
Substituent-Controllable Cascade Regioselective Annulation of β-Enaminones with N-Sulfonyl Triazoles for Modular Access to Imidazoles and Pyrroles



Abstract
:1. Introduction
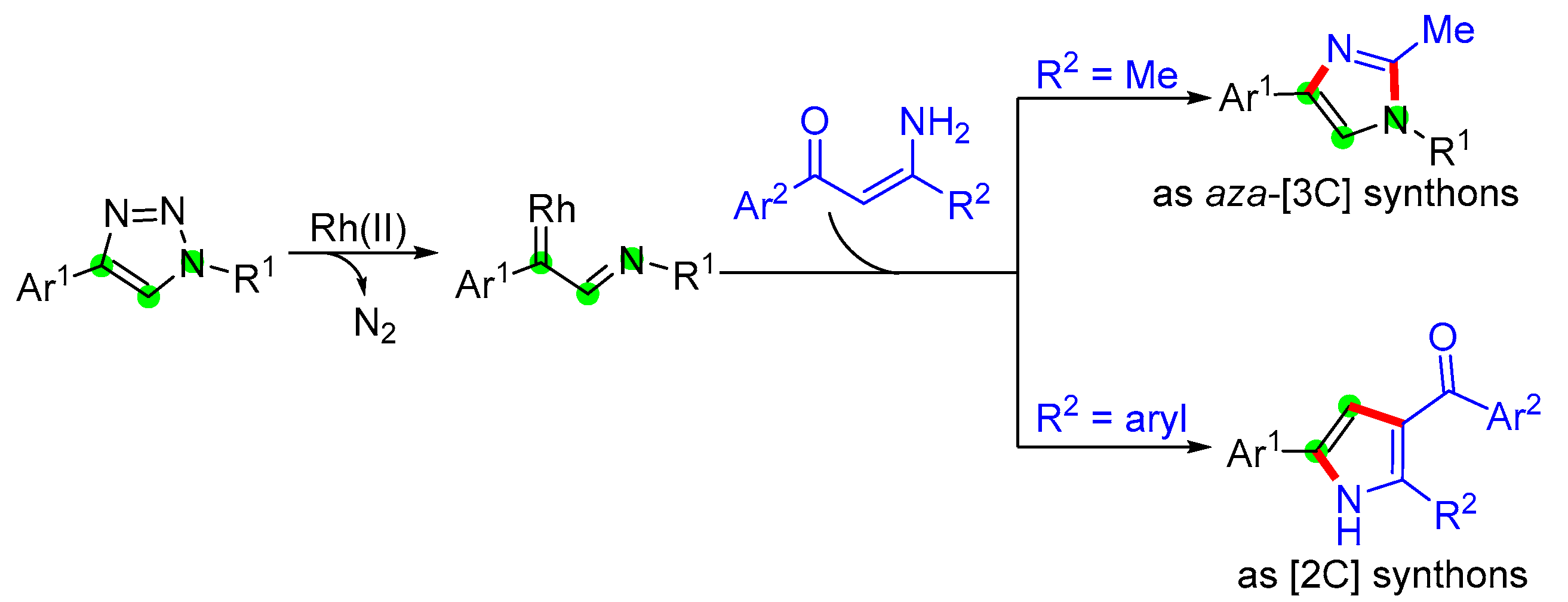
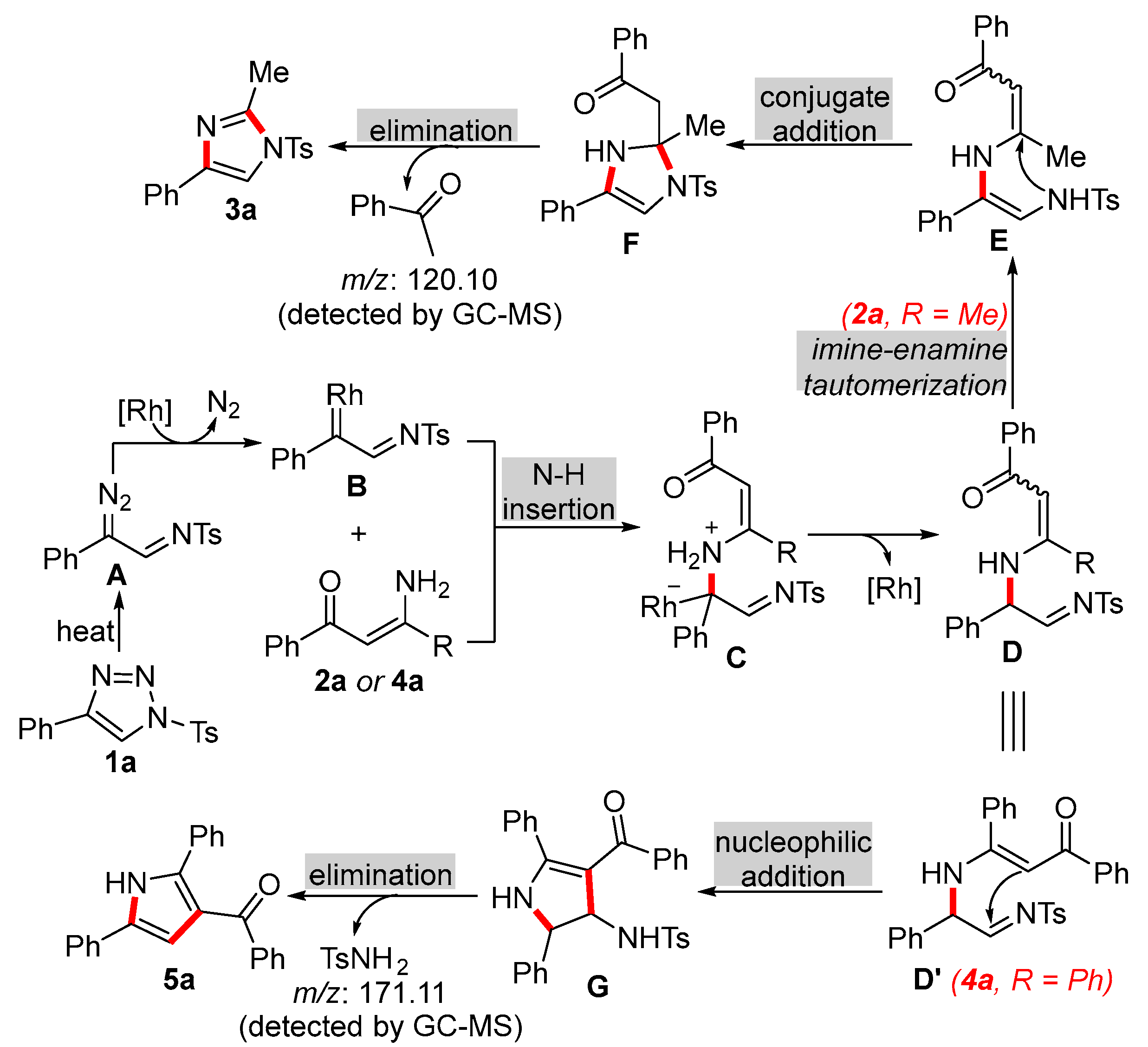
2. Results and Discussion
3. Materials and Methods
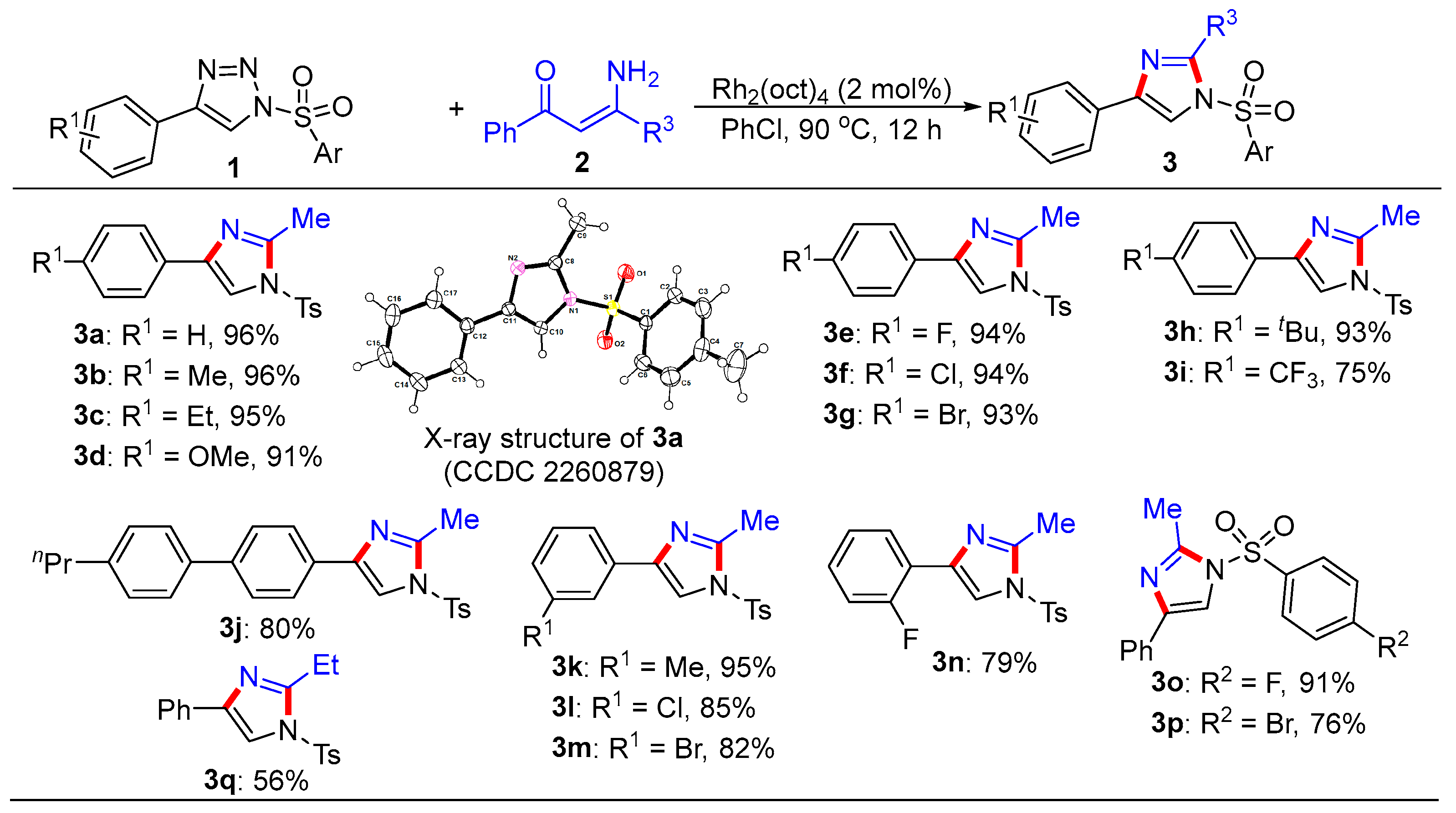
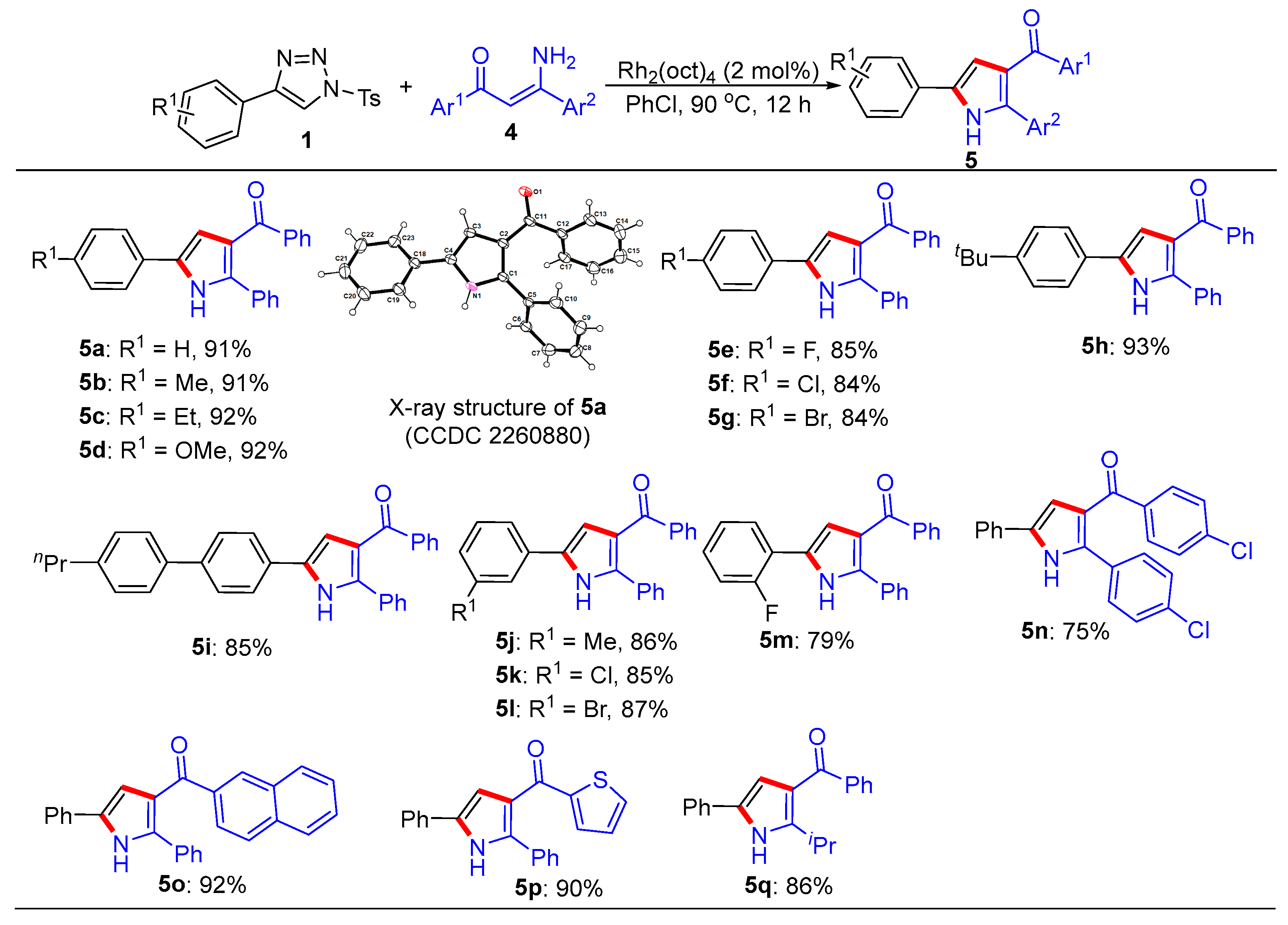
3.1. General Procedure for the Synthesis of Trisubstituted Imidazoles 3 and Pyrroles 5
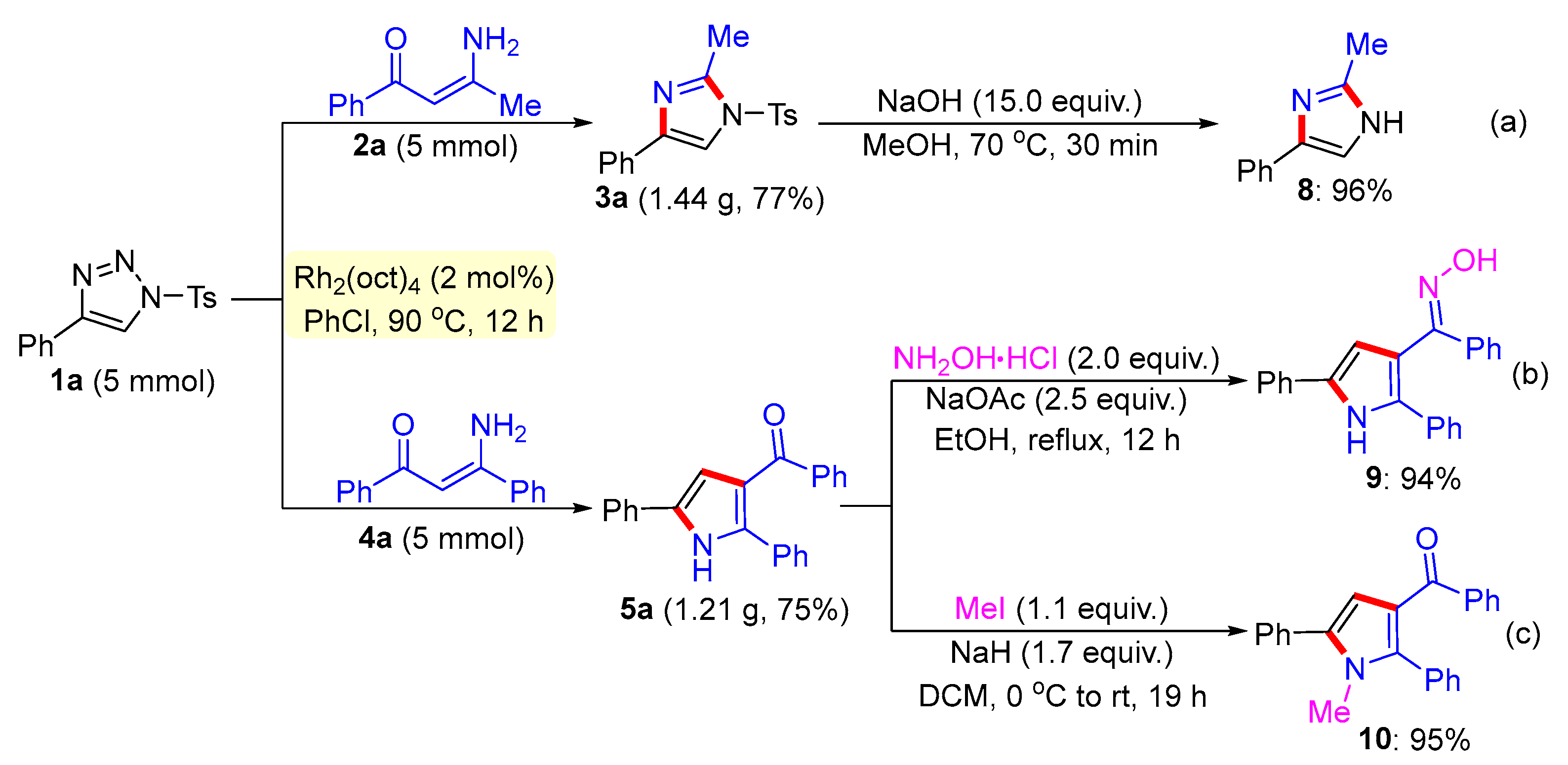
3.2. General Procedure for the Synthesis of Compound 8
3.3. General Procedure for the Synthesis of Compound 9
3.4. General Procedure for the Synthesis of Compound 10
- 2-Methyl-4-phenyl-1-tosyl-1H-imidazole (3a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 96% yield (60 mg); mp 122–124 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.39–7.35 (m, 4H), 7.25 (d, J = 7.5 Hz, 1H), 2.57 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.4, 141.0, 135.5, 132.7, 130.8, 129.1, 128.2, 127.8, 125.6, 114.4, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H17N2O2S 313.1005; found 313.1006.
- 2-Methyl-4-(p-tolyl)-1-tosyl-1H-imidazole (3b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 96% yield (62 mg); mp 60–62 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.0 Hz, 3H), 7.35 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 2.57 (s, 3H), 2.44 (s, 3H), 2.35 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.4, 146.3, 141.0, 138.0, 135.4, 130.7, 129.9, 129.8, 127.7, 125.5, 113.9, 22.1, 21.7, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; found 327.1170.
- 4-(4-Ethylphenyl)-2-methyl-1-tosyl-1H-imidazole (3c). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 95% yield (64 mg); mp 77–79 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.0 Hz, 3H), 7.34 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 2.64 (q, J = 7.5 Hz, 2H), 2.57 (s, 3H), 2.42 (s, 3H), 1.23 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.4, 146.3, 144.4, 141.0, 135.4, 130.8, 130.1, 128.6, 127.7, 125.5, 113.9, 29.1, 22.1, 15.9, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H21N2O2S 341.1318; found 341.1319.
- 4-(4-Methoxyphenyl)-2-methyl-1-tosyl-1H-imidazole (3d). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 91% yield (62 mg); mp 66–68 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 9.0 Hz, 2H), 7.57 (s, 1H), 7.34 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 9.0 Hz, 2H), 3.81 (s, 3H), 2.56 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.8, 146.4, 146.3, 140.8, 135.4, 130.7, 127.7, 126.9, 125.5, 114.5, 113.2, 55.7, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O3S 343.1111; found 343.1112.
- 4-(4-Fluorophenyl)-2-methyl-1-tosyl-1H-imidazole (3e). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 94% yield (62 mg); mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.69 (dd, J = 8.5, 5.0 Hz, 2H), 7.61 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.06 (t, J = 8.5 Hz, 2H), 2.56 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 162.9 (d, JC-F = 246.3 Hz), 146.6, 146.5, 140.1, 135.3, 130.8, 129.0, 127.8, 127.3 (d, JC-F = 8.0 Hz), 116.0 (d, JC-F = 21.6 Hz), 114.0, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16FN2O2S 331.0911; found 331.0909.
- 4-(4-Chlorophenyl)-2-methyl-1-tosyl-1H-imidazole (3f). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 94% yield (65 mg); mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.5 Hz, 2H), 7.66–7.65 (m, 3H), 7.36–7.32 (m, 4H), 2.56 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.6, 146.6, 139.9, 135.2, 133.8, 131.3, 130.8, 129.2, 127.8, 126.8, 114.5, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16ClN2O2S 347.0616; found 347.0612.
- 4-(4-Bromophenyl)-2-methyl-1-tosyl-1H-imidazole (3g). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 93% yield (72 mg); mp 104–106 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.60 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 2.56 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 146.6, 139.9, 135.2, 132.2, 131.7, 130.8, 127.8, 127.1, 122.0, 114.6, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16BrN2O2S 391.0110; found 391.0109.
- 4-(4-(tert-Butyl)phenyl)-2-methyl-1-tosyl-1H-imidazole (3h). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 93% yield (68 mg); mp 69–71 °C; 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 7.0 Hz, 2H), 7.69–7.64 (m, 3H), 7.39 (d, J = 7.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 2.57 (s, 3H), 2.42 (s, 3H), 1.32 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 151.3, 146.4, 146.4, 141.0, 135.5, 130.7, 129.9, 127.7, 126.0, 125.3, 114.0, 35.0, 31.7, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H25N2O2S 369.1631; found 369.1634.
- 2-Methyl-1-tosyl-4-(4-(trifluoromethyl)phenyl)-1H-imidazole (3i). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 75% yield (57 mg); mp 76–78 °C; 1H NMR (500 MHz, CDCl3) δ 7.84–7.82 (m, 4H), 7.76 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 2.58 (s, 3H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.8, 139.5, 136.2, 135.1, 130.9, 129.9 (q, JC-F = 32.5 Hz), 128.5, 127.9, 126.6, 126.1(q, JC-F = 3.8 Hz), 124.6(q, JC-F = 270.0 Hz), 115.6, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H] + Calcd for C18H16F3N2O2S 381.0879; found 381.0878.
- 2-Methyl-4-(4’-propyl-[1,1’-biphenyl]-4-yl)-1-tosyl-1H-imidazole (3j). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in an 80% yield (69 mg); mp 94–96 °C; 1H NMR (500 MHz, CDCl3) δ 7.83–7.79 (m, 4H), 7.71(s, 1H), 7.61 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 2.59 (s, 3H), 1.72–1.64 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.5, 142.4, 140.9, 140.7, 138.4, 135.4, 131.4, 130.8, 129.3, 127.8, 127.6, 127.1, 125.9, 114.3, 38.1, 25.0, 22.1, 15.6, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H27N2O2S 431.1788; found 431.1781.
- 2-Methyl-4-(m-tolyl)-1-tosyl-1H-imidazole (3k). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 95% yield (62 mg); mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.58 (s, 1H), 7.51 (d, J = 7.5 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.09 (d, J = 7.5 Hz, 1H), 2.57 (s, 3H), 2.43 (s, 3H), 2.37 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.4, 141.0, 138.8, 135.4, 132.5, 130.8, 129.0, 127.8, 126.2, 122.6, 114.3, 22.1, 21.8, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; found 327.1163.
- 4-(3-Chlorophenyl)-2-methyl-1-tosyl-1H-imidazole (3l). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in an 85% yield (59 mg); mp 83–85 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.0 Hz, 2H), 7.73 (s, 1H), 7.68 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.29 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 2.56 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 139.7, 135.1, 134.6, 130.8, 130.3, 128.1, 127.8, 125.7, 123.6, 115.0, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16ClN2O2S 347.0616; found 347.0625.
- 4-(3-Bromophenyl)-2-methyl-1-tosyl-1H-imidazole (3m). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in an 82% yield (64 mg); mp 79–81 °C; 1H NMR (500 MHz, CDCl3) δ 7.89 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.39–7.35 (m, 3H), 7.22 (t, J = 8.0 Hz, 1H), 2.56 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 146.6, 139.5, 135.2, 134.8, 131.0, 130.8, 130.6, 128.6, 127.8, 124.1, 123.3, 115.0, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16BrN2O2S 391.0110; found 391.0119.
- 4-(2-Fluorophenyl)-2-methyl-1-tosyl-1H-imidazole (3n). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 79% yield (52 mg); mp 57–59 °C; 1H NMR (500 MHz, CDCl3) δ 8.03 (t, J = 7.5 Hz, 1H), 7.85 (d, J = 4.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 7.0 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 7.10 (t, J = 10 Hz, 1H), 2.58 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 160.2 (d, JC-F = 247.9 Hz), 146.5, 145.9, 135.4, 134.5, 130.8, 129.1 (d, JC-F = 8.5 Hz), 128.2 (d, JC-F = 3.6 Hz), 127.8, 124.7 (d, JC-F = 3.6 Hz), 120.6 (d, JC-F = 12.5 Hz), 118.4 (d, JC-F = 15.4 Hz), 116.0 (d, JC-F = 21.5 Hz), 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16FN2O2S 331.0911; found 331.0914.
- 1-((4-Fluorophenyl)sulfonyl)-2-methyl-4-phenyl-1H-imidazole (3o). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 91% yield (57 mg); mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.97–7.94 (m, 2H), 7.73 (d, J = 7.5 Hz, 2H), 7.67 (s, 1H), 7.38 (t, J = 8.0 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.24 (t, J = 8.0 Hz, 2H), 2.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.6 (d, JC-F = 257.5 Hz), 146.4, 141.3, 134.4 (d, JC-F = 2.8 Hz), 132.5, 130.7 (d, JC-F = 9.8 Hz), 129.1, 128.4, 125.6, 117.7 (d, JC-F = 22.9 Hz), 114.2, 15.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14FN2O2S 317.0755; found 317.0760.
- 1-((4-Bromophenyl)sulfonyl)-2-methyl-4-phenyl-1H-imidazole (3p). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 76% yield (57 mg); mp 97–99 ℃; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 9.0 Hz, 2H), 7.72 (t, J = 9.0 Hz, 4H), 7.65 (s, 1H), 7.38 (d, J = 7.5 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 2.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 141.4, 137.3, 133.6, 132.4, 130.6, 129.1, 129.1, 128.4, 125.6, 114.2, 15.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14BrN2O2S 376.9954; found 376.9952.
- 2-Ethyl-4-phenyl-1-tosyl-1H-imidazole (3q). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 56% yield (37 mg); mp 49–51 °C; 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.39-7.34 (m, 4H), 7.28 (d, J = 7.5 Hz, 1H), 2.90 (q, J = 7.5 Hz, 2H), 2.43 (s, 3H), 1.32 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.4, 146.4, 140.9, 135.7, 132.9, 130.7, 129.0, 128.1, 127.7, 125.6, 114.3, 22.4, 22.1, 12.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; found 327.1163.
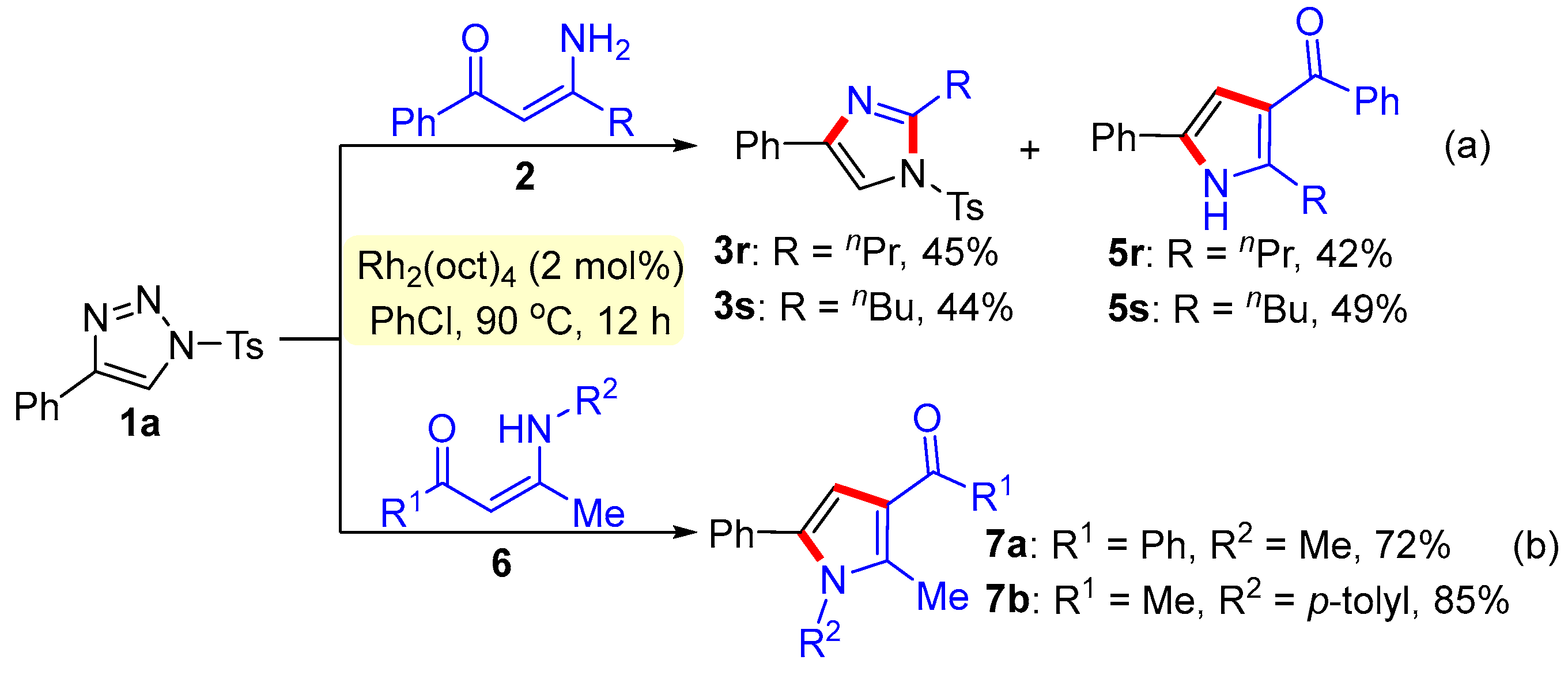
- 4-Phenyl-2-propyl-1-tosyl-1H-imidazole (3r). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a colorless oil in a 45% yield (31 mg); 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.0 Hz, 2H), 7.66 (s, 1H), 7.38–7.33 (m, 4H), 7.27 (t, J = 7.5 Hz, 1H), 2.85 (t, J = 7.5 Hz, 2H), 2.43 (s, 3H), 1.81–1.74 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 150.4, 146.4, 141.0, 135.8, 132.9, 130.7, 129.0, 128.1, 127.6, 125.6, 114.3, 30.8, 22.1, 21.9, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H21N2O2S 341.1318; found 341.1312.
- 2-Butyl-4-phenyl-1-tosyl-1H-imidazole (3s). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a colorless oil in a 44% yield (31 mg); 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.5 Hz, 2H), 7.67 (s, 1H), 7.38–7.33 (m, 4H), 7.28 (d, J = 7.5 Hz, 1H), 2.87 (t, J = 8 Hz, 2H), 2.43 (s, 3H), 1.72–1.69 (m, 2H), 1.42–1.37 (m, 2H), 0.91 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 150.5, 146.4, 140.9, 135.7, 132.9, 130.7, 129.1, 128.2, 127.7, 125.6, 114.3, 30.5, 28.6, 22.9, 22.1, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H23N2O2S 355.1475; found 355.1482.
- (2,5-Diphenyl-1H-pyrrol-3-yl)(phenyl)methanone (5a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 91% yield (59 mg); mp 81–83 °C; 1H NMR (500 MHz, CDCl3) δ 8.98 (s, 1H), 7.80 (d, J = 7.0 Hz, 2H), 7.54 (d, J = 7.5 Hz, 2H), 7.45-7.42 (m, 3H), 7.39 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.29–7.24 (m, 4H), 6.84 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.8, 138.3, 132.3, 132.1, 131.8, 130.1, 129.5, 128.9, 128.8, 128.5, 128.3, 127.5, 124.5, 122.3, 110.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H18NO 324.1383; found 324.1382.
- Phenyl(2-phenyl-5-(p-tolyl)-1H-pyrrol-3-yl)methanone (5b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 91% yield (61 mg); mp 81–83 °C; 1H NMR (500 MHz, CDCl3) δ 9.12 (s, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.47–7.38 (m, 5H), 7.31 (t, J = 7.7 Hz, 2H), 7.21–7.17 (m, 5H), 6.78 (s, 1H), 2.36 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 139.8, 138.1, 137.3, 132.5, 132.2, 132.1, 130.1, 130.1, 129.1, 128.9, 128.7, 128.4, 128.3, 124.6, 122.2, 110.4, 21.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; found 338.1545.
- (5-(4-Ethylphenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5c). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 92% yield (64 mg); mp 71–73 °C; 1H NMR (500 MHz, CDCl3) δ 8.92 (s, 1H), 7.80 (d, J = 7.0 Hz, 2H), 7.47-7.42 (m, 5H), 7.32 (t, J = 7.5 Hz, 2H), 7.26–7.22 (m, 5H), 6.81 (d, J = 3.0 Hz, 1H), 2.67 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 193.0, 143.8, 139.8, 138.0, 132.5, 132.2, 132.1, 130.1, 129.3, 128.9, 128.9, 128.7, 128.4, 128.3, 124.6, 122.2, 110.4, 29.0, 15.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H22NO 352.1693; found 352.1689.
- (5-(4-Methoxyphenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5d). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 92% yield (65 mg); mp 111–113 °C; 1H NMR (500 MHz, CDCl3) δ 8.96 (s, 1H), 7.79 (d, J = 7.5 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 7.0 Hz, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.22 (d, J = 7.5 Hz, 3H), 6.92 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 3.0 Hz, 1H), 3.82 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 159.3, 139.9, 137.9, 132.4, 132.3, 132.1, 130.1, 128.9, 128.7, 128.3, 128.3, 126.0, 124.8, 122.2, 114.9, 109.8, 55.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 354.1489; found 354.1489.
- (5-(4-Fluorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5e). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 85% yield (58 mg); mp 104–106 °C; 1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.50 (dd, J = 9.0, 5.0 Hz, 2H), 7.45–7.41 (m, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.24–7.22 (m, 3H), 7.08 (t, J = 8.5 Hz, 2H), 6.76 (d, J = 7.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 162.4 (d, JC-F = 245.0 Hz), 139.7, 138.3, 132.2, 132.1, 131.5, 130.1, 128.8 (d, JC-F = 6.3 Hz), 128.6, 128.3, 128.2, 126.4, 126.3, 122.4, 116.5 (d, JC-F = 22.5 Hz), 110.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17FNO 342.1289; found 342.1287.
- (5-(4-Chlorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5f). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 84% yield (60 mg); mp 112–114 ℃; 1H NMR (500 MHz, CDCl3) δ 9.04 (s, 1H), 7.77 (d, J = 7.0Hz, 2H), 7.46–7.42 (m, 3H), 7.39 (dd, J = 6.5, 3.0 Hz, 2H), 7.35–7.30 (m, 4H), 7.23–7.19 (m, 3H), 6.79 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.6, 138.6, 133.1, 132.2, 131.9, 131.3, 130.4, 130.1, 129.6, 128.9, 128.8, 128.6, 128.3, 125.8, 122.4, 111.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17ClNO 358.0993; found 358.1002.
- (5-(4-Bromophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5g). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 84% yield (67 mg); mp 127–129 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (s, 1H), 7.79 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.46–7.43 (m, 3H), 7.40 (d, J = 8.5 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.29-7.27 (m, 3H), 6.85 (d, J = 7.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 193.0, 139.6, 138.7, 132.5, 132.3, 131.9, 131.3, 130.8, 130.1, 128.9, 128.7, 128.6, 128.3, 126.1, 122.4, 121.1, 111.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17BrNO 402.0488; found 402.0489.
- (5-(4-(tert-Butyl)phenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5h). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 93% yield (70 mg); mp 98–100 °C; 1H NMR (500 MHz, CDCl3) δ 8.89 (s, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.49–7.40 (m, 7H), 7.32 (t, J = 8.0 Hz, 2H), 7.28–7.23 (m, 3H), 6.82 (s, 1H), 1.34 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 192.9, 150.7, 139.8, 138.0, 132.4, 132.3, 132.1, 130.1, 129.1, 128.9, 128.8, 128.4, 128.3, 126.4, 124.3, 122.3, 110.5, 35.0, 31.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H26NO 380.2009; found 380.2008.
- Phenyl(2-phenyl-5-(4’-propyl-[1,1’-biphenyl]-4-yl)-1H-pyrrol-3-yl)methanone (5i). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 85% yield (72 mg); mp 149–151 °C; 1H NMR (500 MHz, CDCl3) δ 9.43 (s, 1H), 7.80 (d, J = 7.5 Hz, 2H), 7.59 (s, 4H), 7.52 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.5 Hz, 1H), 7.41–7.36 (m, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 7.20–7.15 (m, 3H), 6.85 (d, J = 3.0 Hz, 1H), 2.64 (d, J = 8.0 Hz, 2H), 1.69 (m, 2H), 0.99 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 193.3, 142.5, 140.0, 139.8, 138.7, 138.2, 132.3, 132.2, 132.1, 130.5, 130.2, 129.4, 129.0, 128.7, 128.4, 128.3, 127.8, 127.1, 125.0, 122.3, 110.9, 38.1, 25.0, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C32H28NO 442.2165; found 442.2170.
- Phenyl(2-phenyl-5-(m-tolyl)-1H-pyrrol-3-yl)methanone (5j). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 86% yield (58 mg); mp 118–120 °C; 1H NMR (500 MHz, CDCl3) δ 9.08 (s, 1H), 7.80 (d, J = 7.5 Hz, 2H), 7.43 (d, J = 6.0 Hz, 3H), 7.37–7.31 (m, 4H), 7.27 (d, J = 7.5 Hz, 1H), 7.22 (t, J = 6.0 Hz, 3H), 7.08 (d, J = 7.5 Hz, 1H), 6.82 (d, J = 2.5 Hz, 1H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 139.8, 139.0, 138.4, 132.5, 132.2, 132.1, 131.8, 130.1, 129.3, 128.9, 128.7, 128.4, 128.3, 125.4, 122.2, 121.7, 110.8, 21.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; found 338.1531.
- (5-(3-Chlorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5k). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 85% yield (60 mg); mp 83–85 °C; 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 1H), 7.78 (d, J = 7.5 Hz, 2H), 7.51 (s, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.41–7.38 (m, 3H), 7.34–7.27 (m, 3H), 7.23–7.18 (m, 4H), 6.80 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 193.0, 139.6, 138.9, 135.4, 133.6, 132.3, 131.8, 130.9, 130.6, 130.1, 128.9, 128.7, 128.6, 128.4, 127.3, 124.6, 122.6, 122.3, 111.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17ClNO 358.0993; found 358.0992.
- (5-(3-Bromophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5l). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 87% yield (69 mg); mp 99–101 °C; 1H NMR (500 MHz, CDCl3) δ 9.10 (s, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.46–7.40 (m, 4H), 7.37 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 2H), 7.25–7.21 (m, 4H), 6.81 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.6, 138.9, 133.9, 132.3, 131.8, 130.9, 130.7, 130.2, 130.1, 128.9, 128.8, 128.7, 128.4, 127.4, 123.6, 123.1, 122.4, 111.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17BrNO 402.0488; found 402.0489.
- (5-(2-Fluorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5m). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 79% yield (54 mg); mp 77–79 °C; 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 7.81 (d, J = 7.0 Hz, 2H), 7.64 (t, J = 8.0 Hz, 1H), 7.48–7.44 (m, 3H), 7.34 (t, J = 8.0 Hz, 2H), 7.29–7.26 (m, 3H), 7.23–7.13 (m, 3H), 6.98 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ192.8, 159.2 (d, JC-F = 124.1 Hz), 139.7, 138.3, 132.2, 132.0, 130.1, 128.8, 128.6, 128.57 (d, JC-F = 8.5 Hz), 128.4, 127.3 (d, JC-F = 4.0 Hz), 127.1, 125.3 (d, JC-F = 3.0 Hz), 121.7, 119.4, 119.3, 116.8 (d, JC-F = 23.8 Hz), 112.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17FNO 342.1289; found 342.1281.
- (4-Chlorophenyl)(2-(4-chlorophenyl)-5-phenyl-1H-pyrrol-3-yl) methanone (5n). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 75% yield (58 mg); mp 94–96 °C; 1H NMR (500 MHz, CDCl3) δ 8.75 (s, 1H), 7.77 (d, J = 9.0 Hz, 2H), 7.53 (d, J = 7.0 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.42 (t, J = 8.0 Hz, 2H), 7.34 (m, 4H), 6.80 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 191.2, 138.7, 138.0, 136.8, 134.8, 132.7, 131.4, 130.5, 130.0, 129.6, 129.2, 128.8, 127.9, 124.6, 122.3, 110.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H16Cl2NO 392.0603; found 392.0612.
- (2,5-Diphenyl-1H-pyrrol-3-yl) (naphthalen-2-yl) methanone (5o). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 92% yield (68 mg); mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.34 (s, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.83 (t, J = 8.0 Hz, 3H), 7.60–7.52 (m, 5H), 7.49 (d, J = 7.0 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.31–7.26 (m, 3H), 7.22 (d, J = 7.5 Hz, 1H), 6.92 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.7, 138.1, 137.0, 135.4, 132.7, 132.3, 132.2, 131.8, 131.7, 129.7, 129.5, 128.9, 128.8, 128.6, 128.2, 128.2, 128.1, 127.6, 126.8, 126.1, 124.5, 122.6, 111.0; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H20NO 374.1539; found 374.1537.
- (2,5-Diphenyl-1H-pyrrol-3-yl) (thiophen-2-yl) methanone (5p). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 90% yield (59 mg); mp 86–88 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (s, 1H), 7.66 (d, J = 3.5 Hz, 1H), 7.60–7.54 (m, 5H), 7.42 (t, J = 8.0 Hz, 2H), 7.36 (t, J = 7.0 Hz, 2H), 7.33–7.28 (m, 2H), 7.04 (dd, J = 5.0, 4.0 Hz, 1H), 6.99 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 184.1, 145.9, 137.4, 134.1, 133.2, 132.4, 132.1, 131.8, 129.5, 129.0, 128.7, 128.6, 128.0, 127.6, 124.6, 122.3, 110.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H16NOS 330.0947; found 330.0955.
- (2-Isopropyl-5-phenyl-1H-pyrrol-3-yl) (phenyl)methanone (5q). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 86% yield (50 mg); mp 85–87 °C; 1H NMR (500 MHz, CDCl3) δ 8.79 (s, 1H), 7.84 (d, J = 7.0 Hz, 2H), 7.53 (t, J = 7.0 Hz, 1H), 7.48–7.44 (m, 4H), 7.36 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.5 Hz, 1H), 6.64 (d, J = 3.0 Hz, 1H), 3.87 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 192.8, 147.8, 141.1, 132.2, 131.6, 129.8, 129.5, 129.4, 128.5, 127.2, 124.3, 120.2, 110.1, 26.8, 22.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; found 290.1530.
- Phenyl(5-phenyl-2-propyl-1H-pyrrol-3-yl) methanone (5r). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 42% yield (24 mg); mp 74–76 °C; 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 1H), 7.87–7.82 (m, 2H), 7.53 (t, J = 7.5 Hz, 1H), 7.49–7.44 (m, 4H), 7.39–7.34 (m, 2H), 7.23 (t, J = 7.5 Hz, 1H), 6.66 (d, J = 3.0 Hz, 1H), 3.02 (t, J = 7.5 Hz, 2H), 1.76 (m, 2H), 1.60 (s, 3H), 1.01 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 192.7, 142.4, 141.0, 132.1, 131.6, 130.0, 129.5, 129.4, 128.5, 127.1, 124.2, 121.2, 109.8, 30.1, 23.1, 14.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; found 290.1542.
- (2-Butyl-5-phenyl-1H-pyrrol-3-yl) (phenyl)methanone (5s). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in a 49% yield (30 mg); mp 74–76 °C; 1H NMR (500 MHz, CDCl3) δ 9.24 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.55–7.45 (m, 5H), 7.34 (t, J = 7.0 Hz, 2H), 7.21 (t, J = 7.5 Hz, 1H), 6.67 (s, 1H), 3.01 (t, J = 7.5 Hz, 2H), 1.7–1.64 (m, 2H), 1.39–1.32 (m, 2H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 192.7, 142.7, 140.7, 131.8, 131.3, 129.8, 129.1, 129.0, 128.1, 126.6, 123.9, 120.6, 109.4, 31.7, 27.5, 22.6, 14.0, 13.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H22NO 304.1696; found 304.1702.
- (1,2-Dimethyl-5-phenyl-1H-pyrrol-3-yl) (phenyl)methanone (7a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 72% yield (40 mg); mp 96–98 °C; 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J = 7.0 Hz, 2H), 7.26 (t, J = 7.5Hz, 1H), 7.14 (t, J = 7.5 Hz, 2H), 7.04 (q, J = 8.0 Hz, 4H), 6.98 (d, J = 6.5 Hz, 1H), 6.64 (s, 1H), 3.62 (s, 3H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 194.4, 140.0, 135.8, 135.6, 131.9, 130.2, 128.8, 128.2, 128.0, 126.2, 125.9, 120.2, 120.1, 34.2, 11.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H18NO 276.1383; found 276.1388.
- 1-(2-Methyl-5-phenyl-1-(p-tolyl)-1H-pyrrol-3-yl) ethan-1-one (7b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a yellow solid in an 85% yield (49 mg); mp 66–68 °C; 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 4.0 Hz, 4H), 7.32–7.27 (m, 3H), 7.21 (d, J = 8.5 Hz, 2H), 6.64 (s, 1H), 2.42 (s, 3H), 2.39 (s, 3H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.1, 138.5, 136.6, 136.5, 135.8, 130.3, 129.7, 128.7, 127.2, 126.6, 126.4, 122.8, 121.1, 31.5, 21.5, 13.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; found 290.1538.
- 2-Methyl-4-phenyl-1H-imidazole (8). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:3) to afford a white solid in a 96% yield (30 mg); mp 57–59 °C; 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 7.0 Hz, 2H), 7.37 (s, 1H), 7.30 (t, J = 7.5Hz, 2H), 7.13 (t, J = 7.5 Hz, 1H), 3.39 (brs, 1H), 2.29 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 145.7, 138.2, 133.2, 129.1, 127.2, 125.1, 115.6, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H11N2 159.0917; found 159.091.
- (E)-(2,5-Diphenyl-1H-pyrrol-3-yl) (phenyl)methanone oxime (9). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a white solid in a 94% yield (63 mg); mp 95–97 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.44 (s, 1H), 11.19 (s, 1H), 7.79 (d, J = 7.5 Hz, 2H), 7.49 (d, J = 7.5 Hz, 4H), 7.37 (t, J = 8.0 Hz, 2H), 7.26–7.23 (m, 5H), 7.19 (t, J = 7.5 Hz, 1H), 7.12 (t, J = 7.5 Hz, 1H), 6.52 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 153.7, 137.7, 133.2, 132.8, 131.6, 129.6, 129.5, 129.1, 129.0, 128.8, 127.4, 127.2, 127.0, 126.5, 124.8, 115.1, 109.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H19N2O 339.1492; found 339.1496.
- (1-Methyl-2,5-diphenyl-1H-pyrrol-3-yl) (phenyl)methanone (10). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1:8) to afford a colorless oil in a 98% yield (66 mg); 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 8.0 Hz, 2H), 7.40–7.35 (m, 5H), 7.34 (s, 1H), 7.32–7.27 (m, 3H), 6.67 (s, 1H), 3.49 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 192.3, 140.8, 140.1, 135.7, 132.9, 132.3, 131.6, 131.2, 129.8, 129.4, 129.0, 128.6, 128.5, 128.1, 128.1, 122.3, 112.3, 34.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; found 338.1544.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Weinreb, S.M. Some recent advances in the synthesis of polycyclic imidazole-containing marine natural products. Nat. Prod. Rep. 2007, 24, 931–948. [Google Scholar] [CrossRef]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids. Nat. Prod. Rep. 2014, 31, 1721–1788. [Google Scholar] [CrossRef]
- Muñoz, J.; Köck, M. Hybrid pyrrole-imidazole alkaloids from the sponge agelas sceptrum. J. Nat. Prod. 2016, 79, 434–437. [Google Scholar] [CrossRef]
- Clive, D.L.J.; Cheng, P. The marinopyrroles. Tetrahedron 2013, 69, 5067–5078. [Google Scholar] [CrossRef]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [CrossRef]
- Vessally, E.; Soleimani-Amiri, S.; Hosseinian, A.; Edjlali, L.; Bekhradnia, A. New protocols to access imidazoles and their ring fused analogues: Synthesis from N-propargylamines. RSC Adv. 2017, 7, 7079–7091. [Google Scholar] [CrossRef]
- Khajuria, R.; Dham, S.; Kapoor, K.K. Active methylenes in the synthesis of a pyrrole motif: An imperative structural unit of pharmaceuticals, natural products and optoelectronic materials. RSC Adv. 2016, 6, 37039–37066. [Google Scholar] [CrossRef]
- Borah, B.; Dwivedi, K.D.; Chowhan, L.R. Recent approaches in the organocatalytic synthesis of pyrroles. RSC Adv. 2021, 11, 13585–13601. [Google Scholar] [CrossRef]
- Preeti; Singh, K.N. Multicomponent reactions: A sustainable tool to 1,2- and 1,3-azoles. Org. Biomol. Chem. 2018, 16, 9084–9116. [Google Scholar] [CrossRef] [PubMed]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 2014, 43, 4633–4657. [Google Scholar] [CrossRef]
- Shi, S.; Xu, K.; Jiang, C.; Ding, Z. ZnCl2-Catalyzed [3 + 2] Cycloaddition of Benzimidates and 2H-Azirines for the Synthesis of Imidazoles. J. Org. Chem. 2018, 83, 14791–14796. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, Y.; Song, H.; Liu, Y.; Wang, Q. Copper-Catalyzed Aerobic Oxidative [2 + 3] Cyclization/Aromatization Cascade Reaction: Atom-Economical Access to Tetrasubstituted 4,5-Biscarbonyl Imidazoles. Org. Lett. 2017, 19, 6056–6059. [Google Scholar] [CrossRef]
- Liao, J.-Y.; Shao, P.-L.; Zhao, Y. Catalytic Divergent Synthesis of 3H or 1H Pyrroles by [3 + 2] Cyclization of Allenoates with Activated Isocyanides. J. Am. Chem. Soc. 2015, 137, 628–631. [Google Scholar] [CrossRef]
- Vannada, J.; Sulthan, M.; Arun, D.; Dada, R.; Yaragorla, S. Regiodivergent Synthesis of Penta-Substituted Pyrroles through a Cascade [3 + 2] Cyclization of C-Acylimines with Activated Alkynes and Aromatic Nucleophiles. J. Org. Chem. 2020, 85, 6697–6708. [Google Scholar] [CrossRef]
- Dai, L.; Yu, S.; Lv, N.; Ye, X.; Shao, Y.; Chen, Z.; Chen, J. Synthesis of Imidazoles and Oxazoles via a Palladium-Catalyzed Decarboxylative Addition/Cyclization Reaction Sequence of Aromatic Carboxylic Acids with Functionalized Aliphatic Nitriles. Org. Lett. 2021, 23, 5664–5668. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Kiyota, S.; Komine, N.; Hirano, M. Ru(0)-Catalyzed Synthesis of Conjugated Iminotrienes and Subsequent Intramolecular Cyclization Giving Polysubstituted Pyrroles. Org. Lett. 2022, 24, 2973–2977. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Z.; Yuan, Y.; Peng, D.; Li, Y.; Zhang, L.; Wu, Y. Au(I)-Catalyzed Intramolecular Hydroamination of the Fluorinated N′-Aryl-N-Propargyl Amidines: Mild Conditions for the Synthesis of 2-Fluoroalkyl Imidazole Derivatives. Org. Lett. 2012, 14, 1130–1133. [Google Scholar] [CrossRef]
- Li, W.; Shi, R.; Chen, S.; Zhang, X.; Peng, W.; Chen, S.; Li, J.; Xu, X.-M.; Zhu, Y.-P.; Wang, X. Synthesis of Diverse Pentasubstituted Pyrroles by a Gold(I)-Catalyzed Cascade Rearrangement-Cyclization of Tertiary Enamide. J. Org. Chem. 2022, 87, 3014–3024. [Google Scholar] [CrossRef]
- Akter, M.; Rupa, K.; Anbarasan, P. 1,2,3-Triazole and Its Analogues: New Surrogates for Diazo Compounds. Chem. Rev. 2022, 122, 13108–13205. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, H.; Chen, Z. Advances on the Metallocarbene Formation Reactions Based on Triazole Derivatives. Chin. J. Org. Chem. 2016, 36, 1555–1563. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Alford, J.S. Reactions of metallocarbenes derived from N-sulfonyl-1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 5151–5162. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, R.; Tang, X.-Y.; Shi, M. Recent Advancesin the Synthesis of Heterocycles and RelatedSubstances Based on α-Imino Rhodium Carbene ComplexesDerived from N-Sulfonyl-1,2,3-triazoles. Chem. Eur. J. 2016, 22, 17910–17924. [Google Scholar] [CrossRef]
- Chuprakov, S.; Kwok, S.W.; Zhang, L.; Lercher, L.; Fokin, V.V. Rhodium-Catalyzed Enantioselective Cyclopropanation of Olefins with N-Sulfonyl 1,2,3-Triazoles. J. Am. Chem. Soc. 2009, 131, 18034–18035. [Google Scholar] [CrossRef]
- Miura, T.; Nakamuro, T.; Ishihara, Y.; Nagata, Y.; Murakami, M. Chiral Macrocycles Having C3 Symmetry Resulting from Orientation of Thiophene Rings. Angew. Chem. Int. Ed. 2020, 59, 20475–20479. [Google Scholar] [CrossRef]
- Chuprakov, S.; Worrell, B.T.; Selander, N.; Sit, R.K.; Fokin, V.V. Stereoselective 1,3-Insertions of Rhodium(II) Azavinyl Carbenes. J. Am. Chem. Soc. 2014, 136, 195–202. [Google Scholar] [CrossRef]
- He, X.; Wu, Y.; Zhou, T.; Zuo, Y.; Xie, M.; Li, R.; Duan, J.; Shang, Y. Rh-Catalyzed C−N Coupling of N-Sulfonyl-1,2,3-Trizales with Secondary Amines for Regioselective Synthesis of Phenylvinyl-1,2-Diamines. Synth. Commun. 2020, 50, 2685–2697. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Li, Z.; Dong, S.; Liu, X.; Feng, X. Tandem Insertion-[1,3]-Rearrangement: Highly Enantioselective Construction of α-Aminoketones. Angew. Chem. Int. Ed. 2020, 59, 8052–8056. [Google Scholar] [CrossRef]
- Miura, T.; Nakamuro, T.; Miyakawa, S.; Murakami, M. A Syn-Selective Aza-Aldol Reaction of Boron Aza-Enolates Generated fromNSulfonyl-1,2,3-Triazoles and 9-BBN-H. Angew. Chem. Int. Ed. 2016, 55, 8732–8735. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shi, Y.; Cheng, W.; Man, Z.; Yang, D.; Li, C.-Y. Rhodium-Catalyzed Synthesis of 4-Bromo-1,2-Dihydroisoquinolines: Access to Bromonium Ylides by the Intramolecular Reaction of a Benzyl Bromide and an α-Imino Carbene. Angew. Chem. Int. Ed. 2016, 55, 4557–4561. [Google Scholar] [CrossRef]
- Reddy, A.C.S.; Ramachandran, K.; Reddy, P.M.; Anbarasan, P. Rhodium-Catalyzed Sommelet Hauser Type Rearrangement of α-Diazoimines: Synthesis of Functionalized Enamides. Chem. Commun. 2020, 56, 5649–5652. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, L.; Liao, Y.; Mao, Z.; Huang, X. Rhodium(II)-Catalysed Skeletal Rearrangement of Ether Tethered N-Sulfonyl 1,2,3-Triazoles: A Rapid Approach to 2-Aminoindanone and Dihydroisoquinoline Derivatives. Adv. Synth. Catal. 2016, 358, 1059–1064. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Xiu, S.-D.; Li, C.-Y. Rhodium-catalyzed synthesis of multi-substituted furans from N-sulfonyl-1,2,3-triazoles bearing a tethered carbonyl group. Org. Chem. Front. 2015, 2, 47–50. [Google Scholar] [CrossRef]
- Shi, Y.; Yu, X.; Li, C.-Y. Rhodium-Catalyzed Synthesis of 2,5-Epoxybenzo[f][1,4]Oxazepines by Tandem Reaction of 1-Sulfonyl-1,2,3-Triazoles and Salicylaldehydes. Eur. J. Org. Chem. 2015, 2015, 6429–6433. [Google Scholar] [CrossRef]
- Yadagiri, D.; Reddy, A.C.S.; Anbarasan, P. Rhodium Catalyzed Diastereoselective Synthesis of 2,2,3,3-Tetrasubstituted Indolines from N-Sulfonyl-1,2,3-Triazoles and Ortho-Vinylanilines. Chem. Sci. 2016, 7, 5934–5938. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Fujimoto, Y.; Funakoshi, Y.; Murakami, M. A Reaction of Triazoles with Thioesters to Produce β-Sulfanyl Enamides by Insertion of an Enamine Moiety into the Sulfur-Carbonyl Bond. Angew. Chem. Int. Ed. 2015, 54, 9967–9970. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, R.; Choy, P.Y.; Duan, J.; Yin, Z.; Xu, K.; Tang, Q.; Zhong, R.-L.; Shang, Y.; Kwong, F.Y. An expeditious FeCl3-catalyzed cascade 1,4-conjugate addition/annulation/1,5-H shift sequence for modular access of all-pyrano-moiety-substituted chromenes. Chem. Sci. 2022, 13, 13617–13622. [Google Scholar] [CrossRef]
- Zhou, T.; He, X.; Zuo, Y.; Wu, Y.; Hu, W.; Zhang, S.; Duan, J.; Shang, Y. Rh-catalyzed formal [3 + 2] cyclization for synthesis of 5-aryl-2-(quinolin-2-yl)oxazoles and its applications in metal ions probes. Chin. J. Chem. 2021, 39, 621–626. [Google Scholar] [CrossRef]
- He, X.; Xu, K.; Liu, Y.; Wang, D.; Tang, Q.; Hui, W.; Chen, H.; Shang, Y. Radical-Induced Cascade Annulation/Hydrocarbonylation for Construction of 2-Aryl-4H-chromen-4-ones. Molecules 2022, 27, 7412. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, S.; Ye, Q.; Kong, S.; Yang, T.; Tang, K.; He, X.; Shang, Y. Rh-catalyzed Cascade C-H Activation/Annulation of N-Hydroxybenzamides and Propargylic Acetates for Modular Access to Isoquinolones. Molecules 2022, 27, 8553. [Google Scholar] [CrossRef]
- Jeon, H.J.; Jung, D.J.; Kim, J.H.; Kim, Y.; Bouffard, J.; Lee, S. From Triazoles to Imidazolines through the Sequential N−H Insertion of α-Imino Rhodium−Carbenes into β-Enamino Esters/Enamine−Imine Tautomerization/Conjugate Addition Cascade. J. Org. Chem. 2014, 79, 9865–9871. [Google Scholar] [CrossRef]
- Lei, X.; Li, L.; He, Y.-P.; Tang, Y. Rhodium(II)-Catalyzed Formal [3 + 2] Cycloaddition of N-Sulfonyl-1,2,3-Triazoles with Isoxazoles: Entry to Polysubstituted 3-Aminopyrroles. Org. Lett. 2015, 17, 5224–5227. [Google Scholar] [CrossRef] [PubMed]
- Rostovskii, N.V.; Ruvinskaya, J.O.; Novikov, M.S.; Khlebnikov, A.F.; Smetanin, I.A.; Agafonova, A.V. Switchable Synthesis of Pyrroles and Pyrazines via Rh(II)-Catalyzed Reaction of 1,2,3-Triazoles with Isoxazoles: Experimental and DFT Evidence for the 1,4-Diazahexatriene Intermediate. J. Org. Chem. 2017, 82, 256–268. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Catalyst (x mol%) | Solvent | Yield (%) b |
| 1 | Rh2(OAc)4 (4) | DCM | 56 |
| 2 | Rh2(oct)4 (4) | DCM | 58 |
| 3 | CuI (4) | DCM | 44 |
| 4 | Sc(OTf)3 (4) | DCM | trace |
| 5 | Co2(CO)8 (4) | DCM | nr |
| 6 | Ni(acac)2 (4) | DCM | nr |
| 7 | Rh2(oct)4 (3) | DCM | 73 |
| 8 | Rh2(oct)4 (2) | DCM | 79 |
| 9 | Rh2(oct)4 (1) | DCM | 41 |
| 10 | / | DCM | nr |
| 11 | Rh2(oct)4 (2) | DCE | 55 |
| 12 | Rh2(oct)4 (2) | toluene | 84 |
| 13 | Rh2(oct)4 (2) | PhCl | 96 |
| 14 | Rh2(oct)4 (2) | CH3OH | trace |
| 15 | Rh2(oct)4 (2) | CH3NO2 | trace |
| 16 | Rh2(oct)4 (2) | DMF | nr |
| 17 c | Rh2(oct)4 (2) | PhCl | nr |
| 18 d | Rh2(oct)4 (2) | PhCl | 87 |
| 19 e | Rh2(oct)4 (2) | PhCl | 94 |
| 20 f | Rh2(oct)4 (2) | PhCl | 56 |
| 21 g | Rh2(oct)4 (2) | PhCl | 78 |
| 22 h | Rh2(oct)4 (2) | PhCl | 96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhou, T.; Wu, M.; Ye, Q.; He, X. Substituent-Controllable Cascade Regioselective Annulation of β-Enaminones with N-Sulfonyl Triazoles for Modular Access to Imidazoles and Pyrroles. Molecules 2023, 28, 4416. https://doi.org/10.3390/molecules28114416
Wang H, Zhou T, Wu M, Ye Q, He X. Substituent-Controllable Cascade Regioselective Annulation of β-Enaminones with N-Sulfonyl Triazoles for Modular Access to Imidazoles and Pyrroles. Molecules. 2023; 28(11):4416. https://doi.org/10.3390/molecules28114416
Chicago/Turabian StyleWang, Hua, Tongtong Zhou, Mengdi Wu, Qingqing Ye, and Xinwei He. 2023. "Substituent-Controllable Cascade Regioselective Annulation of β-Enaminones with N-Sulfonyl Triazoles for Modular Access to Imidazoles and Pyrroles" Molecules 28, no. 11: 4416. https://doi.org/10.3390/molecules28114416