were grown to confluence at 37 °C in a humidified atmosphere with 5% CO2. Antibodies for HSF-1 (1:1000; rabbit anti-human mAb; cat. no. 12972) and β-actin (1:1000; rabbit anti-human mAb; cat. no. 4970) were purchased from Cell Signaling Technology (Beverly, MA, USA).
3.5. MTS Colorimetric Assay
MDA-MB-231 cells were seeded at 2 × 103 cells per well in a clear 96-well plate, the medium volume was brought to 100 µL, and the cells were allowed to attach overnight. The cells were then incubated with the indicated concentration of compound 14, 15, or 16 at 37 ℃ for 24 h. Cell viability was determined using the Promega Cell Titer 96 Aqueous One Solution cell proliferation assay. Absorbance at 490 nm was read on Tecan Infinite F200 Pro plate reader, and values were expressed as percent of absorbance from cells incubated in DMSO alone.
3.6. Western Blot
Cells were seeded in 100 mm culture dishes (1 × 106 cells/dish) and allowed to attach overnight. The compound was added at varying concentrations, and the cells were incubated for an additional 24 h. For comparison, cells were also incubated with DMSO (0.5%) for 24 h. The cells were harvested in ice-cold lysis buffer (23 mM Tris-HCl pH 7.6, 130 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS), and 30 µg of lysate per lane was separated by SDS-PAGE, which was transferred to a PVDF membrane (Bio-Rad). The membrane was blocked with 5% skim milk in TBST and then incubated with the corresponding antibody (HSF-1 or β-actin). After binding of an appropriate secondary antibody coupled to horseradish peroxidase, proteins were visualized by ECL chemiluminescence according to the instructions of the manufacturer (GE healthcare, Chicago, IL, USA).
4. Conclusions
Keeping in mind the importance of HSF1 as a master regulator of heat shock protein and as a potential target for the development of anticancer drugs, we designed and synthesized new HSF1-based proteolysis-targeting chimeric molecules HSF1-PROTACs, in which the pomalidomide is tethered with HSF1 binder KRIBB11 to unlock the untapped potential and expand the repertoire of target warhead that can be recruited by PROTACs for anticancer drug development. Further evaluation to find better PROTACs with newer targets is underway in our lab and will be published in a due course of time.
Supplementary Materials
Figure S1: 1H spectrum of compound 3 (500 MHz, DMSO-D6), Figure S2: 1H spectrum of compound 4 (500 MHz, DMSO-D6), Figure S3: 1H spectrum of compound 6 (500 MHz, DMSO-D6), Figure S4: 1H spectrum of compound 7 (500 MHz, DMSO-D6, Figure S5: 1H spectrum of compound 9a (500 MHz, DMSO-D6), Figure S6: 1H spectrum of compound 9b (500 MHz, DMSO-D6), Figure S7: 1H spectrum of compound 9c (500 MHz, DMSO-D6), Figure S8: 1H spectrum of compound 10b (500 MHz, CDCl3), Figure S9: 1H spectrum of compound 12a (500 MHz, CDCl3), Figure S10: 1H spectrum of compound 12b (500 MHz, CDCl3), Figure S11: 1H spectrum of compound 11 (500 MHz, CDCl3), Figure S12: 1H spectrum of compound 13a (500 MHz, CDCl3), Figure S13: 1H spectrum of compound 13b (500 MHz, CD3OD), Figure S14: 1H spectrum of compound 14 (500 MHz, Acetone-D6), Figure S15: 13C spectrum of compound 14 (126 MHz, Acetone-D6), Figure S16: 1H spectrum of compound 15 (500 MHz, Acetone-D6), Figure S17: 13C spectrum of compound 15 (126 MHz, CDCl3), Figure S18: 1H spectrum of compound 16 (500 MHz, Acetone-D6), Figure S19: 13C spectrum of compound 16 (126 MHz, Acetone-D6), Figure S20: HPLC Chromatogram of PROTACs 14–16.
Author Contributions
Conceptualization, C.S. and Y.H.S.; synthesis, C.S. and M.A.C.; biology, Y.S.; writing—review and editing, C.S. and Y.H.S.; supervision, Y.H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1A6A1A03011325 and 2016R1D1A1B01009559).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- WHO Factsheet. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 26 December 2021).
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2022, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Shuzhen, T.; Dongpei, L.; Xiao, Z. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother 2020, 124, 109821. [Google Scholar]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. The PROTAC gold rush. Nat. Biotechnol. 2021, 40, 12–16. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Small-Molecule Modulation of Protein Homeostasis. Chem. Rev. 2017, 117, 11269–11301. [Google Scholar] [CrossRef]
- Modell, A.E.; Lai, S.; Nguyen, T.M.; Choudhary, A. Bifunctional modalities for repurposing protein function. Cell Chem. Biol. 2021, 28, 1081–1089. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Gokmen-Polar, Y. HSF1 as a Cancer Biomarker and Therapeutic Target. Curr. Cancer Drug Targets 2019, 19, 515–524. [Google Scholar] [CrossRef]
- Mantso, T.; Vasileiadis, S.; Anestopoulos, I.; Voulgaridou, G.-P.; Lampri, E.; Botaitis, S.; Kontomanolis, E.N.; Simopoulos, C.; Goussetis, G.; Franco, R.; et al. Hyperthermia induces therapeutic effectiveness and potentiates adjuvant therapy with non-targeted and targeted drugs in an in vitro model of human malignant melanoma. Sci. Rep. 2018, 8, 10724. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Seo, Y.H. Small Molecule Inhibitors of HSF1-Activated Pathways as Potential Next-Generation Anticancer Therapeutics. Molecules 2018, 23, 2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, C.; Oh, Y.J.; Park, B.; Lee, S.; Jeong, C.-H.; Lee, S.; Seo, J.H.; Seo, Y.H. Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction. Int. J. Mol. Sci. 2019, 20, 6213. [Google Scholar] [CrossRef] [PubMed]
- A Choi, M.; Park, S.Y.; Chae, H.Y.; Song, Y.; Sharma, C.; Seo, Y.H. Design, synthesis and biological evaluation of a series of CNS penetrant HDAC inhibitors structurally derived from amyloid-β probes. Sci. Rep. 2019, 9, 13187. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.-S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.-M.; Han, D.C. KRIBB11 Inhibits HSP70 Synthesis through Inhibition of Heat Shock Factor 1 Function by Impairing the Recruitment of Positive Transcription Elongation Factor b to the hsp70 Promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Neudegger, T.; Verghese, J.; Hayer-Hartl, M.; Hartl, F.U.; Bracher, A. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat. Struct. Mol. Biol. 2016, 23, 140–146. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.M.; Lee, J.S.; Park, W.J.; Hwang, Y.H.; Kim, Y.K. Novel 2,6-Substituted-3-Nitropyridine Derivative, Method for Preparing Same, and Pharmaceutical Preparation Including Same. WO2010067989A2, 17 June 2017. [Google Scholar]
- Steinebach, C.; Kehm, H.; Lindner, S.; Vu, L.P.; Köpff, S.; Mármol, Á.L.; Weiler, C.; Wagner, K.G.; Reichenzeller, M.; Krönke, J.; et al. PROTAC-mediated crosstalk between E3 ligases. Chem. Commun. 2019, 55, 1821–1824. [Google Scholar] [CrossRef]
- Hou, X.; Du, J.; Zhang, J.; Du, L.; Fang, H.; Li, M. How to improve docking accuracy of AutoDock4.2: A case study using different electrostatic potentials. J. Chem. Inf. Model. 2013, 53, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
(A) Docking pose of KRIBB11 with HSF1 (PDB code: 5d5U). The image is visualized using PyMOL1.3. (B) Design of HSF1-PROTAC.
Figure 1.
(A) Docking pose of KRIBB11 with HSF1 (PDB code: 5d5U). The image is visualized using PyMOL1.3. (B) Design of HSF1-PROTAC.

Scheme 1.
Synthesis of KRIBB11 analogue 4 (a) TEA, EtOH, 90 °C, 5 h, 80.7%; (b) CH3CN, RT, 14 h, 70.3%.
Scheme 1.
Synthesis of KRIBB11 analogue 4 (a) TEA, EtOH, 90 °C, 5 h, 80.7%; (b) CH3CN, RT, 14 h, 70.3%.
Scheme 2.
Synthesis of Pomalidomide 9c (a) CDI, 4-DMAP, THF, 76 °C, 73%; (b) TFA, 30 min, RT, quant. (c) NaOCH3, AcOH, 18 h, 140 °C, 91% for 9a, and 44.5% for 9b (d) 10% Pd/C, H2, 71 h, RT, 90.8%.
Scheme 2.
Synthesis of Pomalidomide 9c (a) CDI, 4-DMAP, THF, 76 °C, 73%; (b) TFA, 30 min, RT, quant. (c) NaOCH3, AcOH, 18 h, 140 °C, 91% for 9a, and 44.5% for 9b (d) 10% Pd/C, H2, 71 h, RT, 90.8%.

Scheme 3.
Synthesis of linkers. (a) Boc2O, TEA, anhyd. DCM, 4 h, RT, 51.5%; (b) 9a, DIPEA, DMF, 140 °C, 12 h, 40%; (c) Chloroacetyl chloride, DIPEA, THF, 6 h, 44.5%; (d) 9c, Cs2CO3, CH3CN, RT, 3 h, 33%; (e) TFA, 30 min., quant.
Scheme 3.
Synthesis of linkers. (a) Boc2O, TEA, anhyd. DCM, 4 h, RT, 51.5%; (b) 9a, DIPEA, DMF, 140 °C, 12 h, 40%; (c) Chloroacetyl chloride, DIPEA, THF, 6 h, 44.5%; (d) 9c, Cs2CO3, CH3CN, RT, 3 h, 33%; (e) TFA, 30 min., quant.

Scheme 4.
Synthesis of HSF1-PROTACs 14–16. (a) DIPEA, DMF, 90 °C, 48 h, 58%; (b) TEA, CH3CN, RT, 24 h, 47% for 15 and 18 h, 38% for 16.
Scheme 4.
Synthesis of HSF1-PROTACs 14–16. (a) DIPEA, DMF, 90 °C, 48 h, 58%; (b) TEA, CH3CN, RT, 24 h, 47% for 15 and 18 h, 38% for 16.
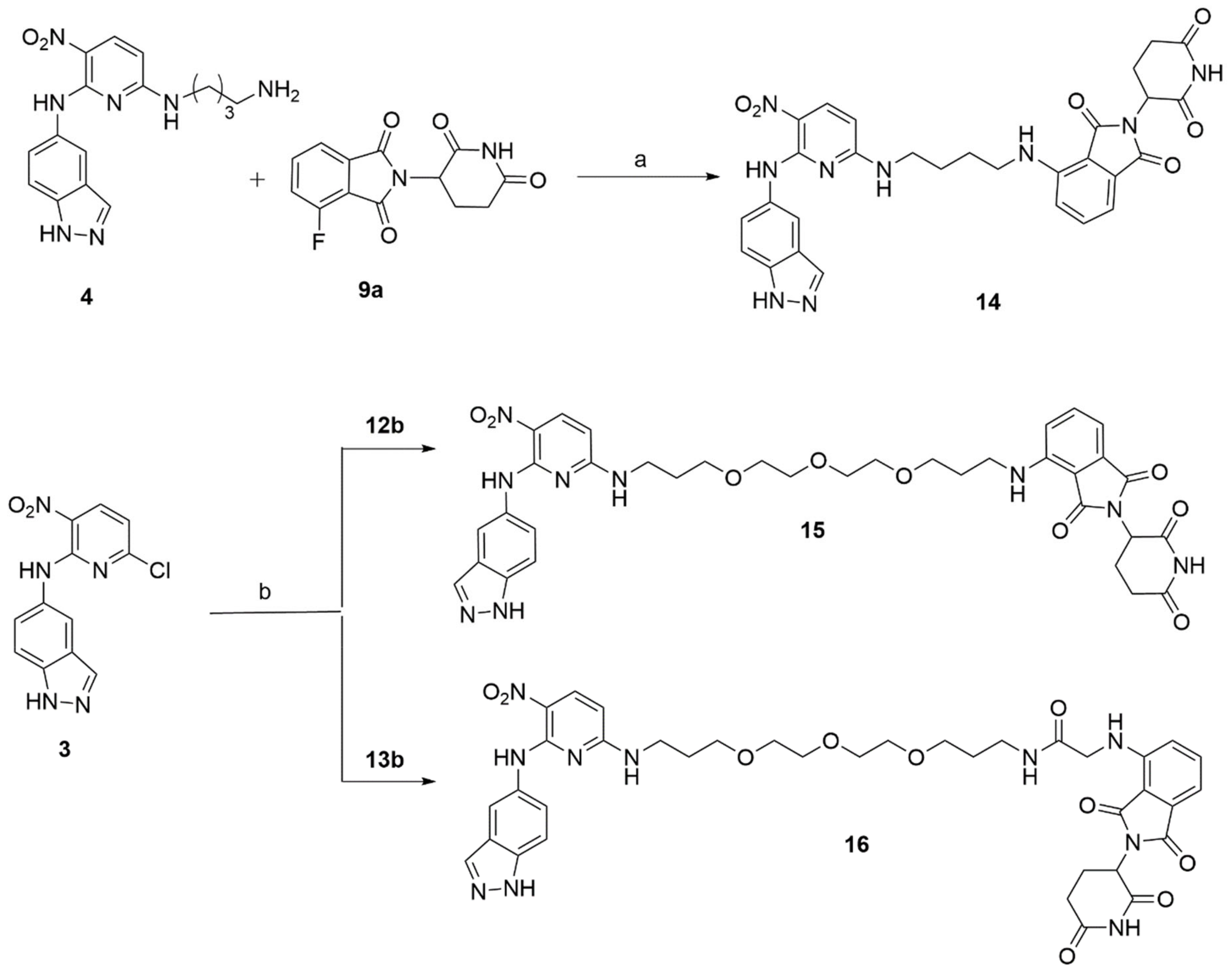
Figure 2.
Effect of compounds 14–16 on the cell viability of triple negative breast cancer cells, MDA-MB-231. MDA-MB-231 cells were incubated with the indicated concentrations of 14, 15, or 16 for 24 h and cell viability was measured by MTS colorimetric assay.
Figure 2.
Effect of compounds 14–16 on the cell viability of triple negative breast cancer cells, MDA-MB-231. MDA-MB-231 cells were incubated with the indicated concentrations of 14, 15, or 16 for 24 h and cell viability was measured by MTS colorimetric assay.
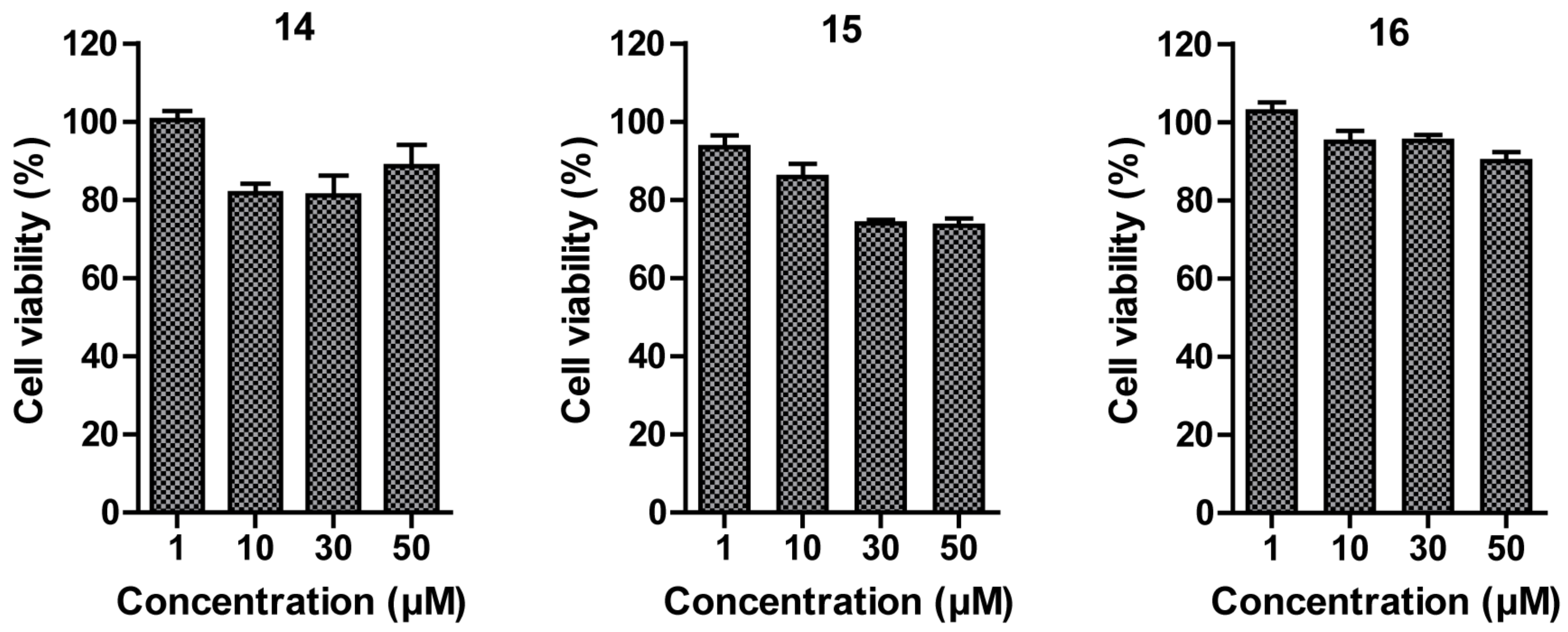
Figure 3.
Western blot analysis of HSF1 protein in MDA-MB-231 cell line. (A) MDA-MB-231 cells were treated with the indicated concentrations of 14, 15, or 16 for 24 h. β-Actine was used as a loading control. (B) MDA-MB-231 cells were treated with the indicated concentrations of 14, 15, or 16, together with an Hsp90 inhibitor (0.5 μM) for 24 h.
Figure 3.
Western blot analysis of HSF1 protein in MDA-MB-231 cell line. (A) MDA-MB-231 cells were treated with the indicated concentrations of 14, 15, or 16 for 24 h. β-Actine was used as a loading control. (B) MDA-MB-231 cells were treated with the indicated concentrations of 14, 15, or 16, together with an Hsp90 inhibitor (0.5 μM) for 24 h.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, C.; Choi, M.A.; Song, Y.; Seo, Y.H. Rational Design and Synthesis of HSF1-PROTACs for Anticancer Drug Development. Molecules 2022, 27, 1655. https://doi.org/10.3390/molecules27051655
AMA Style
Sharma C, Choi MA, Song Y, Seo YH. Rational Design and Synthesis of HSF1-PROTACs for Anticancer Drug Development. Molecules. 2022; 27(5):1655. https://doi.org/10.3390/molecules27051655
Chicago/Turabian StyleSharma, Chiranjeev, Myeong A Choi, Yoojin Song, and Young Ho Seo. 2022. "Rational Design and Synthesis of HSF1-PROTACs for Anticancer Drug Development" Molecules 27, no. 5: 1655. https://doi.org/10.3390/molecules27051655