
Binding and Degradation Reaction of Hydroxide Ions with Several Quaternary Ammonium Head Groups of Anion Exchange Membranes Investigated by the DFT Method

 ,
,  , and
, and
Abstract
:1. Introduction
2. Model and Method
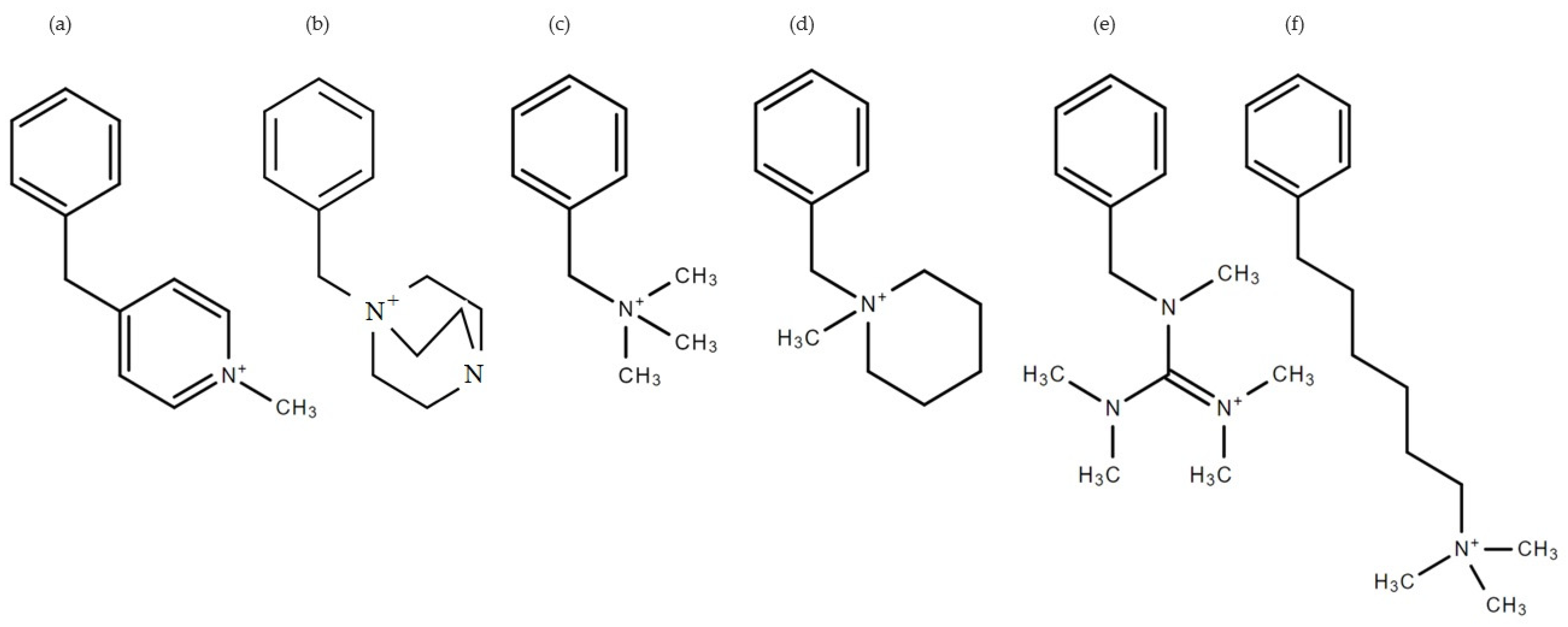
2.1. System of Interest
2.2. DFT Calculations
2.2.1. Molecular Electrostatic Maps, Binding Energies, and LUMO Energies
2.2.2. Degradation Reaction
3. Results and Discussion
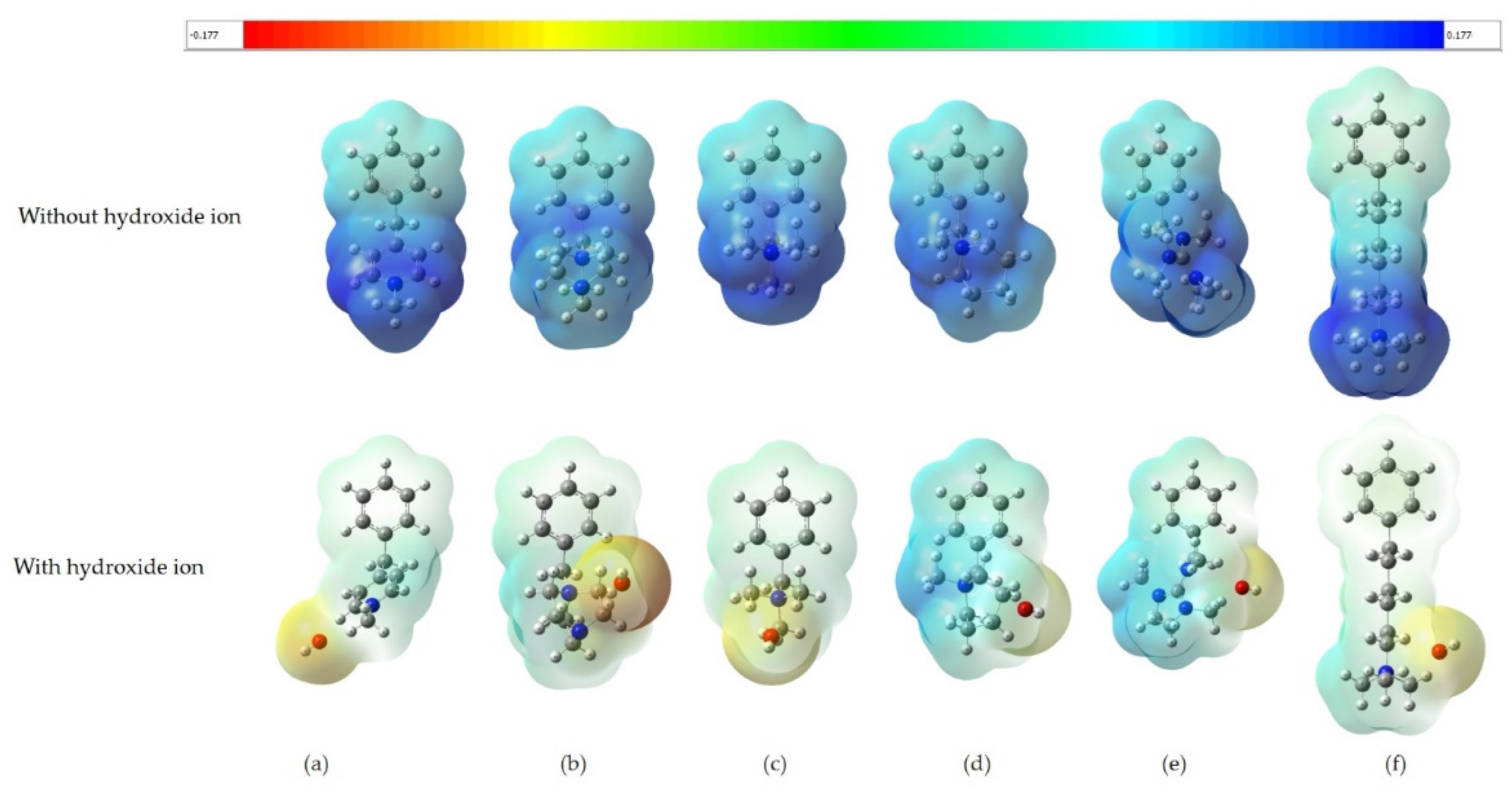
3.1. Molecular Electrostatic Potential Maps
3.2. Binding Energies
3.3. LUMO Distribution and Energy
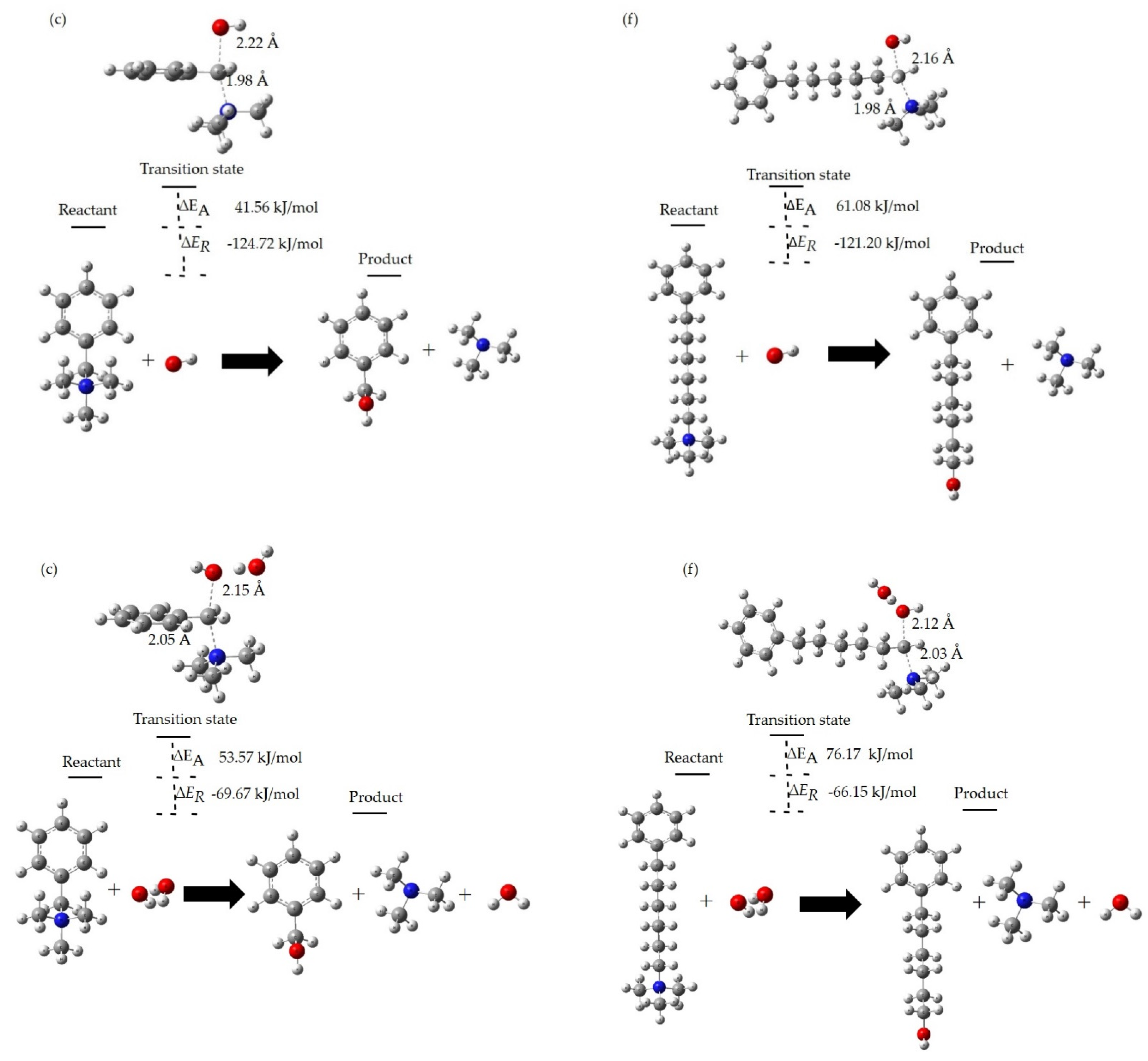
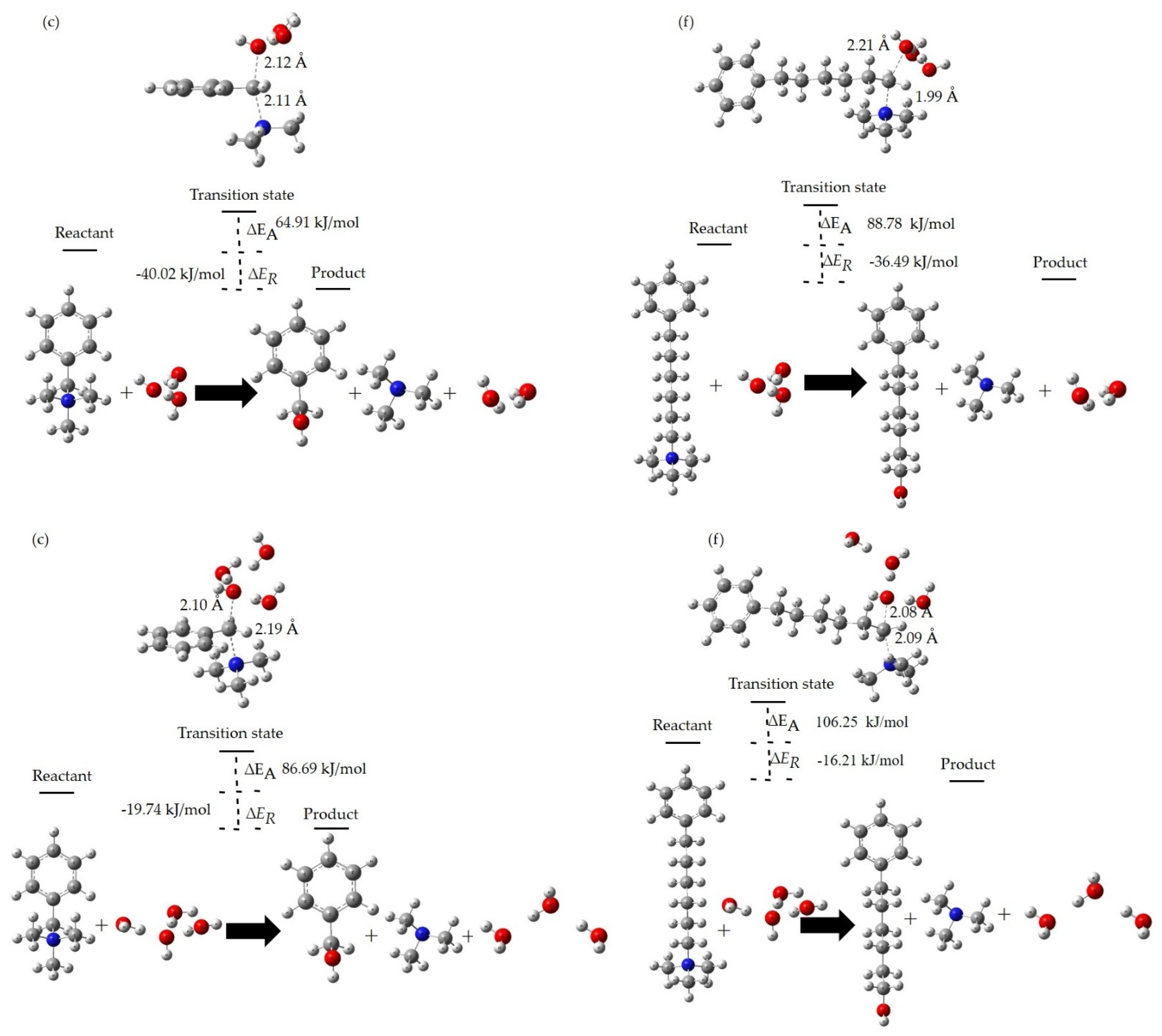
3.4. Degradation Reactions at the Different HLs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AEMFCs | Anion exchange membrane fuel cells |
| BSSE | Basis set superposition error |
| BTMA | Benzyltrimethylammonium |
| DABCO | 1,4-Diazabicyclo [2.2.2] octane |
| DFT | Density functional theory |
| EBinding | Binding energy |
| ER | Reaction energy |
| EA | Activation energy |
| ESP | Electrostatic potential |
| LUMO | Lowest unoccupied molecular orbital |
| OH− | Hydroxide ion |
| PEEK | Poly (ether ether ketone) |
| PEMFCs | Proton exchange membrane fuel cells |
| PPO | Poly phenylene oxide |
| PS | Polystyrene |
| PVBTMA | Poly (vinyl benzyl trimethylammonium) |
| QA | Quaternary ammonium |
| SN2 | Nucleophilic substitution degradation reaction mechanism |
| TMHA | Trimethylhexylammonium |
References
- Osmieri, L.; Pezzolato, L.; Specchia, S. Recent trends on the application of PGM-free catalysts at the cathode of anion exchange membrane fuel cells. Curr. Opin. Electrochem. 2018, 9, 240–256. [Google Scholar] [CrossRef]
- Dekel, D.R. Review of cell performance in anion exchange membrane fuel cells. J. Power Sources 2018, 375, 158–169. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Lin, B.; Yan, F. Anion-Exchange Membranes for Alkaline Fuel-Cell Applications: The Effects of Cations. ChemSusChem 2017, 11, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Dekel, D.R.; Amar, M.; Willdorf, S.; Kosa, M.; Dhara, S.; Diesendruck, C.E. Effect of Water on the Stability of Quaternary Ammonium Groups for Anion Exchange Membrane Fuel Cell Applications. Chem. Mater. 2017, 29, 4425–4431. [Google Scholar] [CrossRef] [Green Version]
- Dekel, D.R.; Willdorf, S.; Ash, U.; Amar, M.; Pusara, S.; Dhara, S.; Srebnik, S.; Diesendruck, C.E. The critical relation between chemical stability of cations and water in anion exchange membrane fuel cells environment. J. Power Sources 2018, 375, 351–360. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Zhang, J.; Lu, S.; Xiang, Y. Effect of side chain on the electrochemical performance of poly (ether ether ketone) based anion-exchange membrane: A molecular dynamics study. J. Membr. Sci. 2020, 605, 118105. [Google Scholar] [CrossRef]
- Lee, M.-T. Designing Anion Exchange Membranes with Enhanced Hydroxide Ion Conductivity by Mesoscale Simulations. J. Phys. Chem. C 2020, 124, 4470–4482. [Google Scholar] [CrossRef]
- Lu, J.; Barnett, A.; Molinero, V. Effect of Polymer Architecture on the Nanophase Segregation, Ionic Conductivity, and Electro-Osmotic Drag of Anion Exchange Membranes. J. Phys. Chem. C 2019, 123, 8717–8726. [Google Scholar] [CrossRef]
- Lee, M.-T. Exploring Side-Chain Designs for Enhanced Ion Conductivity of Anion-Exchange Membranes by Mesoscale Simulations. J. Phys. Chem. C 2019, 123, 10802–10815. [Google Scholar] [CrossRef]
- Lu, J.; Jacobson, L.C.; Sirkin, Y.A.P.; Molinero, V. High-Resolution Coarse-Grained Model of Hydrated Anion-Exchange Membranes that Accounts for Hydrophobic and Ionic Interactions through Short-Ranged Potentials. J. Chem. Theory Comput. 2016, 13, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Paddison, S.J. DPD simulations of anion exchange membrane: The effect of an alkyl spacer on the hydrated morphology. Solid State Ion. 2019, 339, 115012. [Google Scholar] [CrossRef]
- Zhu, Z.; Luo, X.; Paddison, S.J. DPD simulations of anion exchange membranes functionalized with various cationic groups and associated anions. Solid State Ion. 2019, 340, 115011. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, C.H.; Nam, S.Y. Molecular dynamics simulations of modified PEEK polymeric membrane for fuel cell application. Int. J. Hydrogen Energy 2016, 41, 7641–7648. [Google Scholar] [CrossRef]
- Takaba, H.; Hisabe, T.; Shimizu, T.; Alam, K. Molecular modeling of OH− transport in poly(arylene ether sulfone ketone)s containing quaternized ammonio-substituted fluorenyl groups as anion exchange membranes. J. Membr. Sci. 2017, 522, 237–244. [Google Scholar] [CrossRef]
- Zelovich, T.; Vogt-Maranto, L.; Simari, C.; Nicotera, I.; Hickner, M.A.; Paddison, S.J.; Bae, C.; Dekel, D.R.; Tuckerman, M.E. Non-Monotonic Temperature Dependence of Hydroxide Ion Diffusion in Anion Exchange Membranes. Chem. Mater. 2022, 34, 2133–2145. [Google Scholar] [CrossRef]
- Castaneda, S.; Ribadeneira, R.E. Theoretical Description of the Structural Characteristics of the Quaternized SEBS Anion-Exchange Membrane Using DFT. J. Phys. Chem. C 2015, 119, 28235–28246. [Google Scholar] [CrossRef]
- Tsuchitani, R.; Nakanishi, H.; Shishitani, H.; Yamaguchi, S.; Tanaka, H.; Kasai, H. A theoretical study of how C2-substitution affects alkaline stability in imidazolium-based anion exchange membranes. Solid State Ion. 2015, 278, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-L.; Yang, G.; Cheng, J.; Zhang, F.; Hao, C. Hydroxide degradation pathways for guanidimidazolium cation: A density functional theory study. J. Phys. Org. Chem. 2018, 31, e3861. [Google Scholar] [CrossRef]
- Long, H.; Pivovar, B. Hydroxide Degradation Pathways for Imidazolium Cations: A DFT Study. J. Phys. Chem. C 2014, 118, 9880–9888. [Google Scholar] [CrossRef]
- Long, H.; Kim, K.; Pivovar, B.S. Hydroxide Degradation Pathways for Substituted Trimethylammonium Cations: A DFT Study. J. Phys. Chem. C 2012, 116, 9419–9426. [Google Scholar] [CrossRef]
- Van Mourik, T.; Bühl, M.; Gaigeot, M.-P. Density functional theory across chemistry, physics and biology. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2014, 372, 20120488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chempath, S.; Einsla, B.R.; Pratt, L.R.; Macomber, C.S.; Boncella, J.M.; Rau, A.J.A.; Pivovar, B.S. Mechanism of Tetraalkylammonium Headgroup Degradation in Alkaline Fuel Cell Membranes. J. Phys. Chem. C 2008, 112, 3179–3182. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Gill, P.M.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef] [Green Version]
- De Castro, E.V.R.; Jorge, F.E. Accurate universal Gaussian basis set for all atoms of the Periodic Table. J. Chem. Phys. 1998, 108, 5225–5229. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_C01, Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking. Carbon 2020, 171, 514–523. [Google Scholar] [CrossRef]
- Lu, T.; Manzetti, S. Wavefunction and reactivity study of benzo [a] pyrene diol epoxide and its enantiomeric forms. Struct. Chem. 2014, 25, 1521–1533. [Google Scholar] [CrossRef]
- Zeng, Q.H.; Liu, Q.L.; Broadwell, I.; Zhu, A.M.; Xiong, Y.; Tu, X.P. Anion exchange membranes based on quaternized polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene for direct methanol alkaline fuel cells. J. Membr. Sci. 2010, 349, 237–243. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Zhang, X.; Wang, W.; Xie, X.; Pei, P. Degradation of guanidinium-functionalized anion exchange membrane during alkaline environment. Int. J. Hydrogen Energy 2014, 39, 13710–13717. [Google Scholar] [CrossRef]
- Wang, J.; He, G.; Wu, X.; Yan, X.; Zhang, Y.; Wang, Y.; Du, L. Crosslinked poly (ether ether ketone) hydroxide exchange membranes with improved conductivity. J. Membr. Sci. 2014, 459, 86–95. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. van der Waals potential: An important complement to molecular electrostatic potential in studying intermolecular interactions. J. Mol. Modeling 2020, 26, 315. [Google Scholar] [CrossRef]
- Noh, S.; Jeon, J.Y.; Adhikari, S.; Kim, Y.S.; Bae, C. Molecular Engineering of Hydroxide Conducting Polymers for Anion Exchange Membranes in Electrochemical Energy Conversion Technology. Acc. Chem. Res. 2019, 52, 2745–2755. [Google Scholar] [CrossRef]
- Mohanty, A.D.; Bae, C. Mechanistic analysis of ammonium cation stability for alkaline exchange membrane fuel cells. J. Mater. Chem. A 2014, 2, 17314–17320. [Google Scholar] [CrossRef]
- Mohanty, A.D.; Tignor, S.E.; Sturgeon, M.R.; Long, H.; Pivovar, B.S.; Bae, C. Thermochemical Stability Study of Alkyl Tethered Quaternary Ammonium Cations for Anion Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2017, 164, F1279–F1285. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QA | Conductivity (mS/cm) | |
|---|---|---|
| (a) | ||
| (c) | 5.12 [34] | |
| (f) | ||
| (d) | ||
| (e) | 6.40 [35] | |
| (b) | 8.60 [36] |
| QA | HL | BSSE | ||
|---|---|---|---|---|
| (c) | 0 | |||
| 1 | ||||
| 2 | ||||
| 3 | ||||
| (f) | 0 | |||
| 1 | ||||
| 2 | ||||
| 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karibayev, M.; Myrzakhmetov, B.; Kalybekkyzy, S.; Wang, Y.; Mentbayeva, A. Binding and Degradation Reaction of Hydroxide Ions with Several Quaternary Ammonium Head Groups of Anion Exchange Membranes Investigated by the DFT Method. Molecules 2022, 27, 2686. https://doi.org/10.3390/molecules27092686
Karibayev M, Myrzakhmetov B, Kalybekkyzy S, Wang Y, Mentbayeva A. Binding and Degradation Reaction of Hydroxide Ions with Several Quaternary Ammonium Head Groups of Anion Exchange Membranes Investigated by the DFT Method. Molecules. 2022; 27(9):2686. https://doi.org/10.3390/molecules27092686
Chicago/Turabian StyleKaribayev, Mirat, Bauyrzhan Myrzakhmetov, Sandugash Kalybekkyzy, Yanwei Wang, and Almagul Mentbayeva. 2022. "Binding and Degradation Reaction of Hydroxide Ions with Several Quaternary Ammonium Head Groups of Anion Exchange Membranes Investigated by the DFT Method" Molecules 27, no. 9: 2686. https://doi.org/10.3390/molecules27092686