
Massive Solubility Changes in Neuronal Proteins upon Simulated Traumatic Brain Injury Reveal the Role of Shockwaves in Irreversible Damage

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Dynamic Impact Experiments
2.2. Acceleration during Impact
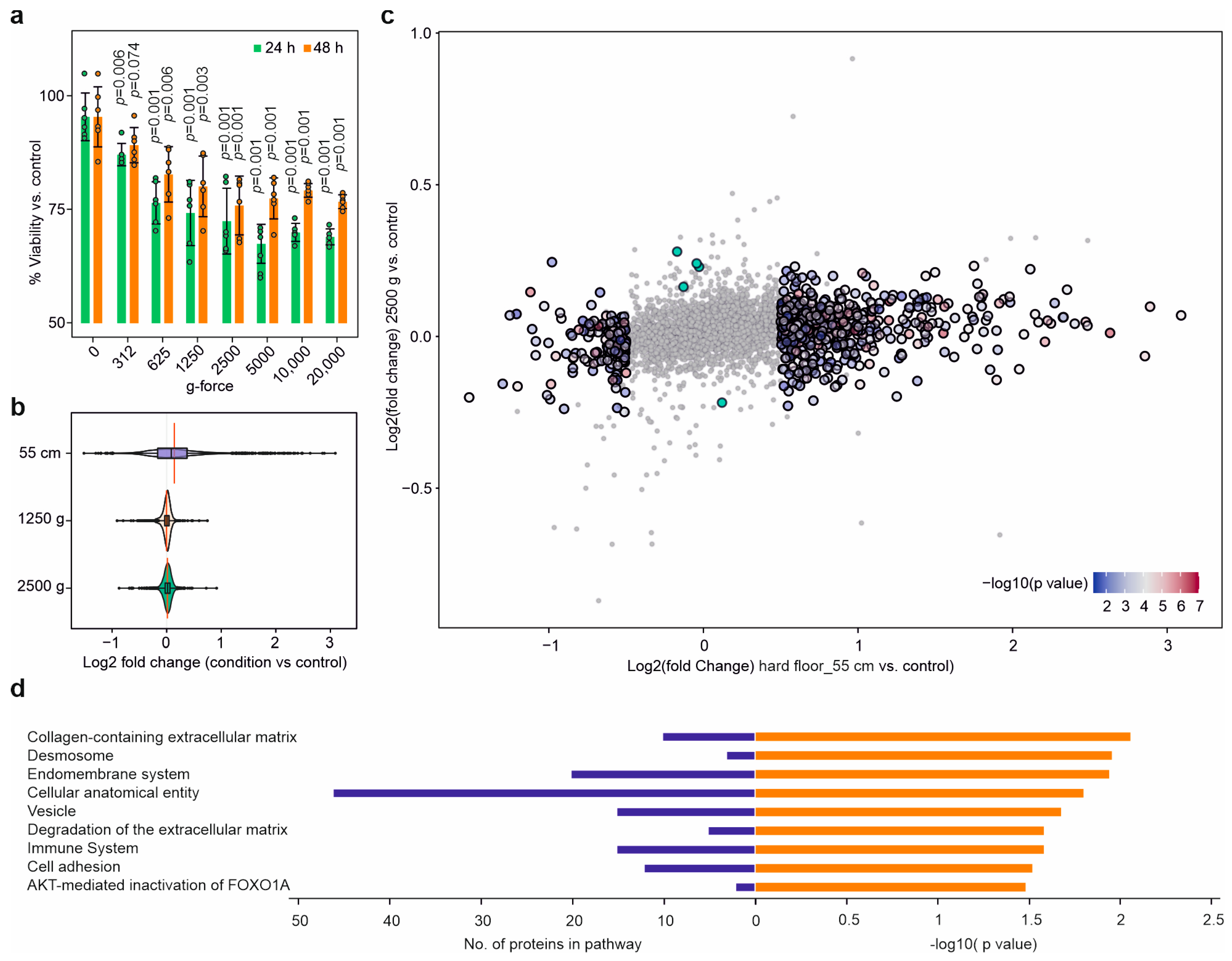
2.3. Dynamic Impact Reduced Cell Viability
2.4. Dynamic Impact Induced Massive Solubility Changes in the Cellular Proteome
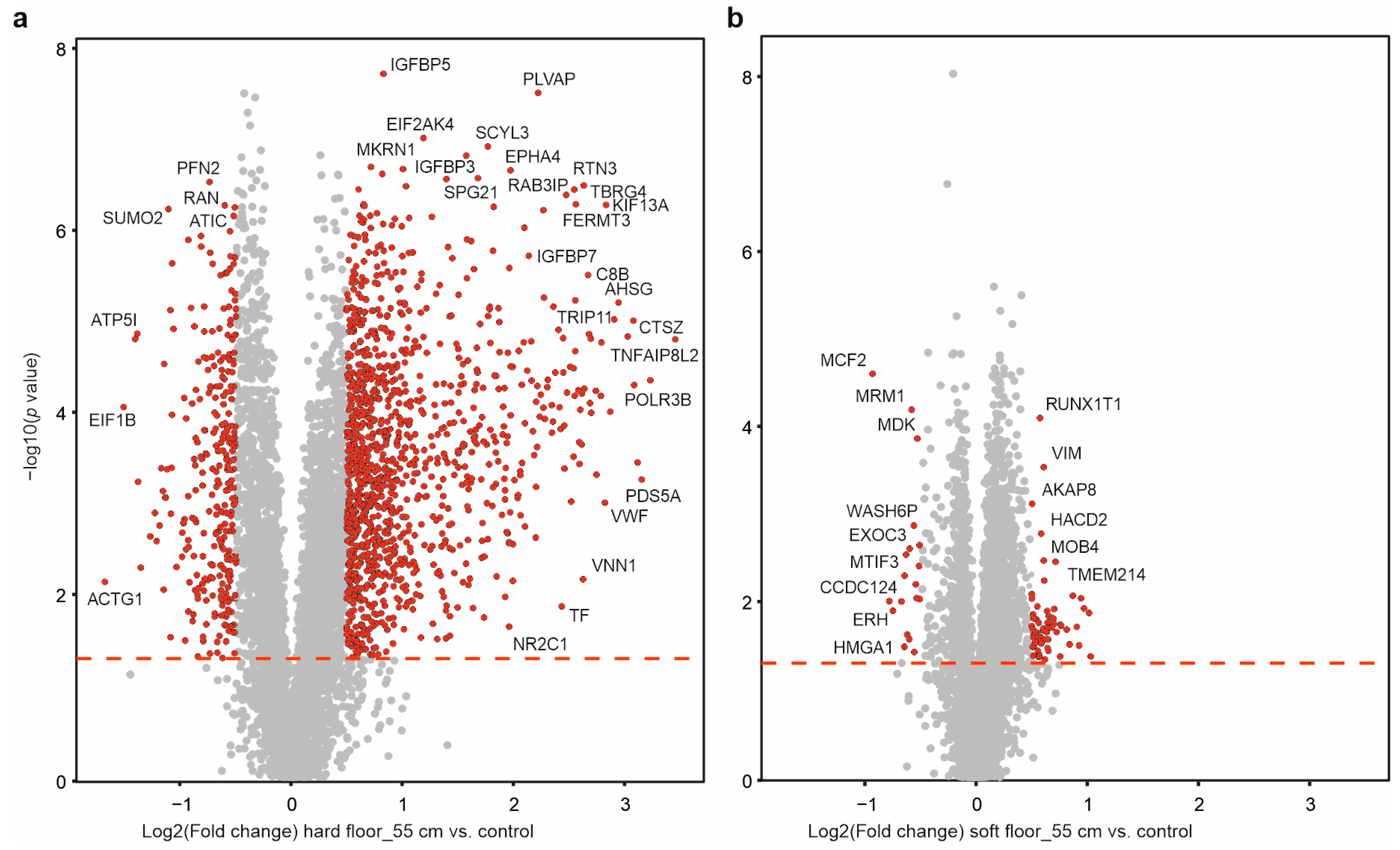
2.5. Individual Protein Solubility Changes after Dynamic Impact
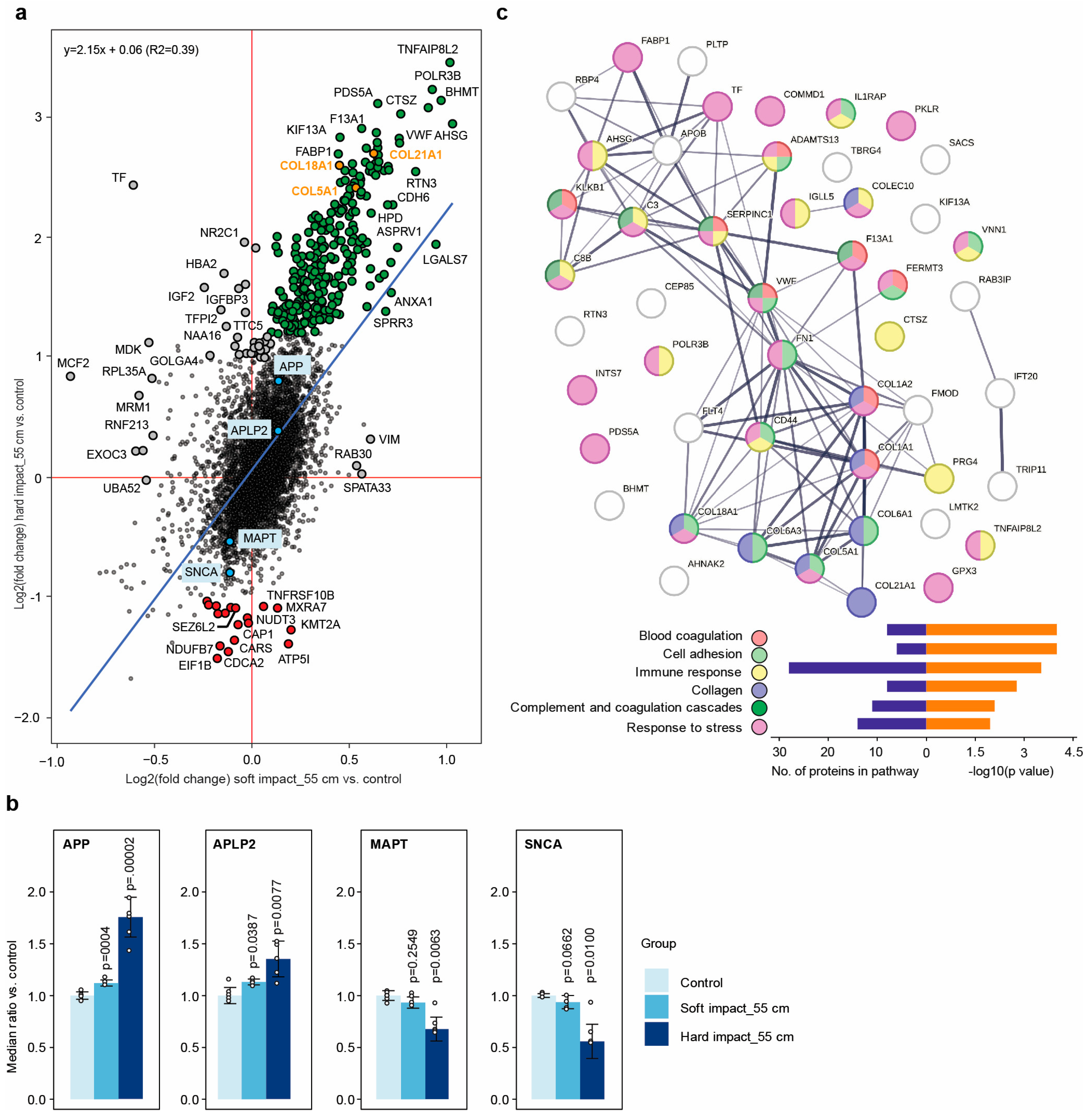
2.6. Dynamic Impact Modulated the Solubility of Proteins Involved in Neurodegenerative Disorders
2.7. Dynamic Impact Affected Essential Cellular Processes
2.8. The Cellular Effects Were Mainly Due to the Impact’s Shockwaves
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Simulation of Dynamic Impacts at Different Heights
4.3. Cell Viability Measurements
4.4. PISA Assay
4.5. LC-MS/MS
4.6. Data Processing
4.7. Network Mapping
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Von Holst, H. Traumatic Brain Injury in Handbook of Clinical Neuroepidemiology; Nova Publishers: Hauppauge, NY, USA, 2007; ISBN 978-1-60021-511-7. [Google Scholar]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, M. Traumatic Brain Injury: Giving Voice to a Silent Epidemic. Nat. Rev. Neurol. 2013, 9, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M. Traumatic Brain Injury: A Silent Epidemic. Ann. Neurol. 1990, 27, 327. [Google Scholar] [CrossRef] [PubMed]
- Perlesz, A.; Kinsella, G.; Crowe, S. Impact of Traumatic Brain Injury on the Family: A Critical Review. Rehabil. Psychol. 1999, 44, 6–35. [Google Scholar] [CrossRef]
- Degeneffe, C.E. Family Caregiving and Traumatic Brain Injury. Health Soc. Work. 2001, 26, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Kaup, A.; Barnes, D.E.; Yaffe, K. Dementia Risk after Traumatic Brain Injury vs. Nonbrain Trauma: The Role of Age and Severity. JAMA Neurol. 2014, 71, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Nordström, A.; Nordström, P. Traumatic Brain Injury and the Risk of Dementia Diagnosis: A Nationwide Cohort Study. PLoS Med. 2018, 15, e1002496. [Google Scholar] [CrossRef]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and Brain Trauma. Neuroscience 2004, 129, 1019–1027. [Google Scholar] [CrossRef]
- Kolias, A.G.; Kirkpatrick, P.J.; Hutchinson, P.J. Decompressive Craniectomy: Past, Present and Future. Nat. Rev. Neurol. 2013, 9, 405–415. [Google Scholar] [CrossRef]
- Stiver, S.I. Complications of Decompressive Craniectomy for Traumatic Brain Injury. Neurosurg. Focus. 2009, 26, E7. [Google Scholar] [CrossRef]
- Honeybul, S.; Ho, K.M. Long-Term Complications of Decompressive Craniectomy for Head Injury. J. Neurotrauma 2011, 28, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Tang-Schomer, M.D.; Patel, A.R.; Baas, P.W.; Smith, D.H. Mechanical Breaking of Microtubules in Axons during Dynamic Stretch Injury Underlies Delayed Elasticity, Microtubule Disassembly, and Axon Degeneration. FASEB J. 2010, 24, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial Dysfunction and Calcium Perturbation Induced by Traumatic Brain Injury. J. Neurotrauma 1997, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Sullivan, P.G.; Deng, Y.; Mbye, L.H.; Hall, E.D. Time Course of Post-Traumatic Mitochondrial Oxidative Damage and Dysfunction in a Mouse Model of Focal Traumatic Brain Injury: Implications for Neuroprotective Therapy. J. Cereb. Blood Flow Metab. 2006, 26, 1407–1418. [Google Scholar] [CrossRef]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of Extracellular Glutamate Measured by Cerebral Microdialysis in Severe Traumatic Brain Injury: Clinical Article. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The Role of Excitatory Amino Acids and NMDA Receptors in Traumatic Brain Injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, G.; De Rivero Vaccari, J.P.; Perez, E.R.; Sanchez-Molano, J.; Furones-Alonso, O.; Bramlett, H.M.; Dietrich, W.D. Alterations in Blood-Brain Barrier Permeability to Large and Small Molecules and Leukocyte Accumulation after Traumatic Brain Injury: Effects of Post-Traumatic Hypothermia. J. Neurotrauma 2009, 26, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Kohen, R. The Role of Reactive Oxygen Species in the Pathogenesis of Traumatic Brain Injury. In Oxidative Stress and Free Radical Damage in Neurology; Springer: New York, NY, USA, 2011. [Google Scholar]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative Stress and Modification of Synaptic Proteins in Hippocampus after Traumatic Brain Injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed Head Injury Induces Upregulation of Beclin 1 at the Cortical Site of Injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef]
- Clark, R.S.B.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy Is Increased in Mice after Traumatic Brain Injury and Is Detectable in Human Brain after Trauma and Critical Illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fukuda, T.; Iwadate, K. Immunohistochemical Analysis of the Ubiquitin Proteasome System and Autophagy Lysosome System Induced after Traumatic Intracranial Injury: Association with Time between the Injury and Death. Am. J. Forensic Med. Pathol. 2014, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Au, A.K.; Aneja, R.K.; Bayır, H.; Bell, M.J.; Janesko-Feldman, K.; Kochanek, P.M.; Clark, R.S.B. Autophagy Biomarkers Beclin 1 and P62 Are Increased in Cerebrospinal Fluid after Traumatic Brain Injury. Neurocrit. Care 2017, 26, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Beer, R.; Franz, G.; Srinivasan, A.; Hayes, R.L.; Pike, B.R.; Newcomb, J.K.; Zhao, X.; Schmutzhard, E.; Poewe, W.; Kampfl, A. Temporal Profile and Cell Subtype Distribution of Activated Caspase-3 Following Experimental Traumatic Brain Injury. J. Neurochem. 2000, 75, 1264–1273. [Google Scholar] [CrossRef]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and Glial Cell Number in the Hippocampus after Experimental Traumatic Brain Injury: Analysis by Stereological Estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef]
- Smith, D.H.; Chen, X.H.; Pierce, J.E.S.; Wolf, J.A.; Trojanowski, J.Q.; Graham, D.I.; Mcintosh, T.K. Progressive Atrophy and Neuron Death for One Year Following Brain Trauma in the Rat. J. Neurotrauma 1997, 14, 715–727. [Google Scholar] [CrossRef]
- Veenith, T.; Goon, S.S. Molecular Mechanisms of Traumatic Brain Injury: The Missing Link in Management. World J. Emerg. Surg. 2009, 4, 4. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Kim, Y.S.; Randolph, T.W.; Seefeldt, M.B.; Carpenter, J.F. High-Pressure Studies on Protein Aggregates and Amyloid Fibrils. Methods Enzymol. 2006, 413, 237–253. [Google Scholar]
- Von Holst, H.; Purhonen, P.; Lanner, D.; Balakrishnan Kumar, R.; Hebert, H. White Shark Protein Metabolism May Be a Model to Improve the Outcome of Cytotoxic Brain Tissue Edema and Cognitive Deficiency after Traumatic Brain Injury and Stroke. J. Neurol. Neurobiol. 2018, 4. [Google Scholar] [CrossRef]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundström, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking Cancer Drugs in Living Cells by Thermal Profiling of the Proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arnér, E.S.J.; Zubarev, R.A. Comprehensive Chemical Proteomics for Target Deconvolution of the Redox Active Drug Auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef]
- Saei, A.A.; Beusch, C.M.; Sabatier, P.; Wells, J.A.; Gharibi, H.; Meng, Z.; Chernobrovkin, A.; Rodin, S.; Näreoja, K.; Thorsell, A.-G.; et al. System-Wide Identification and Prioritization of Enzyme Substrates by Thermal Analysis. Nat. Commun. 2021, 12, 1296. [Google Scholar] [CrossRef]
- Sabatier, P.; Beusch, C.M.; Saei, A.A.; Aoun, M.; Moruzzi, N.; Coelho, A.; Leijten, N.; Nordenskjöld, M.; Micke, P.; Maltseva, D.; et al. An Integrative Proteomics Method Identifies a Regulator of Translation during Stem Cell Maintenance and Differentiation. Nat. Commun. 2021, 12, 6558. [Google Scholar] [CrossRef]
- Becher, I.; Andrés-Pons, A.; Romanov, N.; Stein, F.; Schramm, M.; Baudin, F.; Helm, D.; Kurzawa, N.; Mateus, A.; Mackmull, M.T.; et al. Pervasive Protein Thermal Stability Variation during the Cell Cycle. Cell 2018, 173, 1495–1507.e18. [Google Scholar] [CrossRef]
- Dai, L.; Zhao, T.; Bisteau, X.; Sun, W.; Prabhu, N.; Lim, Y.T.; Sobota, R.M.; Kaldis, P.; Nordlund, P. Modulation of Protein-Interaction States through the Cell Cycle. Cell 2018, 173, 1481–1494.e13. [Google Scholar] [CrossRef]
- Potel, C.M.; Kurzawa, N.; Becher, I.; Typas, A.; Mateus, A.; Savitski, M.M. Impact of Phosphorylation on Thermal Stability of Proteins. Nat. Methods 2021, 18, 757–759. [Google Scholar] [CrossRef]
- Saei, A.A.; Lundstrom, S.L.; Lyu, H.; Gharibi, H.; Lu, W.; Fang, P.; Zhang, X.; Meng, Z.; Wang, J.; Gaetani, M. Mapping the GALNT1 Substrate Landscape with Versatile Proteomics Tools. bioRxiv 2022. [Google Scholar] [CrossRef]
- Courant, R.; Friedrichs, K.O. Supersonic Flow and Shock Waves; Springer Science & Business Media: Berlin, Germany, 1999; Volume 21, ISBN 0387902325. [Google Scholar]
- Sun, T.-P.; Álvarez-Novoa, F.; Andrade, K.; Gutiérrez, P.; Gordillo, L.; Cheng, X. Stress Distribution and Surface Shock Wave of Drop Impact. Nat. Commun. 2022, 13, 1703. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gong, S.; Carmody, R.J.; Hilliard, A.; Li, L.; Sun, J.; Kong, L.; Xu, L.; Hilliard, B.; Hu, S.; et al. TIPE2, a Negative Regulator of Innate and Adaptive Immunity That Maintains Immune Homeostasis. Cell 2008, 133, 415–426. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, X.; Liu, L.; Liu, S.; Wang, Z.; Zhang, B.; Fan, B.; Yang, F.; Huang, S.; Jiang, F.; et al. TIPE2, a Novel Regulator of Immunity, Protects against Experimental Stroke. J. Biol. Chem. 2012, 287, 32546–32555. [Google Scholar] [CrossRef]
- Yaguchi, H.; Yabe, I.; Takahashi, H.; Watanabe, M.; Nomura, T.; Kano, T.; Matsumoto, M.; Nakayama, K.I.; Watanabe, M.; Hatakeyama, S. Sez6l2 Regulates Phosphorylation of ADD and Neuritogenesis. Biochem. Biophys. Res. Commun. 2017, 494, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Boonen, M.; Staudt, C.; Gilis, F.; Oorschot, V.; Klumperman, J.; Jadot, M. Cathepsin D and Its Newly Identified Transport Receptor SEZ6L2 Can Modulate Neurite Outgrowth. J. Cell Sci. 2016, 129, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, H.; Yabe, I.; Takahashi, H.; Okumura, F.; Takeuchi, A.; Horiuchi, K.; Kano, T.; Kanda, A.; Saito, W.; Matsumoto, M.; et al. Identification of Anti-Sez6l2 Antibody in a Patient with Cerebellar Ataxia and Retinopathy. J. Neurol. 2014, 261, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Hahn, S.; Hanssen, H.; Münchau, A.; Wandinger, K.P.; Brüggemann, N. Sez6l2-Antibody-Associated Progressive Cerebellar Ataxia: A Differential Diagnosis of Atypical Parkinsonism. J. Neurol. 2019, 266, 522–524. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy Failure in Alzheimer’s Disease-Locating the Primary Defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and Its Role in Innate and Adaptive Immune Responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Tam, J.C.H.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular Sensing of Complement C3 Activates Cell Autonomous Immunity. Science 2014, 345, 1256070. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Atkinson, J.P. Evolution of the Complement System: From Defense of the Single Cell to Guardian of Intravascular Space. Immunol. Rev. 2016, 274, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F.; Schad, P.E.; Kleinman, H.K. Cross-Linking of Fibronectin to Collagen by Blood Coagulation Factor XIIIa. J. Clin. Investig. 1979, 64, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Zillmann, A.; Luther, T.; Müller, I.; Kotzsch, M.; Spannagl, M.; Kauke, T.; Oelschlägel, U.; Zahler, S.; Engelmann, B. Platelet-Associated Tissue Factor Contributes to the Collagen-Triggered Activation of Blood Coagulation. Biochem. Biophys. Res. Commun. 2001, 281, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Niewiarowski, S.; Stuart, R.K.; Thomas, D.P.; Chalmers, T.C. Activation of Intravascular Coagulation by Collagen. Proc. Soc. Exp. Biol. Med. 1966, 123, 196–200. [Google Scholar] [CrossRef]
- Mayer, A.R.; Quinn, D.K.; Master, C.L. The Spectrum of Mild Traumatic Brain Injury: A Review. Neurology 2017, 89, 623–632. [Google Scholar] [CrossRef]
- Gregoire, S.; Irwin, J.; Kwon, I. Techniques for Monitoring Protein Misfolding and Aggregation in Vitro and in Living Cells. Korean J. Chem. Eng. 2012, 29, 693–702. [Google Scholar] [CrossRef]
- Roche, J.; Royer, C.A. Lessons from Pressure Denaturation of Proteins. J. R. Soc. Interface 2018, 15, 20180244. [Google Scholar] [CrossRef]
- Kuffel, A.; Zielkiewicz, J. Why the Solvation Water around Proteins Is More Dense than Bulk Water. J. Phys. Chem. B 2012, 116, 12113–12124. [Google Scholar] [CrossRef]
- Saei, A.A.; Beusch, C.M.; Chernobrovkin, A.; Sabatier, P.; Zhang, B.; Tokat, Ü.G.; Stergiou, E.; Gaetani, M.; Végvári, Á.; Zubarev, R.A. ProTargetMiner as a Proteome Signature Library of Anticancer Molecules for Functional Discovery. Nat. Commun. 2019, 10, 5715. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING Database in 2017: Quality-Controlled Protein–Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange Provides Globally Coordinated Proteomics Data Submission and Dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saei, A.A.; Gharibi, H.; Lyu, H.; Nilsson, B.; Jafari, M.; Von Holst, H.; Zubarev, R.A. Massive Solubility Changes in Neuronal Proteins upon Simulated Traumatic Brain Injury Reveal the Role of Shockwaves in Irreversible Damage. Molecules 2023, 28, 6768. https://doi.org/10.3390/molecules28196768
Saei AA, Gharibi H, Lyu H, Nilsson B, Jafari M, Von Holst H, Zubarev RA. Massive Solubility Changes in Neuronal Proteins upon Simulated Traumatic Brain Injury Reveal the Role of Shockwaves in Irreversible Damage. Molecules. 2023; 28(19):6768. https://doi.org/10.3390/molecules28196768
Chicago/Turabian StyleSaei, Amir Ata, Hassan Gharibi, Hezheng Lyu, Brady Nilsson, Maryam Jafari, Hans Von Holst, and Roman A. Zubarev. 2023. "Massive Solubility Changes in Neuronal Proteins upon Simulated Traumatic Brain Injury Reveal the Role of Shockwaves in Irreversible Damage" Molecules 28, no. 19: 6768. https://doi.org/10.3390/molecules28196768