3.1. Chemistry
All reactions were conducted in oven-dried glassware in an atmosphere of nitrogen. Melting points were measured with a Buchi B-520 melting point apparatus and were not corrected. The NMR spectra were recorded on a Bruker MSL-300 spectrometer at 25 °C (
1H: 300 MHz;
13C: 75 MHz; chemical shifts are reported as parts per million (δ, ppm)) in dimethyl sulfoxide (DMSO)-
d6, CDCl
3 or D
2O; the residual solvent peaks were used as internal standards: 7.28 ppm for
1H in CDCl
3, 2.50 ppm for
1H in DMSO-
d6 and 4.79 ppm for
1H in D
2O, 40.01 and 77.02 ppm for
13C in DMSO-
d6 and CDCl
3. Copies of the NMR spectra of synthesized compounds are presented in
Supplementary Materials. Mass spectra were recorded using the Shimadzu LCMS-2020 system with ESI. High-resolution mass spectra (HRMS) were recorded using a Bruker microTOF spectrometer (ionization by electrospray, positive ions detection). Analytical thin-layer chromatography was carried out on Sorbfil UV-254 silica gel plates (Imid Ltd., Krasnodar, Russia) using appropriate mixtures of solvent. The compounds were visualized with short-wavelength UV light. Column chromatography was performed on silica gel 60 (230–400 mesh). All reagents and solvents were obtained from commercial sources and used without purification. 5-tert-butyl 3-ethyl 1,4,6,7-tetrahydro-5
H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (
5) was synthesized according to the procedure reported by Arrington et al. [
22] with 52% yield over two steps.
3.1.1. General Procedure for the Synthesis of Compounds 6a–h
To the suspension of the 60% sodium hydride (62.5 mmol, 2.5 g) in anhydrous toluene (150 mL), 5-tert-butyl 3-ethyl 1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c] pyridine-3,5-dicarboxylate (5) (33.9 mmol, 10 g) was added followed by alkyl halide (42.4 mmol). The reaction progress was monitored by TLC (chloroform/methanol 97:3). After stirring at room temperature for 6 h, the reaction mass was washed with water (2 × 50 mL), then 5% aqueous potassium carbonate solution (50 mL) and 5% aqueous citric acid solution (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated by rotary evaporation under vacuum.
5-tert-butyl 3-ethyl 1-methyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6a). Yield 10 g (95%), white solid, m.p. 124–124.5 °C. 1H NMR (300 MHz, CDCl3) δ 4.58 (s, 2H), 4.37 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.70 (t, J = 5.7 Hz, 2H), 2.66 (t, J = 5.6 Hz, 2H), 1.46 (s, 9H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 162.5, 155.1, 138.7, 138.1, 117.8, 80.3, 60.8, 41.5, 40.1, 36.5, 28.5, 22.0, 14.5. LCMS (ESI): m/z (M + H+) calcd, 310.2; found, 310.4.
5-tert-butyl 3-ethyl 1-ethyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6b). Yield 9.4 g (86%), white solid, m.p. 168–169 °C. 1H NMR (300 MHz, DMSO-d6) δ 4.47 (s, 2H), 4.25 (q, J = 7.0 Hz, 2H), 4.09 (q, J = 7.2 Hz, 2H), 3.61 (t, J = 5.5 Hz, 2H), 2.71 (t, J = 5.3 Hz, 2H), 1.41 (s, 9H), 1.35–1.24 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ 161.98, 154.01, 138.01, 136.20, 116.67, 79.21, 59.99, 44.14, 41.13, 40.34, 28.01, 21.00, 14.90, 13.80. LCMS (ESI): m/z (M + H+) calcd, 324.2; found, 324.4.
5-tert-butyl 3-ethyl 1-(propan-2-yl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6c). Yield 8.6 g (75%), white solid, m.p. 119–120 °C. 1H NMR (300 MHz, DMSO-d6) δ 4.56–4.45 (m, 3H), 4.26 (q, J = 7.1 Hz, 2H), 3.62 (t, J = 5.7 Hz, 2H), 2.73 (t, J = 5.5 Hz, 2H), 1.42 (s, 9H), 1.38 (d, J = 6.6 Hz, 6H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 161.86, 153.97, 137.39, 136.63, 116.41, 79.09, 59.84, 50.40, 40.96, 27.95, 22.03, 21.17, 14.16. LCMS (ESI): m/z (M + H+) calcd, 338.2; found, 338.4.
5-tert-butyl 3-ethyl 1-(2-methylpropyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6d). Yield 9.3 g (78%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 4.48 (s, 2H), 4.25 (q, J = 7.1 Hz, 2H), 3.87 (d, J = 7.4 Hz, 2H), 3.60 (t, J = 5.7 Hz, 2H), 2.69 (t, J = 5.6 Hz, 2H), 2.10 (hept, J = 6.8 Hz, 1H), 1.41 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H), 0.84 (d, J = 6.7 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 161.82, 154.04, 138.59, 136.87, 116.28, 79.13, 59.90, 55.94, 40.72, 28.84, 27.95, 21.42, 19.50, 14.12. LCMS (ESI): m/z (M + H+) calcd, 352.2; found, 352.4.
5-tert-butyl 3-ethyl 1-(cyclopropylmethyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6e). Yield 7.9 g (67%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 4.49 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 3.96 (d, J = 7.0 Hz, 2H), 3.62 (t, J = 5.6 Hz, 2H), 2.73 (t, J = 5.5 Hz, 2H), 1.42 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H), 1.25–1.15 (m, 1H), 0.55–0.46 (m, 2H), 0.38–0.30 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 161.92, 154.07, 138.05, 136.76, 116.69, 79.21, 59.99, 53.56, 41.20, 40.62, 28.00, 21.36, 14.21, 11.11, 3.65. LCMS (ESI): m/z (M + H+) calcd, 350.2; found, 350.4.
5-tert-butyl 3-ethyl 1-propyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6f). Yield 10.4 g (91%), white solid, m.p. 64–65 °C. 1H NMR (300 MHz, DMSO-d6) δ 4.59 (s, 2H), 4.37 (q, J = 7.1 Hz, 2H), 4.01 (t, J = 7.3 Hz, 2H), 3.70 (t, J = 5.4 Hz, 2H), 2.66 (t, J = 5.1 Hz, 2H), 1.84 (h, J = 7.3 Hz, 2H), 1.47 (s, 9H), 1.37 (t, J = 7.1 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 162.26, 154.43, 138.76, 137.17, 116.84, 79.56, 60.35, 50.90, 41.54, 40.98, 28.35, 21.51, 14.55. LCMS (ESI): m/z (M + H+) calcd, 338.2; found, 338.4.
5-tert-butyl 3-ethyl 1-(2-methoxyethyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6g). Yield 8.15 g (68%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 4.48 (s, 2H), 4.32–4.17 (m, 4H), 3.65 (t, J = 5.2 Hz, 2H), 3.60 (t, J = 5.6 Hz, 2H), 3.20 (s, 3H), 2.71 (t, J = 5.4 Hz, 2H), 1.41 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 162.21, 154.42, 139.63, 137.47, 116.78, 79.54, 71.00, 60.37, 58.48, 49.47, 41.50, 39.86, 28.33, 21.68, 14.52. LCMS (ESI): m/z (M + H+) calcd, 354.2; found, 354.6.
5-tert-butyl 3-ethyl 1-(2-methoxypropyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-3,5-dicarboxylate (6h). Yield 7.2 g (58%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 4.49 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 4.10 (t, J = 6.9 Hz, 2H), 3.61 (t, J = 5.6 Hz, 2H), 3.25 (t, J = 6.0 Hz, 2H), 3.21 (s, 3H), 2.69 (t, J = 5.3 Hz, 2H), 2.03–1.90 (m, 2H), 1.42 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 162.23, 154.42, 138.83, 137.45, 116.81, 79.50, 71.00, 60.38, 58.37, 50.49, 41.50, 41.07, 29.69, 28.33, 21.68, 14.53. LCMS (ESI): m/z (M + H+) calcd, 368.2; found, 368.4.
3.1.2. Synthesis of Compounds 9a–d
1-methyl-3-(5-methyl-1,3-oxazol-2-yl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridinehydrochloride (9a). A 50 mL round-bottomed flask was charged with a solution of compound 6a (5.0 g, 16.16 mmol) in methanol (30 mL). A solution of KOH (2.26 g, 40.4 mmol) in water (6.0 mL) was added and the resulting mixture was stirred at room temperature for 12 h. Methanol was removed in vacuo and the residue was dissolved in water (100 mL). The aqueous solution was extracted with ethyl acetate (3 × 50 mL) and the organic extracts were discarded. The pH of the aqueous phase was carefully adjusted to 5.0 with 5% aqueous HCl and the solution was again extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give 2.98 g (65%, assuming analytical purity) of product 7a as a white solid, which was used in the next step without further purification.
To a solution of 2.98 g (10.6 mmol) of 7a in 25 mL of CH2Cl2, 1.89 g (11.6 mmol) of N,N-carbonyldiimidazole was added and stirred for 30 min at room temperature. Then, 0.70 g (13 mmol) of propargylamine was added dropwise to the reaction mixture and stirred overnight. The reaction mixture was poured into water, the organic layer was separated and washed sequentially with 5% aqueous citric acid solution (2 × 50 mL) and 10% aqueous K2CO3 solution (2 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed on a vacuum rotary evaporator to give 1.49 g (44%, assuming analytical purity) of amide 8a as a colorless oil, which was used in the next step without further purification.
To a solution of 1.49 g (4.7 mmol) of amide 8a in absolute DMSO (10 mL), Cs2CO3 (3.00 g, 9.2 mmol) was added and stirred for 2 h at 100 °C. The reaction mixture was poured into 100 mL of water and extracted with EtOAc (3 × 50 mL), and the organic phases were combined and washed with 5% aqueous citric acid solution (2 × 50 mL) followed 10% aqueous K2CO3 solution (2 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed on a vacuum rotary evaporator. Column chromatography on silica gel using 0 → 5% MeOH in CHCl3 as eluent afforded the target compound. Fractions containing the target compound were combined and evaporated under vacuum. The residue was dissolved in 10 mL of 1,4-dioxane and 5 mL of a 4 N solution of HCl in 1,4-dioxane was added dropwise. The precipitate that formed was filtered off, washed with ether, and dried to give compound 9a. Yield 0.84 g (56%, 16% over 3 steps), white solid, m.p. 219–220 °C. 1H NMR (300 MHz, D2O) δ 7.21 (s, 1H), 4.50 (s, 2H), 3.87 (s, 3H), 3.63 (t, J = 5.8 Hz, 2H), 3.13 (t, J = 5.8 Hz, 2H), 2.43 (s, 3H); 13C NMR (75 MHz, DMSO) δ 155.17, 148.68, 136.93, 135.02, 123.92, 108.85, 39.99, 39.70, 36.50, 18.52, 10.73. LCMS (ESI): m/z (M + H+) calcd, 219.2; found, 219.4.
1-ethyl-3-(5-methyl-1,3-oxazol-2-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (9b). Prepared using 6b as described for 6a. Yield 0.68 g (38% over 3 steps), white solid, m.p. 215–216 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.98 (s, 2H), 6.97 (d, J = 1.0 Hz, 1H), 4.21 (s, 2H), 4.12 (q, J = 7.2 Hz, 2H), 3.38 (d, J = 5.0 Hz, 2H), 3.02 (t, J = 5.8 Hz, 2H), 2.35 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 155.10, 148.46, 135.99, 135.12, 123.81, 108.66, 44.00, 39.90, 39.64, 18.33, 14.98, 10.53. LCMS (ESI): m/z (M + H+) calcd, 233.2; found, 233.4.
3-(5-methyl-1,3-oxazol-2-yl)-1-(propan-2-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (9c). Prepared using 6c as described for 6a. Yield 0.77 g (35% over 3 steps, approx. 14% dioxane), white solid, m.p. 261–266 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.96 (s, 2H), 6.97 (s, 1H), 4.54 (q, J = 6.4 Hz, 1H), 4.21 (s, 2H), 3.37 (s, 2H), 3.04 (t, J = 5.3 Hz, 2H), 2.36 (s, 3H), 1.39 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 155.19, 148.38, 135.48, 135.04, 123.77, 108.50, 50.34, 39.87, 39.62, 22.17, 18.42, 10.54. LCMS (ESI): m/z (M + H+) calcd, 247.2; found, 247.2.
3-(5-methyl-1,3-oxazol-2-yl)-1-(2-methylpropyl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (9d). Prepared using 6d as described for 6a. Yield 0.85 g (39% over 3 steps, approx. 6% dioxane), white solid, m.p. 121–122 °C. 1H NMR (300 MHz, D2O) δ 7.01 (s, 1H), 4.49 (s, 2H), 3.95 (d, J = 7.5 Hz, 2H), 3.62 (t, J = 6.0 Hz, 2H), 3.13 (t, J = 6.0 Hz, 2H), 2.40 (s, 3H), 2.19 (hept, J = 6.6 Hz, 1H), 0.88 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, D2O) δ 155.61, 151.58, 137.84, 134.99, 121.98, 109.00, 57.13, 41.55, 40.95, 29.66, 19.45, 19.39, 10.59. LCMS (ESI): m/z (M + H+) calcd, 261.2; found, 261.4.
3.1.3. General Procedure for the Synthesis of Compounds 11e–h
tert-butyl 1-(cyclopropylmethyl)-3-formyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxylate (11e). A total of 5 g (14.3 mmol) of ester 6e was dissolved in 50 mL of anhydrous tetrahydrofuran and LiAlH4 (13.1 mmol, 0.5 g) was added in portions at 0 °C. The reaction progress was monitored by TLC (chloroform/methanol 98:2) and it took about 1 h. After the reaction was completed, 0.5 mL of H2O, 0.5 mL of 15% NaOH aqueous solution, and 1.5 mL of H2O were successively added dropwise. The mixture was stirred for 2 h. The formed precipitate was filtered off, the filtrate was evaporated under vacuum. The residue was dissolved in 25 mL of dry methylene chloride and 2.5 g (28.7 mmol) of MnO2 was added. The mixture was stirred overnight, then filtered, and the solvent was evaporated under vacuum. Column chromatography on silica gel using 0 → 5% MeOH in CHCl3 as eluent afforded the corresponding aldehyde 11e. Yield 2.66 g (61%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 9.87 (s, 1H), 4.50 (s, 2H), 4.01 (d, J = 7.1 Hz, 2H), 3.62 (t, J = 5.7 Hz, 2H), 2.76 (t, J = 5.7 Hz, 2H), 1.42 (s, 9H), 1.29–1.20 (m, 1H), 0.56–0.48 (m, 2H), 0.41–0.33 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 187.24, 154.01, 145.09, 138.88, 114.98, 79.30, 53.77, 40.59, 28.02, 22.76, 21.33, 10.95, 3.66. LCMS (ESI): m/z (M + H+) calcd, 306.2; found, 306.4.
tert-butyl 3-formyl-1-propyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxylate (11f). Yield 2.43 g (58%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 9.85 (s, 1H), 4.49 (s, 2H), 4.07 (t, J = 7.1 Hz, 2H), 3.62 (t, J = 5.7 Hz, 2H), 2.73 (t, J = 5.6 Hz, 2H), 1.79 (h, J = 7.3 Hz, 2H), 1.41 (s, 9H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 187.22, 154.01, 145.18, 139.12, 114.84, 79.40, 50.77, 40.95, 28.02, 21.35, 11.28. LCMS (ESI): m/z (M + H+) calcd, 294.2; found, 294.4.
tert-butyl 3-formyl-1-(2-methoxyethyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxylate (11g). Yield 2.12 g (48%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 9.86 (s, 1H), 4.49 (s, 2H), 4.29 (t, J = 5.1 Hz, 2H), 3.69 (t, J = 5.1 Hz, 2H), 3.61 (t, J = 5.7 Hz, 2H), 3.21 (s, 3H), 2.74 (t, J = 5.5 Hz, 2H), 1.41 (s, 9H).; 13C NMR (75 MHz, DMSO-d6) δ 187.20, 154.02, 145.35, 139.99, 114.74, 79.27, 70.42, 58.10, 49.31, 40.63, 27.99, 21.34. LCMS (ESI): m/z (M + H+) calcd, 310.2; found, 310.2.
tert-butyl 3-formyl-1-(3-methoxypropyl)-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxylate (11h). Yield 1.66 g (36%), clear oil. 1H NMR (300 MHz, DMSO-d6) δ 9.86 (s, 1H), 4.49 (s, 2H), 4.15 (t, J = 7.0 Hz, 2H), 3.62 (t, J = 5.7 Hz, 2H), 3.28 (t, J = 6.0 Hz, 2H), 3.22 (s, 3H), 2.72 (t, J = 5.6 Hz, 2H), 2.01 (p, J = 6.5 Hz, 2H), 1.42 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 187.23, 154.02, 145.27, 139.44, 114.79, 79.28, 68.46, 57.87, 46.27, 40.63, 29.21, 28.00, 21.02. LCMS (ESI): m/z (M + H+) calcd, 324.2; found, 324.6.
3.1.4. General Procedure for the Synthesis of Compounds 12e–h
To a solution of aldehyde 11 (3.2 mmol) in 25 mL of dry MeOH was added 1.81 g (13 mmol) of K2CO3 and TosMic (0.83 g, 4.2 mmol) and stirred for 6 h at reflux. The reaction mixture was poured into 100 mL of water and extracted with EtOAc (2 × 50 mL). The organic phases were combined and washed with 5% aqueous citric acid solution (2 × 50 mL) and 10% aqueous K2CO3 solution (2 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and evaporated under vacuum. Column chromatography on silica gel using 0 → 5% MeOH in CHCl3 as eluent afforded target oxazoles. Fractions containing the target compound were combined and evaporated under vacuum. The residue was dissolved in 10 mL of 1,4-dioxane and a 4N solution of HCl in 1,4-dioxane was added dropwise. The precipitate that formed was filtered off, washed with ether, and dried.
1-(cyclopropylmethyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (12e). Yield 0.43 g (48%), white solid, m.p. 126–127 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.29 (s, 1H), 7.36 (s, 1H), 4.43 (s, 2H), 3.98 (d, J = 7.0 Hz, 2H), 3.61 (t, J = 6.2 Hz, 2H), 3.13 (t, J = 6.1 Hz, 2H), 1.27–1.19 (m, 1H), 0.63–0.54 (m, 2H), 0.40–0.32 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 151.65, 145.28, 135.95, 135.22, 122.10, 107.01, 53.22, 40.08, 39.13, 18.71, 11.37, 3.79. LCMS (ESI): m/z (M + H+) calcd, 245.1; found, 245.2.
3-(1,3-oxazol-5-yl)-1-propyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (12f). Yield 0.45 g (45%, approx. 13% dioxane), white solid, m.p. 142–143 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.00 (s, 2H), 8.44 (s, 1H), 7.42 (s, 1H), 4.22 (s, 2H), 4.03 (t, J = 6.9 Hz, 2H), 3.37 (d, J = 5.9 Hz, 2H), 3.01 (t, J = 5.7 Hz, 2H), 1.75 (h, J = 7.2 Hz, 2H), 0.85 (t, J = 7.4 Hz, 3H).; 13C NMR (75 MHz, DMSO-d6) δ 151.56, 145.25, 136.24, 135.21, 122.05, 106.70, 50.29, 40.07, 39.12, 23.00, 18.55, 10.99. LCMS (ESI): m/z (M + H+) calcd, 233.1; found, 233.4.
1-(2-methoxyethyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (12g). Yield 0.35 g (30%, approx. 20% dioxane), white solid, m.p. 119–120 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.03 (s, 2H), 8.46 (s, 1H), 7.43 (s, 1H), 4.28–4.18 (m, 4H), 3.65 (t, J = 5.0 Hz, 2H), 3.35 (d, J = 4.5 Hz, 2H), 3.20 (s, 3H), 3.01 (t, J = 5.7 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 151.95, 145.54, 137.39, 135.85, 122.53, 107.14, 71.05, 58.65, 49.34, 40.38, 39.34, 18.97. LCMS (ESI): m/z (M + H+) calcd, 249.1; found, 249.2.
1-(3-methoxypropyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine hydrochloride (12h). Yield 0.4 g (36%, approx. 14% dioxane), white solid, m.p. 84–85 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.12 (s, 2H), 8.45 (s, 1H), 7.42 (s, 1H), 4.20 (s, 2H), 4.08 (t, J = 6.7 Hz, 2H), 3.23 (t, J = 6.0 Hz, 2H), 3.21 (s, 3H), 2.99 (t, J = 5.4 Hz, 2H), 1.99–1.90 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 151.70, 145.31, 136.61, 135.53, 122.19, 106.76, 68.49, 58.13, 45.82, 40.22, 39.24, 29.69, 18.52. LCMS (ESI): m/z (M + H+) calcd, 263.2; found, 263.4.
3.1.5. General Procedure for the Synthesis of Compounds 10a–d and 13e–h
To a solution of 5-nitro-2-furoic acid (0.1 g, 0.6 mmol) in dry DMF (5 mL) CDI (0.12 g, 0.70 mmol) was added and the mixture was stirred at r.t. for 30 min. This one was added dropwise to the mixture of hydrochloride 9 (for 10 synthesis) or 12 (for 13) (0.7 mmol) and triethylamine (0.1 mL, 0.8 mmol) in dry DMF (5 mL) and the stirring continued for 18 h. The resulting mixture was poured into water (30 mL) and extracted with ethyl acetate (3 × 50 mL). The organic phase was successively washed with 10% aqueous K2CO3 (2×10 mL) and dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was suspended in diethyl ether and filtered, then dried under vacuum.
1-methyl-3-(5-methyl-1,3-oxazol-2-yl)-5-(5-nitro-2-furoyl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01510 (10a). Yield 0.121 g (54%), brown solid, m.p. 245 ÷ 247 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79 (br.s, 1H), 7.34 (br.s, 1H), 6.95 (br.s, 1H), 5.02–4.66 (m, 2H), 3.95 (s, 2H), 3.79 (s, 3H), 2.92 (s, 2H), 2.35 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 157.66, 155.34, 151.31, 148.14, 147.54, 138.55, 134.60, 123.80, 117.03, 112.94, 112.05, 43.35, 36.16, 21.95, 21.03, 10.51. HRMS (ESI), m/z calcd C16H15N5O5Na [M + Na+] 380.0966, found 380.0965.
1-ethyl-3-(5-methyl-1,3-oxazol-2-yl)-5-(5-nitro-2-furoyl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01511 (10b). Yield 0.124 g (52%), brown solid, m.p. 180 ÷ 181 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 2.8 Hz, 1H), 7.36 (d, J = 2.8 Hz, 1H), 6.97 (s, 1H), 5.02–4.70 (m, 2H), 4.12 (q, J = 6.4 Hz, 2H), 3.96 (br.s, 2H), 2.97 (br.s, 2H), 2.36 (s, 3H), 1.37 (t, J = 5.8 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) 13C NMR (75 MHz, DMSO) δ 157.68, 155.51, 151.36, 148.25, 147.63, 137.57, 134.78, 125.10, 118.25, 114.21, 111.75, 45.74, 43.91, 22.13, 20.23, 14.27, 9.71. HRMS (ESI), m/z calcd C17H17N5O5Na [M + Na+] 394.1122, found 394.1122.
1-isopropyl-3-(5-methyl-1,3-oxazol-2-yl)-5-(5-nitro-2-furoyl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01515 (10c). Yield 0.113 g (46%), brown solid, m.p. 194 ÷ 196 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 3.8 Hz, 1H), 7.36 (d, J = 3.8 Hz, 1H), 6.98 (s, 1H), 5.01–4.70 (m, 2H), 4.60–4.47 (m, 1H), 3.96 (br.s, 2H), 3.07–2.84 (m, 2H), 2.37 (s, 3H), 1.42 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 157.62, 155.52, 151.31, 148.09, 147.62, 136.95, 134.68, 123.77, 116.94, 112.92, 111.72, 50.18, 43.97, 43.66, 22.24, 22.07, 20.76, 10.54. HRMS (ESI), m/z calcd C18H19N5O5Na [M + Na+] 408.1279, found 408.1278.
1-isobutyl-3-(5-methyl-1,3-oxazol-2-yl)-5-(5-nitro-2-furoyl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01516 (10d). Yield 0.130 g (51%), brown solid, m.p. 123 ÷ 124 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 3.9 Hz, 1H), 7.37 (d, J = 3.9 Hz, 1H), 6.99 (s, 1H), 5.03–4.71 (m, 2H), 3.95 (t, J = 5.3 Hz, 2H), 3.90 (d, J = 6.9 Hz, 2H), 3.03–2.79 (m, 2H), 2.36 (s, 3H), 2.21–2.07 (m, 1H), 0.87 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 157.62, 155.37, 151.29, 148.15, 147.63, 138.13, 134.87, 123.80, 117.06, 112.92, 111.64, 55.87, 43.69, 28.98, 22.36, 21.05, 19.67, 10.51. HRMS (ESI), m/z calcd C19H21N5O5Na [M + Na+] 422.1435, found 422.1435.
1-(cyclopropylmethyl)-5-(5-nitro-2-furoyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01512 (13e). Yield 0.152 g (62%), brown solid, m.p. 144 ÷ 145 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.79 (d, J = 3.6 Hz, 1H), 7.38 (s, 2H), 5.01–4.69 (m, 2H), 4.01–3.93 (m, 4H), 2.97 (s, 2H), 1.31–1.14 (m, 1H), 0.57–0.47 (m, 2H), 0.41–0.33 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 157.60, 151.37, 147.55, 145.69, 137.38, 134.79, 121.65, 117.55, 117.05, 112.97, 110.15, 53.18, 43.77, 43.22, 22.36, 21.15, 11.33, 3.70. HRMS (ESI), m/z calcd C18H17N5O5Na [M + Na+] 406.1122, found 406.1130.
5-(5-nitro-2-furoyl)-3-(1,3-oxazol-5-yl)-1-propyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine, LK01513 (13f). Yield 0.137 g (58%), brown solid, m.p. 149 ÷ 150 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.78 (s, 1H), 7.38 (s, 2H), 5.04–4.64 (m, 2H), 4.11–3.90 (m, 4H), 2.94 (br.s, 2H), 1.78 (br.s, 2H), 0.86 (br.s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 157.65, 151.34, 147.53, 145.62, 137.75, 134.75, 121.61, 117.58, 117.06, 112.94, 109.91, 50.21, 43.74, 22.94, 22.23, 21.01, 11.00. HRMS (ESI), m/z calcd C17H17N5O5Na [M + Na+] 394.1122, found 394.1128.
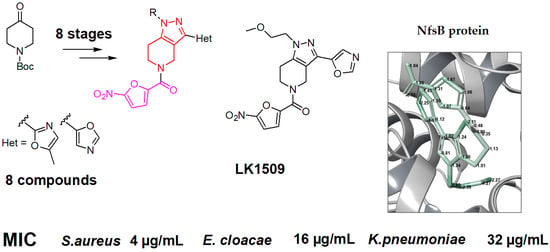
1-(2-methoxyethyl)-5-(5-nitro-2-furoyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01509 (13g). Yield 0.104 g (42%), brown solid, m.p. 130 ÷ 131 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.78 (d, J = 3.7 Hz, 1H), 7.38 (s, 2H), 5.03–4.66 (m, 2H), 4.24 (t, J = 4.4 Hz, 2H), 3.95 (br.s, 2H), 3.68 (t, J = 4.5 Hz, 2H), 3.23 (s, 3H), 2.95 (br.s, 2H); 13C NMR (75 MHz, DMSO-d6) δ 157.68, 151.38, 147.55, 145.61, 138.58, 135.09, 121.73, 117.61, 117.08, 112.94, 109.95, 70.70, 58.16, 48.75, 43.71, 22.36, 21.13. HRMS (ESI), m/z calcd C17H17N5O6 Na [M + Na+] 410.1072, found 410.1071.
1-(3-methoxypropyl)-5-(5-nitro-2-furoyl)-3-(1,3-oxazol-5-yl)-4,5,6,7-tetrahydro-1H-pyrazolo [4,3-c]pyridine, LK01514 (13h). Yield 0.100 g (39%), brown solid, m.p. 97÷98 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.78 (d, J = 3.3 Hz, 1H), 7.38 (s, 2H), 5.00–4.66 (m, 2H), 4.11 (t, J = 5.8 Hz, 2H), 3.96 (br.s, 2H), 3.28 (t, J = 6.0 Hz, 2H), 3.23 (s, 3H), 2.91 (br.s, 2H), 2.06–1.92 (m, 2H).; 13C NMR (75 MHz, DMSO-d6) δ 157.63, 151.34, 147.55, 145.66, 137.99, 135.00, 121.67, 117.55, 117.05, 112.91, 109.91, 68.55, 57.92, 45.65, 43.71, 29.53, 22.13, 20.81. HRMS (ESI), m/z calcd C18H19N5O6Na [M + Na+] 424.1228, found 424.1228.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}