
Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y

Department of Chemistry, Ochanomizu University, 2-1-1 Otsuka, Bukyo-ku, Tokyo 104-8610, Japan
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(22), 7577; https://doi.org/10.3390/molecules28227577
Submission received: 22 October 2023
/
Revised: 11 November 2023
/
Accepted: 12 November 2023
/
Published: 14 November 2023
(This article belongs to the Special Issue Recent Advances in Photocatalytic Organic Synthesis)
Abstract
:Fluoroalkyl compounds are widely used, underscoring a pressing need for the development of methods for their synthesis. However, reports on perfluoroalkylation to styrenes have been sparse. In this study, both hydroxy- and hydro-perfluoroalkylation of styrene were achieved using visible light reactions, catalyzed by eosin Y, by selecting appropriate additives and controlling the eosin Y quenching cycle. These reactions are heavy-metal free, use water as the hydroxyl or hydrogen source, and employ inexpensive and readily available reagents.
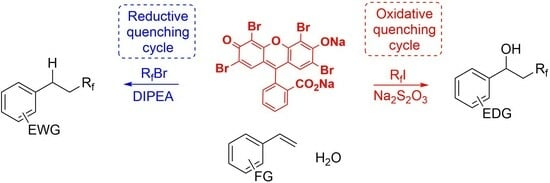
1. Introduction
Fluorine-containing organic compounds play a crucial role in agricultural and pharmaceutical products [1,2,3,4,5,6] and the development of functional materials [7,8]. However, very few fluorine-containing organic compounds exist naturally, and they must be synthesized artificially. The incorporation of perfluoroalkyl groups using perfluoroalkyl halides into organic compounds is an effective and widely used method for the synthesis of fluorine-containing compounds [9,10,11,12]. It is achieved using perfluoroalkyl halides, which are upstream raw materials in the fluorine industry, offering a diverse array of fluoroalkyl groups. Perfluoroalkyl halides possess low homolysis energies and redox potentials, causing them to readily form perfluoro radicals. Indeed, many examples of perfluoroalkylation to electron-rich olefins have been reported. However, few studies have reported on perfluoroalkylation with conjugated olefins [13], particularly styrene, due to the likelihood of side reactions such as oligomerization, polymerization, and nucleophilic trapping.
Hydroxy- [14,15,16,17,18,19,20,21,22,23,24,25] and hydro-perfluoroalkylation [26,27,28,29,30,31,32,33] have been reported as examples of perfluoroalkylation to styrene. Hydro-perfluoroalkylation was reported in 1999 using light irradiation with a metal-halogen lamp in the presence of (Bu3Sn)2 [14]. In 2018, visible-light-induced hydroxy-perfluoroalkylation by Jiao et al. was realized using ruthenium complexes as a photoredox catalyst [17]. Chen et al. [19] and our group [24,25] have reported visible-light-induced hydroxy-perfluoroalkylation. During such reactions, catalyzation is achieved by amines or enamines and the process is free of heavy metals. Oxygen molecules trap the intermediate benzyl radicals (Scheme 1a). For hydro-perfluoroalkylation, a reaction using a radical initiator was reported in 2010 by Postigo et al. In this type of reaction, the benzyl radicals produced are hydrogen-trapped by tris(trimethylsilyl)silane [26]. Noël et al. reported a visible light reaction in which an iridium-catalyzed reaction used 4-hydroxythiophen as a hydrogen donor [29] (Scheme 1b).
Here, we have developed a series of eosin-Y-catalyzed [34,35] visible-light-induced fluoroalkylations using perfluoroalkyl halides [36,37,38]. Eosin Y is an inexpensive, safe, and readily available organic dye. The reactions proceed by both reductive and oxidative quenching cycles. We have reported the iodoperfluoroalkylation of olefins by oxidative formation of perfluoroalkyl radicals with the addition of sodium thiosulfate [36], and their hydro-perfluoroalkylation, under reductive conditions, in the presence of amines [37]. We hypothesized that if this reaction were applied to styrene in the presence of water, hydroxy-perfluoroalkyl products would be obtained via benzyl cation intermediates under oxidative reaction conditions, and hydro-perfluoroalkylated products would be produced via benzyl anion intermediates under reductive reaction conditions (Scheme 1c). Such reactions via benzyl anions and cations have only been reported using trifluoromethylation reagents, and we have found no reports using perfluoroalkyl halides.
2. Results and Discussion
Based on our previous reaction conditions, perfluoroalkylations to styrenes were attempted (Table 1). Following our previously reported methodology on iodoperfluoroalkylation [36], we proceeded with the oxidative quenching cycle of eosin Y. Firstly, we reacted p-methoxystyrene with perfluorohexyl iodide (1.2 equiv.). This reaction uses eosin Y (10 mol%) as the catalyst in the presence of aqueous Na2S2O3. It requires photoirradiation for 3 h using a 12 W white light-emitting diode (LED) lamp. As expected, it yielded a 40% hydroxy-perfluorohexylated product (Entry 1). The addition of potassium carbonate to trap the byproduct HI improved the yield (Entry 2). When the amount of Na2S2O3 was reduced to 0.25 eq., the yield was further improved (Entry 3). However, the yield decreased when the reaction was performed without Na2S2O3 (Entry 4). The yield was not improved using 3 eq. of perfluorohexyl bromide (Entry 5). According to our previously reported hydro-perfluoroalkylation methodology [38], the reaction proceeded via the reductive quenching cycle of eosin Y (Entry 6). To test this, p-cyanostylene was reacted with perfluorohexyl iodide (3.0 equiv.) in the presence of diisopropylethylamine (DIPEA, 2.0 equiv.). As a result, a 70% hydro-perfluoroalkylated product was obtained as the sole product. When perfluoroalkyl bromide was used under reaction conditions in the presence of sodium thiosulfate, or conversely when perfluoroalkyl iodide was used in the presence of amine, no product was obtained (Scheme S1).
Subsequently, reactions using oxidative (Table 1, Entry 3) or reductive (Table 1, Entry 6) quenching cycles were applied to substituted styrenes (Scheme 2). First, we tried oxidative reaction conditions using various p-substituted styrenes (1c–i). Styrene with a tert-butoxy group (1c) gave good yields of hydroxy-perfluorohexylated product (4c). The yields were lowered with tert-butyl (1d) and methyl (1d) substituents or unsubstituted styrene (1f); even lower yields were obtained for halogen-substituted styrene (1g, h). The reaction of m-methoxy (1j) and o-methyl (1k) styrene gave 33% yields whereas o,p-dimethoxystyrene (1l) improved the yield to 68%. The hydroxy-perfluorohexylation proceeded with 1,2-dihydronaphthalene (1m), giving a 31% yield with a 62:38 diastereomer ratio. Reactions with different perfluoroalkyl iodides were also investigated. The reaction with gaseous CF3I yielded a 35% hydroxy-trifluoromethylated product (4aa). Perfluoroethyl, propyl, i-propyl, and butyl octyl all yielded the corresponding hydro-perfluoroalkyalted products (4ab–f).
Next, p-(trifluorometyl)stylene (1n), 2,3,4,5,6-pentafluorostylene (1o), and 1,1-diphenylethylene (1p) were tested, indicating that reactions proceeded under both oxidative and reductive conditions for these substrates. In particular, 1,1-diphenylethylene (1p) produced good yields of hydroxy- (4p) and hydro-perfluoroalkylated product (5p). In contrast, previous radical perfluoroalkylation of 1p yielded olefination products [39,40]. With p-nitrostyrene (1q), the yield was only 21%. With styrene, the yield was also 21%, but this may be due to the radical inhibitory effect of the nitro compound. The introduction of perfluoropropyl and butyl was investigated using p-cyanostylene (1b), producing high yields of the products 5bc and 5be. The effect of substituents in para-substituted styrene is correlated with the Hammett constant [41,42]. Hydroxy-perfluoroalkylation (4) was dominant with electron-donating substituents, which have a smaller Hamett σ parameter. In contrast, hydro-perfluoroalkylation (4) was dominant with electron-withdrawing substituents (Table S1).
We attempted reactions utilizing the respective properties of benzyl cations and anions (Scheme 3). Under oxidative reaction conditions via benzyl cations, alkoxyperfluoroalkylation was investigated by the addition of alcohols instead of water. Adding methanol, ethanol, or butanol produced corresponding methoxy (6a), ethoxy (6b), and butoxy (6c) products. Under reductive reaction conditions via benzyl anions, the synthesis of fluoro-olefins using α-trifluoromethylstyrene (7) as a substrate was performed [43,44,45]. The reaction produced a 51% yield of fluoro-olefins (9) along with 20% of hydro-perfluoroalkylated product (8).
Based on our experimental results and previous reports [36,37,38,46], we propose the following reaction mechanism (Scheme 4). Under oxidative reaction conditions, photo-irradiated eosin Y (EY*) is reacted with perfluoroalkyl iodide to give a perfluoroalkyl radical. This radical is added to styrene to give intermediate radical A. Radical A is converted to benzyl cation B via the radical cation of eosin Y. At this time, the presence of an excess amount of Na2S2O3 would consume the radical cation of eosin Y, thus reducing the yield of the desired product. Then we investigated the equimolar amount of Na2S2O3 and 0.25 equivalents were found to be optimal (Table S2). Then, cation B is reacted with water to give the hydroxy-perfluoroalkylated product 4 (Scheme S2). When the time course of the reaction was followed without potassium carbonate, iodide D was also observed during the reaction (Table S3). This suggests a possible pathway in which radical intermediate A is trapped by iodine once and then reverts back to a radical or is directly hydroxylated. Under a reductive quenching cycle, the radical anion of eosin Y is produced by the reaction of DIPEA and photo-irradiated eosin Y [47,48,49]. The radical anion of eosin Y reduces perfluoroalkyl bromide to produce the perfluoroalkyl radical, which is reacted with styrene. The subsequent reaction with the radical anion of eosin Y gives benzyl anion C. In this case, two equivalents of amine are required because the radical anion of eosin Y reacts with both the perfluoroalkyl bromide and the intermediate radical A. The benzyl anion C reacts with water to produce the hydro-perfluoroalkylated product 5. This is suggested by the experimental results as the reaction in the absence of water does not yield product 5n, and the yield is not improved even in the presence of THF, which can act as a hydrogen radical donor (Table S2). The stability of the intermediate benzyl cation (B) and anion (C) is influenced by the electronic properties of the substituent on styrene, which is linked to the correlation between the product ratio and the Hammett constant.
3. Materials and Methods
3.1. General Information
All reactions were performed under the argon atmosphere. 1H, 13C, and 19F NMR spectra were recorded on a JEOL GSX-400 spectrometer (Jeol, Akishima, Tokyo, Japan) (400 MHz for 1H, 126 MHz for 13C, and 376 MHz for 19F), a JEOL ECX-500 spectrometer (Jeol, Akishima, Tokyo, Japan) (500 MHz for 1H and 471 MHz for 19F), or a BRUKER AVANCE 600 spectrometer (Bruker, Billerica, MA, USA) (600 MHz for 1H and 151 MHz for 13C) with CDCl3 as the solvent, tetramethylsilane (TMS: δ 0 ppm for 1H), chloroform-d (CDCl3: δ 76.9 ppm for 13C), and hexafluorobenzene (C6F6: −162.2 ppm for 19F) as an internal standard. IR spectra were taken on SHIMADZU IRSpirit-T, and HRMS were obtained with a JEOL JMS-T100TD (FD) (Jeol, Akishima, Tokyo, Japan), JEOL JMS-700 (EI) (Jeol, Akishima, Tokyo, Japan), or BRUKER Compact (ESI) (Bruker, Billerica, MA, USA). Precoated Merck Kieselgel 60 F254 and Kanto silica gel 60 (spherical neutral) were used for thin-layer chromatography and flash chromatography, respectively. Visualization was accomplished by UV light (254 nm). All commercially available reagents and solvents were used as received without further purification.
3.2. General Procedure
3.2.1. Oxidative Reaction Conditions
To a solution of styrene (0.300 mmol) in CH3CN/H2O (7.50 mL/1.00 mL) mixed solvent were added Eosin Y-2Na (20.7 mg, 0.0299 mmol), Na2S2O3 (11.9 mg, 0.0753 mmol), K2CO3 (82.9 mg, 0.600 mmol), and perfluoroalkyl iodide (0.360 mmol) under an Ar atmosphere. Then, the mixture was stirred under irradiation with 12 W white LEDs at room temperature for 3 h. Then, the mixture was extracted with dichloromethane, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to yield hydroxy-perfluoroalkylation products as a colorless oil.
3.2.2. Reductive Reaction Conditions
To a solution of styrene (0.200 mmol) in dry CH3CN/H2O (5.00 mL/1.00 mL) mixed solvent were added Eosin Y-2Na (13.8 mg, 0.020 mmol), DIPEA (0.07 mL, 0.402 mmol), and perfluoroalkyl bromide (0.600 mmol) under an Ar atmosphere. Then, the mixture was stirred under irradiation with 12 W white LEDs at room temperature for 3 h. Then, the mixture was extracted with dichloromethane, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to yield hydro-perfluoroalkylation products as a colorless oil.
3.3. Characterization Data of Products
3.3.1. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-methoxyphenyl)octan-1-ol (4a)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (70.5 mg, 50%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 7.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.68–2.56 (1H, m), 2.45–2.33 (1H, m), 2.17 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.2, 127.1 (2C), 119.7–110.3 (6C, m), 114.3 (2C), 67.7, 55.5, 39.8 (t, J = 21.0 Hz), 32.7, 18.3, 14.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, d, J = 282.3 Hz), −114.4 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.2 (2F, s), −126.7 (2F, s); IR; 3431,3007, 2960, 2914, 1614, 1515, 1232, 1182, 1144, 1036, 812, 707; HRMS (EI+) calcd for C15H11O2F13 [M]+: 470.0551, found 470.0528.
3.3.2. 3,3,3-Trifluoro-1-(4-methoxyphenyl) propan-1-ol (4aa)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (23.3 mg, 35%); 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, d, J = 8.4 Hz), 6.90 (2H, d, J = 8.8 Hz), 5.02 (1H, d, J = 8.8 Hz), 3.81 (3H, s), 2.70–2.56 (1H, m), 2.49–2.36 (1H, m), 2.16 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.6, 130.8–112.3 (m), 127.1 (2C), 114.3 (2C), 68.5 (dd, J = 3.0, 6.0 Hz), 55.5, 42.8 (dd, J = 27.2, 54.3 Hz); 19F NMR (376 MHz, CDCl3) δ −64.2 (3F, s); IR; 3425, 3007, 2911, 2840, 1613, 1514, 1241, 1123, 1102, 1032, 824; HRMS (EI+) calcd for C10H11O2F3 [M]+: 220.0711, found 220.0718.
3.3.3. 3,3,4,4,4-Pentafluoro-1-(4-methoxyphenyl) tetran-1-ol (4ab)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow solid (25.9 mg, 32%); 1H NMR (400 MHz, CDCl3) δ 7.31 (2H, d, J = 8.8 Hz), 6.86 (2H, d, J = 8.8 Hz), 5.15 (1H, dt, J = 8.8, 3.2 Hz), 3.81 (3H, s), 2.66–2.51 (1H, m), 2.41–2.12 (1H, m), 2.11 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 127.1, 120.0–113.4 (2C, m), 114.3, 67.7, 55.5, 39.7 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −86.3 (3F, s), −117.1 (1F, dd, J = 263.3, 37.6 Hz), −118.3 (1F, dd, J = 263.3, 37.6 Hz); IR; 3441, 3007, 2914, 2842, 1614, 1515, 1248, 1186, 1065, 1033; HRMS (EI+) calcd for C11H11O2F5 [M]+: 270.0679, found 270.0678.
3.3.4. 3,3,4,4,5,5,5-Heptafluoro-1-(4-methoxyphenyl) pentan-1-ol (4ac)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (50.3 mg, 52%); 1H NMR (500 MHz, CDCl3) δ 7.30 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 9.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.67–2.54 (1H, m), 2.44–2.32 (1H, m), 2.18 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 134.9–108.6 (3C, m), 127.1 (2C), 114.3 (2C), 67.6, 55.5, 39.5 (t, J = 21.0); 19F NMR (471 MHz, CDCl3) δ −80.9 (3F, s), −114.0 (1F, dd, J = 282.3, 47.1 Hz), −115.4 (1F, dd, J = 282.3, 47.1 Hz), −128.4 (2F, s); IR; 3455, 3007, 2842, 1613, 1515, 1304, 1155, 832, 713; HRMS (EI+) calcd for C12H11O2F7 [M]+: 320.0647, found 320.0657.
3.3.5. 3,4,4,4-Tetrafluoro-3-trifluoromethyl-1-(4-methoxyphenyl) tetran-1-ol (4ad)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (57.8 mg, 60%); 1H NMR (500 MHz, CDCl3) δ 7.30 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 8.5 Hz), 5.12 (1H, d, J = 22.5 Hz), 3.81 (3H, s), 2.66–2.57 (1H, m), 2.41–2.33 (1H, m), 2.13 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 153.73, 128.9–113.9 (2C, m), 126.9 (2C), 114.3 (2C), 92.3–90.6 (m), 68.3 (d, J = 3.0 Hz), 55.5, 38.2 (t, J = 18.0 Hz); 19F NMR (471 MHz, CDCl3) δ −76.9 (3F, s), −77.5 (3F, s), −186.0 (1F, s); IR; 3455, 3007, 2917, 2842, 1613, 1515, 1304, 1217, 1155, 1036, 832, 713; HRMS (EI+) calcd for C12H11O2F7 [M]+: 320.0647, found 320.0652.
3.3.6. 3,3,4,4,5,5,6,6,6-Nonafluoro-1-(4-methoxyphenyl) hexan-1-ol (4ae)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (46.4 mg, 42%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, dd, J = 9.0 Hz), 5.16 (1H, dd, J = 8.5, 3.5 Hz), 3.81 (3H, s), 2.68–2.55 (1H, m), 2.45–2.34 (1H, m), 2.17 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.7, 134.9, 127.1 (2C), 119.5–108.7 (4C, m), 114.3 (2C), 67.7, 55.5, 39.7 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, dd, J = 235.3, 47.1 Hz), −114.4 (1F, dd, J = 235.3, 47.1 Hz), −124.9 (2F, s), −126.2; IR; 3426, 2842, 2248, 1614, 1515, 1132, 1132, 1034, 881; HRMS (EI+) calcd for C13H11O2F9 [M]+: 370.0615, found 370.0617.
3.3.7. 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-Heptadecafluoro-1-(4-methoxyphenyl) decan-1-ol (4af)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (78.1 mg, 47%); 1H NMR (500 MHz, CDCl3) δ 7.31 (2H, d, J = 8.5 Hz), 6.91 (2H, dd, J = 8.5 Hz), 5.17 (1H, dt, J = 5.5, 3.0 Hz), 3.81 (3H, s), 2.68–2.56 (1H, m), 2.45–2.34 (1H, m), 2.15 (1H, s); 13C NMR (151 MHz, CDCl3) δ 159.3, 134.9, 127.1 (2C), 119.7–108.4 (8C, m), 114.3 (2C), 67.7, 55.5, 39.9 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.0 (1F, d, J = 282.3 Hz), −114.3 (1F, d, J = 282.3 Hz), −122.1 (2F, s), −122.4 (4F, s), −123.2 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3326, 1517, 1199, 1145, 832; HRMS (EI+) calcd for C17H11O2F17 [M]+: 570.0487, found 570.0480.
3.3.8. 1-(4-tert-Buthoxyphenyl)3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4c)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (88.9 mg, 58%); 1H NMR (500 MHz, CDCl3) δ 7.27 (2H, d, J = 8.5 Hz), 6.98 (2H, d, J = 8.5 Hz), 5.18 (1H, d, J = 9.0 Hz), 2.68–2.56 (1H, m), 2.46–2.35 (1H, m), 2.28 (1H, s), 1.34 (3H, s); 13C NMR (151 MHz, CDCl3) δ 155.5, 137.6, 126.4 (2C), 124.6 (2C), 120.2–110.4 (6C, m), 79.0, 67.7, 39.9 (t, J = 21.5 Hz), 29.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.4 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.7 (2F, s); IR; 3432, 2981, 1608, 1508, 1234, 1191, 707; HRMS (ESI+) calcd for C18H17O2F13 [M+Na]+: 535.0913, found 535.0913.
3.3.9. 1-(4-(tert-butyl)phenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4d)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (52.3 mg, 35%); 1H NMR (500 MHz, CDCl3) δ 7.41 (2H, d, J = 9.0 Hz), 7.32 (2H, d, J = 8.0 Hz), 5.20 (1H, dd, J = 9.0, 3.0 Hz), 2.69–2.56 (1H, m), 2.47–2.36 (1H, m), 2.18 (1H), 1.32 (9H, s); 13C NMR (151 MHz, CDCl3) δ 151.7, 139.8, 130.2, 128.4, 126.0, 125.5, 119.7–109.4 (6C, m), 67.9, 39.9 (J = 20.4 Hz), 34.7, 31.4 (3C); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.5 (1F, d, J = 282.3 Hz), −123.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3400, 2966, 2908, 2872, 1512, 1364, 1235, 1143, 836, 731, 707; HRMS (EI+) calcd for C18H17OF13 [M]+: 496.1072, found 496.1085.
3.3.10. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-toyl) octan-1-ol (4e)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (30.6 mg, 22%); 1H NMR (400 MHz, CDCl3) δ 7.29 (2H, d, J = 8.4 Hz), 7.20 (2H, d, J = 8.0 Hz), 5.20 (1H, dd, J = 8.8, 3.2 Hz), 2.70–2.55 (1H, m), 2.47–2.32 (1H, m), 2.36 (3H, s), 2.11 (1H, s); 13C NMR (151 MHz, CDCl3) δ 139.9, 138.5, 130.3, 129.7, 125.7, 122.8, 122.8–108.7 (6C, m), 68.0, 39.9 (d, J = 20.4 Hz), 21.3; 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 263.3 Hz), −114.4 (1F, d, J = 263.3 Hz) (1F, s), −123.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3423, 3030, 2928, 1517, 1364, 1232, 1144, 814, 707; HRMS (EI+) calcd for C15H10OF13 [M + H]+: 454.0602, found 454.0602.
3.3.11. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-phenyl octan-1-ol (4f)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (47.5 mg, 36%); 1H NMR (500 MHz, CDCl3) δ 7.39 (4H, d, J = 4.5 Hz), 7.36–7.32 (1H, m), 5.22 (1H, dd, J = 9.0, 3.5 Hz), 2.69–2.57 (1H, m), 2.48–2.36 (1H, m), 2.27 (1H, br); 13C NMR (151 MHz, CDCl3) δ 142.8, 129.0 (2C), 128.6, 125.8 (2C), 120.2–108.7 (6C, m), 68.1, 40.0 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.3 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3409, 3069, 3036, 1232, 1189, 1070, 607; HRMS (EI+) calcd for C14H9OF13 [M]+: 440.0446, found 440.0450.
3.3.12. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-fluorophenyl) octan-1-ol (4g)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (27.4 mg, 20%); 1H NMR (500 MHz, CDCl3) δ 7.40–7.36 (2H, m), 7.10–7.06 (2H, m), 5.23 (1H, dd, J = 8.5, 3.5 Hz), 2.67–2.55 (1H, m), 2.45–2.33 (1H, m), 2.19 (1H, br); 13C NMR (151 MHz, CDCl3) δ 162.7 (d, J = 247.5 Hz), 138.5 (d, J = 3.0 Hz), 127.6 (2C), 118.5–100.3 (6C, m), 118.2 (2C), 67.5, 40.1 (t, J = 21.0 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 282.3 Hz), −114.0 (1F, s), −114.2 (1F, d, J = 282.3 Hz), −122.3 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3436, 1607, 1512, 1228, 1187, 1142, 838, 707; HRMS (EI+) calcd for C14H8O2F14 [M]+: 458.0352, found 458.0334.
3.3.13. 1-(4-Chlorophenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4h)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (36.3 mg, 25%); 1H NMR (400 MHz, CDCl3) δ 7.35 (4H, dd, J = 13.2, 8.8 Hz), 5.22 (1H, d, J = 8.8 Hz), 2.63–2.57 (1H, m), 2.44–2.33 (1H, m), 2.20 (1H, s); 13C NMR (151 MHz, CDCl3) δ 141.1, 134.3, 129.2 (2C), 127.2 (2C), 119.6–108.4 (6C, m), 67.5, 40.0 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 263.3 Hz), −114.2 (1F, d, J = 263.3 Hz), 122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3413, 1494, 1364, 1232, 1187, 1144, 831, 699; HRMS (EI+) calcd for C14H8OClF13 [M]+: 474.0056, found 474.0062.
3.3.14. 1-([1,1′-biphenyl]-4-yl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4i)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). White solid (35.0 mg, 27%); 1H NMR (400 MHz, CDCl3) δ7.62–7.57 (4H, m), 7.50–7.43 (4H, m), 7.38–7.34 (1H, m), 5.29–5.25 (1H, m), 2.74–2.56 (1H, m), 2.53–2.39 (1H, m), 2.29 (1H, d, J = 1.8 Hz); 13C NMR (151 MHz, CDCl3) δ 141.7, 141.6, 140.6, 132.6, 130.2, 129.0, 128.4, 127.8, 127.7, 127.3, 126.2, 120.2–108.4 (6C, m), 67.9, 40.0 (t, J = 20.4 Hz); 19F NMR (471 MHz, CDCl3) δ −81.2 (3F, s), −112.8 (1F, d, J = 134.9 Hz), −114.2 (1F, d, J = 134.9 Hz), −122.2 (2F, s), −123.3 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 3299, 1234, 1189, 1144, 1007, 839, 766, 692, 651; HRMS (ESI+) calcd for C20H13OF13 [M+Na]+: 539.0651, found 539.0644.
3.3.15. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-methoxyphenyl) octan-1-ol (4j)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (46.8 mg, 33%); 1H NMR (400 MHz, CDCl3) δ 7.40 (1H, t, J = 7.3 Hz), 6.96 (1H, d, J = 3.2 Hz), 6.95 (1H, s), 5.19 (1H, dd, J = 8.8, 3.2), 3.82 (3H, s), 2.72–2.53 (2H, m), 2.48–2.31 (1H, m); 13C NMR (151 MHz, CDCl3) δ 160.1, 144.4, 130.1, 120.3–109.2 (6C, m), 117.9, 113.9, 111.3, 68.0, 55.4, 40.0 (t, J = 21.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.2 (3F, s), −112.8 (1F, d, J = 263.3 Hz), −114.3 (1F, d, J = 263.3 Hz), −122.2 (2F, s), −123.2 (2F, s), −124.0 (2F, s), −126.5 (2F, s); IR; 3495, 2963, 2842, 1603, 1364, 1221, 1144, 1047, 786, 696; HRMS (EI+) calcd for C15H11O2F13 [M]+: 470.0551, found 470.0559.
3.3.16. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-toyl) octan-1-ol (4k)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (56.6 mg, 42%); 1H NMR (400 MHz, CDCl3) δ 7.54 (1H, dd, J = 7.6, 1.2 Hz), 7.23 (1H, m), 5.48 (1H, dd, J = 8.8, 1.6), 2.68–2.49 (2H, m), 2.42–2.52 (1H, m), 2.34 (1H, s), 2.15 (1H, br); 13C NMR (151 MHz, CDCl3) δ 140.9, 134.0, 130.9, 128.2, 126.9, 125.2, 120.2–108.4 (6C, m), 64.3, 39.1, 18.9; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.3 (1F, d, J = 263.3 Hz), −117.7 (1F, d, J = 263.3 Hz), −122.2 (2F, s), −123.2 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3416, 3029, 2957, 1364, 1232, 1187, 1144, 812, 707; HRMS (EI+) calcd for C15H11OF13 [M]+: 454.0602, found 454.0601.
3.3.17. 1-(3,4-Dimethoxyphenyl)-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-1-ol (4l)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (85.0 mg, 68%); 1H NMR (400 MHz, CDCl3) δ 6.93–6.85 (3H, m), 5.18 (1H, dd, J = 6.18, 2.52 Hz), 3.91 (3H, s), 3.89 (3H, s), 2.70–2.55 (1H, m), 2.47–2.33 (1H, m), 2.18 (1H, s); 13C NMR (151 MHz, CDCl3) δ149.4, 149.1, 135.4, 119.9–109.1 (6C, m), 118.0, 111.2, 108.6, 68.0, 56.1, 56.0, 40.0 (t, J = 21.1 Hz), 29.9; 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.9 (1F, d, J = 289.0 Hz), −114.4 (1F, d, J = 289.0 Hz), −122.3 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3498, 2841, 1518, 1231, 1188, 1141, 1116, 1026, 809, 765, 735, 707; HRMS (ESI+) calcd for C16H13O3F13 [M + Na]+: 523.0549, found 523.0557.
3.3.18. 2-(1,1,2,2,3,3,4,4,5,5,6,6,6-Tridecafluorohexyl)-1,2,3,4-tetrahydronaphthalen-1-ol (4m)
- The compound was synthesized as described in oxidative reaction conditions (62:38 diastereomer mixture, dr was measured by crude 1H NMR). The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (43.5 mg, 31%); 1H NMR (400 MHz, CDCl3) δ 7.50–7.11 (major and minor, 4H, m), 5.14 (major, 1H, t, J = 6.5 Hz), 5.10 (minor, 1H, s), 3.07–2.52 (major and minor, 3H, m), 2.33–2.09 (major and minor, 2H, m), 1.92–1.83 (major and minor, 1H, m); 13C NMR (151 MHz, CDCl3) δ 136.7 (minor), 136.4 (major), 132.6 (minor), 130.2 (major), 129.3 (minor), 129.0 (minor), 128.8 (major), 128.5 (major), 128.4 (minor), 128.2 (major), 127.0 (major), 126.8 (minor), 121.0–106.9 (major and minor, 6C, m), 66.9 (major), 66.1 (minor), 45.6 (major, t, J = 19.6 Hz), 42.4 (minor, t, J = 19.6 Hz), 28.5 (minor), 27.5 (major), 20.6 (major), 16.2 (minor); 19F NMR (471 MHz, CDCl3) δ −81.2 (major and minor, 3F, s), −113.5 (major, 1F, d, J = 288.8 Hz), −114.4 (minor, 1F, d, J = 288.8 Hz), −116.0 (minor, 1F, d, J = 288.8 Hz), −117.0 (major, 1F, d, J = 288.8 Hz), −120.5–−124.1 (major and minor, 6F, m), −125.7–−127.4 (major and minor, 2F, m); IR; 3447, 3063, 3013, 2986, 2936, 2884, 1491, 1451, 1227, 1190, 1179, 1142, 1121, 1045, 743; HRMS (EI+) calcd for C16H11OF13 [M]+: 466.0602, found 466.0565.
3.3.19. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(4-(trifluoromthyl)phenyl) octan-1-ol (4n)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (25.9 mg, 17%); 1H NMR (400 MHz, CDCl3) δ 7.66 (2H, d, J = 8.0 Hz), 7.54 (2H, d, J = 8.0 Hz), 5.31 (1H, d, J = 8.6 Hz), 2.68–2.56 (1H, m), 2.48–2.36 (1H, m), 2.28 (1H, s); 13C NMR (151 MHz, CDCl3) δ: 146.3, 132.6, 130.8 (dd, J = 64.9, 33.2 Hz), 130.2, 128.3, 126.2, 126.0 (q, J = 3.0 Hz), 121.3–106.5 (6C, m), 67.5, 40.1 (t, J = 21.1 Hz); 19F NMR (376 MHz, CDCl3) δ −63.1 (3F, s), −81.2 (3F, s), −112.8 (1F, d, J = 265.8 Hz), −114.0 (1F, d, J = 265.8 Hz), −122.2 (2F, s), −123.3 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 3443, 1326, 1237, 1129, 1069, 1017, 844, 707; HRMS (EI+) calcd for C15H8OF16 [M]+: 508.0320, found 508.0320.
3.3.20. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-(2,3,4,5,6-pentafluorophenyl) octan-1-ol (4o)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (27.2 mg, 17%); 1H NMR(500 MHz, CDCl3) δ 5.60 (1H, dd, J = 12.5, 6.0 Hz), 3.00–2.89 (1H, m), 2.72–2.61 (1H, m), 2.47 (1H, d, J = 6.5 Hz); 13C NMR (151 MHz, CDCl3) δ; 145.7, 144.1, 138.6 (2C), 137.0 (2C), 119.2–106.7 (6C, m), 59.4, 37.3 (t, J = 19.5 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.8 (1F, d, J = 282.3 Hz), −114.7 (1F, d, J = 282.3 Hz) −122.3 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s), −143.7 (2F, d, J = 47.1 Hz), −153.4 (1F, d, J = 47.1 Hz), 161.2 (2F, s); IR; 3486, 1656, 1523, 1504, 1235, 1192, 1142, 996, 947, 700; HRMS (EI+) calcd for C14H4OF18 [M]+: 529.9975, found 529.9976.
3.3.21. 3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1,1-diphenyloctan-1-ol (4p)
- The compound was synthesized as described in oxidative reaction conditions. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Yellow oil (60.3 mg, 43%); 1H NMR (400 MHz, CDCl3) δ 7.45–7.42 (4H, m), 7.36–7.31 (4H, m), 7.28–7.24 (2H, m), 3.17 (2H, t, J = 18.3 Hz), 2.74 (1H, t, J = 2.1 Hz); 13C NMR (151 MHz, CDCl3) δ: 145.5 (2C), 137.7, 132.6, 130.2, 128.6 (2C), 128.4, 127.7 (2C), 125.5 (2C), 120.1–108.4 (6C, m), 76.6, 41.0 (t, J = 19.6 Hz); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −109.5 (2F, s), −122.1 (2F, s), −123.3 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 3470, 3088, 3061, 2971, 3028, 1655, 1495, 1233, 1188, 1142, 1121, 812, 696; HRMS (ESI−) calcd for C20H13OF13 [M − H]−: 515.0681, found 515.0641.
3.3.22. 4-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl)benzonitrile (5b)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (96.5 mg, 72%); 1H NMR (400 MHz, CDCl3) δ 7.64–7.61 (2H, m), 7.34 (2H, d, J = 8.2 Hz), 3.01–2.98 (2H, m), 2.46–2.33 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.7, 132.8 (2C), 129.3 (2C), 119.8–108.4 (6C, m), 118.8, 111.0, 32.4 (t, J = 22.7 Hz), 26.8 (t, J = 4.5 Hz); 19F NMR (376 MHz, CDCl3) δ −81.2 (3F, s) −115.0 (2F, s), −122.3 (2F, s), −123.3 (2F, s), −123.9 (2F, s), −126.6 (2F, s); IR; 3062, 2227, 1185, 1165, 1119, 1086, 1072, 979, 908, 862, 812, 736, 704; HRMS (ESI+) calcd for C15H8F13N [M + Na]+: 472.0347, found 472.0347.
3.3.23. 4-(3,3,4,4,5,5,5-Heptafluoropentyl)benzonitrile (5bc)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (57.4 mg, 64%); 1H NMR (400 MHz, CDCl3) δ 7.63 (2H, dd, J = 6.6, 1.6 Hz), 7.33 (2H, d, J = 8.2 Hz), 3.02–2.97 (2H, m), 2.45–2.32 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.6, 132.7 (2C), 129.3 (2C), 120.7–107.0 (3C, m), 118.8, 111.0, 32.1 (t, J = 22.7 Hz), 26.7 (t, J = 3.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.0 (3F, s) −116.0 (2F, s), −128.2 (2F, s); IR; 2961, 2231, 1352, 1220, 1169, 1112, 1085, 947, 910, 824, 743, 680; HRMS (ESI+) calcd for C12H8F7N [M + Na]+: 322.0442, found 322.0443.
3.3.24. 4-(3,3,4,4,5,5,6,6,6-Nonafluorohexyl)benzonitrile (5be)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (76.0 mg, 73%); 1H NMR (400 MHz, CDCl3) δ 7.65–7.62 (2H, m), 7.34 (2H, d, J = 8.2 Hz), 3.02–2.98 (2H, m), 2.46–2.33 (2H, d); 13C NMR (151 MHz, CDCl3) δ: 144.6, 132.8 (2C), 129.3 (2C), 120.3–108.7 (4C, m), 118.8, 111.0, 32.3 (t, J = 22.7 Hz), 26.8 (t, J = 3.0 Hz); 19F NMR (376 MHz, CDCl3) δ −81.5 (3F, s) −115.2 (2F, s), −124.9 (2F, s), −126.5 (2F, s); IR; 2960, 2231, 1357, 1217, 1131, 1088, 1009, 976, 881, 824, 722, 692; HRMS (ESI+) calcd for C13H8F9N [M + Na]+: 372.0411, found 372.0411.
3.3.25. 1-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl)-4-(trifluoromethyl)benzene (5n)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (63.8 mg, 65%); 1H NMR (500 MHz, CDCl3) δ 7.59 (2H, d, J = 8.0 Hz), 7.34 (2H, d, J = 8.0 Hz), 2.99 (2H, tt, J = 8.5, 3.0 Hz), 2.39 (2H, qt, J = 19.0, 18.5 Hz); 13C NMR (151 MHz, CDCl3) δ143.3, 129.3 (q, J = 33.2 Hz), 128.8, 125.9 (q, J = 3.0 Hz), 119.9–108.4 (6C, m), 32.7 (t, J = 21.1 Hz), 26.5 (t, J = 4.5 Hz); 19F NMR (471 MHz, CDCl3) δ −62.3 (3F, s), −81.3 (3F, s) −115.0 (2F, d, J = 272.5), −122.4 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s); IR; 1612, 1422, 1325, 1235, 1166, 1069, 1020, 826, 707; HRMS (APCI+) calcd for C15H8F16 [M]+: 492.0370 found, 492.0370.
3.3.26. 1,2,3,4,5-Pentafluoro-6-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)benzene (5o)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (54.5 mg, 53%); 1H NMR (500 MHz, CDCl3) δ 2.99 (2H, t, J = 7.5 Hz), 2.38 (2H, qt, J = 18.0, 18.0 Hz); 13C NMR (150 MHz, CDCl3) δ 146.1–136.8 (6C, m), 120.2–108.4 (6C, m), 30.2 (t, J = 22.6 Hz), 14.0; 19F NMR (471 MHz, CDCl3) δ −81.2 (3F, s), −115.8 (2F, d, J = 272.5), −122.4 (2F, s), −123.4 (2F, s), −124.0 (2F, s), −126.6 (2F, s), −144.3 (2F,s), −156.0 (2F,s), −162.2 (2F,s); IR; 1659, 1522, 1506, 1232, 1191, 1144, 1013, 968, 697; HRMS (FD+) calcd for C14H4F18 [M − H]+: 512.9947 found 512.9938.
3.3.27. (3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctane-1,1-diyl)dibenzene (5p)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (64.0 mg, 64%); 1H NMR (500 MHz, CDCl3) δ 7.32–7.26 (8H, m), 7.24–7.19 (2H, m), 4.46 (1H, t, J = 7.2), 2.92 (2H, td, J = 28.4, 11.2); 13C NMR (151 MHz, CDCl3) δ 143.2 (2C), 128.9 (4C), 127.6 (4C), 127.0 (2C), 120.2–108.4 (6C, m), 43.8, 36.3 (t, J = 19.6 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −113.3 (2F, s), −122.2 (2F, s), −123.3 (2F, s), 124.0 (2F, s), −126.6 (2F, s); IR; 3090, 3066, 3032, 1601, 1495, 1364, 1235, 1186, 1144, 696; HRMS (ESI+) calcd for C20H13F18 [M + Na]+: 523.0702 found 523.0702.
3.3.28. 1-Nitro-4-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)benzene (5q)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Yellow oil (19.7 mg, 21%); 1H NMR (500 MHz, CDCl3) δ 8.20 (2H, d, J = 8.4, 2.8 Hz), 7.40 (2H, d, J = 8.8 Hz), 3.05 (2H, tt, J = 8.4, 2.4 Hz), 2.42 (2H, qt, J = 17.6, 18.8 Hz); 13C NMR (151 MHz, CDCl3) δ 147.1, 146.7, 129.4 (2C), 124.2 (2C), 119.6–105.8 (6C, m), 32.4 (t, J = 21.0 Hz), 26.6; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s) −115.0 (2F, s), −122.4 (2F, s), −123.4 (2F, s), −123.9 (2F, s), −126.7 (2F, s); IR; 2961, 1611, 1534, 1349, 1198, 1138, 1073, 977, 855, 782, 752, 693; HRMS (ESI+) calcd for C14H8NO2F13 [M + Na]+: 492.0240 found, 492.0240.
3.3.29. 1-Methoxy-4-(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluoro-1-methoxyoctyl)benzene (6a)
- The compound was synthesized as described in oxidative reaction conditions using methanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (71.0 mg, 48%); 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, d, J = 8.8 Hz), 6.92 (2H, d, J = 8.8 Hz), 4.52 (1H, dd, J = 8.4, 4.0 Hz), 3.82 (3H, s), 3.20 (3H, s), 2.71–2.55 (1H, m), 2.39–2.25 (1H, m); 13C NMR (151 MHz, CDCl3) δ 159.7, 132.4, 127.9 (2C), 119.3–111.0 (6C, m), 114.4 (2C), 76.4, 56.5, 55.3, 39.3 (t, J = 21.0); 19F NMR (376 MHz, CDCl3) δ −81.3 (3F, s), −112.7 (1F, d, J = 225.7 Hz), −114.0 (1F, d, J = 225.7 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.2 (2F, s), −126.6 (2F, s); IR; 3002, 2940, 2841, 1613, 1514, 1144, 1036, 707; HRMS (EI+) calcd for C16H13O2F13 [M]+: 484.0708, found 484.0732.
3.3.30. 1-(1-Ethoxy-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-4-methoxybenzene (6b)
- The compound was synthesized as described in oxidative reaction conditions using ethanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (77.3 mg, 52%); 1H NMR (400 MHz, CDCl3) δ 7.26 (2H, d, J = 8.4 Hz), 6.91 (2H, d, J = 8.8 Hz), 4.63 (1H, dd, J = 8.4, 4.0 Hz), 3.82 (3H, s), 3.20 (3H, s), 3.41–3.29 (2H, m), 2.71–2.56 (1H, m), 2.38–2.24 (1H, m), 1.17 (3H, t, J = 6.8 Hz); 13C NMR (151 MHz, CDCl3) δ 159.6, 133.2, 127.7, 119.3–109.2 (6C, m), 114.2, 74.5, 64.2 55.4, 39.4 (t, J = 21.0 Hz), 15.2; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.7 (2F, d, J = 300.9 Hz), −113.9 (2F, d, J = 300.9 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 2981, 1613, 1514, 1235, 1102, 1037, 832, 707; HRMS (APCI+) calcd for C17H15O2F13 [M]+: 498.0864; found 498.0859.
3.3.31. 1-(1-Butoxy-3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)-4-methoxybenzene (6c)
- The compound was synthesized as described in oxidative reaction conditions using n-butanol instead of water. The product was purified by column chromatography (Hexane/EtOAc = 10:1). Colorless oil (75.8 mg, 47%); 1H NMR (500 MHz, CDCl3) δ 7.26 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 8.5 Hz), 4.61 (1H, dd, J = 8.5, 3.5 Hz), 3.82 (3H, s), 3.32 (3H, s), 3.41–3.23 (2H, m), 2.66–2.57 (1H, m), 2.33–2.24 (1H, m), 1.54–1.48 (1H, m), 1.38–1.31 (1H, m), 0.87 (3H, t, J = 7.0 Hz); 13C NMR (151 MHz, CDCl3) δ 159.6, 133.2, 127.7 (2C), 119.3–110.4 (6C, m), 114.2 (2C), 74.8, 68.7, 55.4, 39.4 (t, J = 4.5 Hz), 31.9, 19.4, 13.0; 19F NMR (471 MHz, CDCl3) δ −81.3 (3F, s), −112.6 (2F, d, J = 272.5 Hz), −114.0 (2F, d, J = 272.5 Hz), −122.3 (2F, s), −123.4 (2F, s), −124.1 (2F, s), −126.6 (2F, s); IR; 2963, 2938, 2877, 1613, 1514, 1144, 1102, 1037, 707; HRMS (ESI+) calcd for C19H19O2F13 [M + Na]+: 549.1070 found, 549.1078.
3.3.32. (1,1,1,4,4,5,5,6,6,7,7,8,8,9,9,9-Hexadecafluorononan-2-yl)benzene (8)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (20.0 mg, 20%); 1H NMR (500 MHz, CDCl3) 7.55–7.28 (5H, m), 3.78–3.71 (1H, m), 2.84–2.68 (2H, m); 13C NMR (151 MHz, CDCl3) δ 155.6 (t, J = 294.5 Hz), 132.5 (t, J = 4.5 Hz), 129.1 (t, J = 9.1 Hz), 128.8, 128.7, 128.3, 128.2, 120.6–108.4 (6C, m), 83.7 (t, J = 19.6 Hz), 30.7 (t, J = 22.7 Hz); 19F NMR (471 MHz, CDCl3) −71.0 (3F, s), −81.3 (3F, s), −112.4 (2F, d, J = 272.7 Hz), −114.7 (2F, d, J = 272.7 Hz), −122.4 (2F, s), −123.4 (2F, s), −123.9 (2F, s), −126.7 (2F, s); IR; 1232, 1187, 1141, 1119, 1033, 845, 812, 778, 755, 744; HRMS (FD+) calcd for C15H8F16 [M − H]+: 491.0293, found 491.0289.
3.3.33. (1,1,4,4,5,5,6,6,7,7,8,8,9,9,9-Pentadecafluoronon-1-en-2-yl)benzene (9)
- The compound was synthesized as described in reductive reaction conditions. The product was purified by column chromatography (Hexane). Colorless oil (49.0 mg, 51%); 1H NMR (500 MHz, CDCl3) δ 7.46–7.26 (5H, m), 3.21 (2H, t, J = 17.5 Hz); 13C NMR (151 MHz, CDCl3) δ 129.1 (2C), 128.3 (2C), 128.8, 128.3, 128.2, 120.6–108.2 (7C, m), 43.6 (q, J = 27.2 Hz), 31.0 (t, J = 28.7 Hz); 19F NMR (471 MHz, CDCl3) δ −81.3 (3F), −86.0 (1F, d, J = 57.9 Hz), −86.4 (1F, d, J = 27.3 Hz), −122.7 (2F, s), −122.3 (2F, s), −123.4 (2F, s),−123.7 (2F, s), −126.6 (2F, s); IR; 1736, 1232, 1197, 1139, 868, 847, 812, 799, 778, 755, 707, 696, 663; HRMS (FD+) calcd for C15H7F15 [M]+: 472.0308, found 472.0327.
4. Conclusions
In conclusion, we have achieved the hydroxy- and hydro-perfluoroalkylation of styrenes by controlling the quenching cycle of eosin Y through additive selection. In the oxidative quenching cycle with sodium thiosulfate addition, the reaction of perfluoroalkyl iodide with styrene had electron-donating substituents, producing hydroxy-perfluoroalkylated products. Conversely, in the reductive quenching cycle where amines were added, the reaction of perfluoroalkyl bromide with styrene had electron-withdrawing substituents and yielded hydro-perfluoroalkylated products. These reactions utilize the properties of the organic dye eosin Y and use water as the hydroxyl group or hydrogen source. The reagents used in these reactions are inexpensive and readily available and do not use heavy metals or other harmful reagents, making these reactions environmentally adaptive.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28227577/s1, Scheme S1: Reactions using different perfluoroalkyl halide; Scheme S2: Reactions in the absence of H2O. 1H, 13C and 19F NMR spectra of products 4a, 4c–4p, 4aa–4af, 5b, 5n–5q, 5bc–5be, 6a–c, 8, and 9; Table S1: Oxidative and Reductive reactions with various styrenes; Table S2: Investigation of the number of equivalents of Na2S2O3; Table S3: Investigation of reaction time for oxidative reactions in the absence of K2CO3; Table S4: Investigation of reaction condition for reductive reaction conditions.
Author Contributions
Conceptualization, T.K. and T.Y.; methodology, H.S. and M.N.; validation, H.S. and K.T.; investigation, H.S., M.N. and K.T.; writing—original draft preparation, T.Y.; writing—review and editing, T.Y.; visualization, T.Y.; supervision, T.Y.; project administration, T.Y.; funding acquisition, T.Y. (MEXT) H.S and K.T. (Avanade). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Grant-in-Aid for Transformative Research Areas (A), MEXT, Japan, grant number JP21H05219 and Ochanomizu University-Avanade Research Scholarship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material.
Acknowledgments
We are thankful for financial support from MEXT, Japan (Grant-in-Aid for Transformative Research Areas (A), JP21H05219). We also thank Avanard Japan for providing H.S. and K.T. with Ochanomizu University-Avanade Research Scholarship We are grateful to Tosoh Finechem Co. for their generous donation of trifluoromethyl iodide.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2022, 122, 167–208. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Application; Wiley-VCH: Weinhein, Germany, 2013. [Google Scholar]
- Ameduri, B. Fluoropolymers: The Right Material for the Right Applications. Chem. Eur. J. 2018, 24, 18830–18841. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr. Structure, Reactivity, and Chemistry of Fluoroalkyl Radicals. Chem. Rev. 1996, 96, 1557–1584. [Google Scholar] [CrossRef]
- Tsuchii, K.; Imura, M.; Kamada, N.; Hirao, T.; Ogawa, A. An Efficient Photoinduced Iodoperfluoroalkylation of Carbon−Carbon Unsaturated Compounds with Perfluoroalkyl Iodides. J. Org. Chem. 2004, 69, 6658–6665. [Google Scholar] [CrossRef]
- Murphy, O.M.; Baldwin, C.S.; Buck, R.C. Synthesis utilizing n-perfluorlakyl iodides [RFI, CnF2n+1-I] 2000–2010. J. Fluor. Chem. 2012, 138, 3–23. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Cooke, M.V.; Postigo, A. Radical Fluoroalkylation Reactions. ACS Catal. 2018, 8, 7287–7307. [Google Scholar] [CrossRef]
- Tagami, K.; Yajima, T. Development of Electrophilic Radical Perfluoroalkylation of Electron-Deficient Olefins. Chem. Rec. 2023, 23, e202300037. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ohkoshi, M.; Aoki, N.; Ohnuma, Y.; Iyoda, M. Photochemical Oxyfluoroalkylation of Styrenes by the Addition of Perfluoroalkyl Radicals in an Atomosphere of Oxyfem. Tetrahedron Lett. 1999, 40, 5731–5734. [Google Scholar] [CrossRef]
- Yasu, Y.; Koike, T.; Akita, M. Three-Component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts. Angew. Chem. Int. Ed. 2012, 51, 9567–9571. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.W.; Douglas, J.J.; Cole, K.P.; Stephenson, C.R.J. A Scalable and Operationally Simple Radical Trifluoromethylation. Nat. Commun. 2015, 6, 7919. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Lin, F.; Wang, X.; Jiao, N. Photoredox-Catalyzed Hydroxyfluoroalkylation of Alkene with Simple Fluoroalkyl Iodides. J. Photochem. Photobiol. A Chem. 2018, 355, 194–201. [Google Scholar] [CrossRef]
- Jud, W.; Kappe, C.O.; Cantillo, D. Catalyst-Free Oxytrifluoromethylation of Alkenes through Paired Electrolysis in Organic-Aqueous Media. Chem. Eur. J. 2018, 24, 17234–17238. [Google Scholar] [CrossRef]
- Chen, T.; Guo, Y.; Sun, K.; Wu, L.Z.; Liu, W.Q.; Liu, C.; Huang, Y.; Chen, Q.Y. Photoinduced Hydroxylperfluoroalkylation of Styrenes. Org. Chem. Front. 2018, 5, 1045–1048. [Google Scholar] [CrossRef]
- Su, Z.; Guo, Y.; Chen, Q.-Y.; Zhan, Z.-G.; Nian, B.-Y. Catalyst-Free Hydrozytrifluoromethylation of Alkenes Using Iodotrifluoromethane. Chin. J. Chem. 2019, 37, 597–604. [Google Scholar] [CrossRef]
- Shen, W.G.; Wu, Q.Y.; Gong, X.Y.; Ao, G.Z.; Liu, F. A Facile Method for Hydroxytrifluoromethylation of Alkenes with Langlois Reagent and DMSO. Green Chem. 2019, 21, 2983–2987. [Google Scholar] [CrossRef]
- Li, Q.; Fan, W.; Peng, D.; Meng, B.; Wang, S.; Huang, R.; Liu, S.; Li, S. Cobalt-Tertiary-Amine-Mediated Hydroxytrifluoromethylation of Alkenes with CF3Br and Atmospheric Oxygen. ACS Catal. 2020, 10, 4012–4018. [Google Scholar] [CrossRef]
- Quan, Y.; Shi, W.; Song, Y.; Jiang, X.; Wang, C.; Lin, W. Bifunctional Metal-Organic Layer with Organic Dyes and Iron Centers for Synergistic Photoredox Catalysis. J. Am. Chem. Soc. 2021, 143, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Ofuji, Y.; Kanbaa, T.; Yajima, T. Metal-free visible-light-induced hydroxyperfluoroalkylation of conjugated olefins using enamine catalyst. RSC Adv. 2022, 12, 32790–32795. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Yajima, T. Halogen-Bond-Promoted Hydroxyperfluoroalkylation of Olefins with Molecular Oxygen under Visible-Light Irradiation. Asian J. Org. Chem. 2023, 12, e202300273. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Postigo, A. (Me3Si)3SiH-Mediated Intermolecular Radical Perfluoroalkylation Reactions of Olefins in Water. J. Org. Chem. 2010, 75, 6141–6148. [Google Scholar] [CrossRef] [PubMed]
- Wilger, D.J.; Gesmundo, N.J.; Nicewicz, D.A. Catalytic Hydrotrifluoromethylation of Styrenes and Unactivated Aliphatic Alkenes via an Organic Photoredox System. Chem. Sci. 2013, 4, 3160–3165. [Google Scholar] [CrossRef]
- Choi, S.; Kim, Y.J.; Kim, S.M.; Yang, J.W.; Kim, S.W.; Cho, E.J. Hydrotrifluoromethylation and Iodotrifluoromethylation of Alkenes and Alkynes Using an Inorganic Electride as a Radical Generator. Nat. Commun. 2014, 5, 4881. [Google Scholar] [CrossRef]
- Straathof, N.J.W.; Cramer, S.E.; Hessel, V.; Noël, T. Practical Photocatalytic Trifluoromethylation and Hydrotrifluoromethylation of Styrenes in Batch and Flow. Angew. Chem. Int. Ed. 2016, 128, 15778–15782. [Google Scholar] [CrossRef]
- Peng, D.; Fan, W.; Zhao, X.; Chen, W.; Wen, Y.; Zhang, L.; Li, S. Zinc-Brønsted Acid Mediated Practical Hydrotrifluoromethylation of Alkenes with CF3Br. Org. Chem. Fron. 2021, 8, 6356–6363. [Google Scholar] [CrossRef]
- Jia, H.; Häring, A.P.; Berger, F.; Zhang, L.; Ritter, T. Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3-Reactivity. J. Am. Chem. Soc. 2021, 143, 7623–7628. [Google Scholar] [CrossRef]
- Yang, Y.F.; Lin, J.H.; Xiao, J.C. Starting from Styrene: A Unified Protocol for Hydrotrifluoromethylation of Diversified Alkenes. Org. Lett. 2021, 23, 9277–9282. [Google Scholar] [CrossRef] [PubMed]
- Louvel, D.; Souibgui, A.; Taponard, A.; Rouillon, J.; ben Mosbah, M.; Moussaoui, Y.; Pilet, G.; Khrouz, L.; Monnereau, C.; Vantourout, J.C.; et al. Tailoring the Reactivity of the Langlois Reagent and Styrenes with Cyanoarenes Organophotocatalysts under Visible-Light. Adv. Synth. Catal. 2022, 364, 139–148. [Google Scholar] [CrossRef]
- Haria, D.P.; König, B. Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun. 2014, 50, 6688–6699. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Singh, P.P. Eosin Y Catalysed Photoredox Synthesis: A Review. RSC Adv. 2017, 7, 31377–31392. [Google Scholar] [CrossRef]
- Yajima, T.; Ikegami, M. Metal-Free Visible-Light Radical Iodoperfluoroalkylation of Terminal Alkenes and Alkynes. Eur. J. Org. Chem. 2017, 15, 2126–2129. [Google Scholar] [CrossRef]
- Yajima, T.; Shigenaga, S. Metal-Free Visible Light Hydroperfluoroalkylation of Unactivated Alkenes Using Perfluoroalkyl Bromides. Org. Lett. 2019, 21, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, S.; Shibata, H.; Tagami, K.; Kanbara, T.; Yajima, T. Eosin Y catalyzed Visible-Light-Induced Hydroperfluoroalkylation of Electron-Deficient Alkenes. J. Org. Chem. 2022, 87, 14923–14929. [Google Scholar] [CrossRef]
- Sun, H.; Huang, C.; Bin, X.; Cui, F.; Jiang, G. Photochemical akynylperfluoroalkylation of unactive alkenes mediated by halogen-bonded charge-transfer complexes. Org. Chem. Fron. 2023, 10, 2332–2339. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, L.; Guo, X.; Chen, J.; Liu, Y.; Wang, L.; Ma, Y. VisibleLight-Drivenα-Diazoketonesas Denitrogenated Synthons: Synthesis of Fluorinated N-Heterocyclesvia Multicomponent Cyclization Reactions. Org. Lett. 2023, 25, 3314–3318. [Google Scholar] [CrossRef]
- Jaffé, H.H. A Reëxamination of the Hammett Equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Experimants Related to Thermodynamics and Kinetics. In Modern Physical Organic Chmistry, 1st ed.; University Science Books: Sausalito, CA, USA, 2005; p. 446. [Google Scholar]
- Bégué, J.-P.; Bonnet-Delpon, D.; Rock, M.H. A concise synthesis of functionalised gem-difluoroalkenes, via the addition of organolithium reagents to α-trifluoromethylstyrene. Tetrahedron Lett. 1995, 36, 5003–5006. [Google Scholar]
- Tian, F.; Yan, G.; Yu, J. Recent advances in the synthesis and applications of a-(trifluoromethyl)styrenes in organic synthesis. Chem. Commun. 2019, 55, 13486–13505. [Google Scholar] [CrossRef]
- Yan, G.; Qiu, K.; Guo, M. Recent advance in the C–F bond functionalization of trifluoromethyl-containing compounds. Org. Chem. Front. 2021, 8, 3915–3942. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.U.; Slanina, T.; Tao, C.-J.; König, B. Metal-Free Perfluoroarylation by Visible Light Photoredox Catalysis. ACS Catal. 2016, 6, 369–375. [Google Scholar] [CrossRef]
- Tang, W.-K.; Feng, Z.-W.; Xu, Z.-W.; Cheng, Z.-F.; Xu, J.; Cai, J.-J.; Xu, H.-J. Visible-Light-Enabled Decarboxylative Mono- and Difluoromethylation of Cinnamic Acids under Metal-Free Conditions. Org. Lett. 2017, 19, 5501–5504. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.D.; Bakshi, D.; Shigh, A. Visible Light Mediated Organocatalytic Activation of EthylBromofluoroacetate: Coupling with Indoles and Anilines. J. Org. Chem. 2015, 80, 10187–10196. [Google Scholar] [CrossRef]
Scheme 1.
Hydroxy- and hydro-perfluoroalkylation of styrenes using perfluroalkyl halide as a fluorine source.
Scheme 1.
Hydroxy- and hydro-perfluoroalkylation of styrenes using perfluroalkyl halide as a fluorine source.

Scheme 2.
Hydroxy- and hydro-perfluoroalkylation of various styrenes. a Oxidative reaction conditions: Eosin Y (10 mol%), 1 (0.25 mmol), 2, Na2S2O3 (0.25 equiv.), and K2CO3 (2.0 equiv.) in CH3CN/H2O (7.5 mL/1 mL) under an argon atmosphere with 3 h irradiation with white LED light at 25 °C. b Oxidative reaction conditions: Eosin Y (10 mol%), 1 (0.25 mmol), 3 and DIPEA (2.0 equiv.) in CH3CN/H2O (7.5 mL/1 mL) under an argon atmosphere with 3 h irradiation with white LED light at 25 °C.
Scheme 2.
Hydroxy- and hydro-perfluoroalkylation of various styrenes. a Oxidative reaction conditions: Eosin Y (10 mol%), 1 (0.25 mmol), 2, Na2S2O3 (0.25 equiv.), and K2CO3 (2.0 equiv.) in CH3CN/H2O (7.5 mL/1 mL) under an argon atmosphere with 3 h irradiation with white LED light at 25 °C. b Oxidative reaction conditions: Eosin Y (10 mol%), 1 (0.25 mmol), 3 and DIPEA (2.0 equiv.) in CH3CN/H2O (7.5 mL/1 mL) under an argon atmosphere with 3 h irradiation with white LED light at 25 °C.

Scheme 3.
Types of perfluoroalkylation.

Scheme 4.
Proposed mechanism for hydroxy- and hydro-perfluoroalkylation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of reaction conditions a.
 | |||||
|---|---|---|---|---|---|
| Entry | 1 | 2 or 3 | Additive | Yield of 4 (%) b | Yield of 5 (%) b |
| 1 | 1a | 2 (1.2 equiv.) | Na2S2O3 (5.0 equiv.) | 40 | n.d. c |
| 2 | 1a | 2 (1.2 equiv.) | Na2S2O3 (5.0 equiv.), K2CO3 (2.0 equiv.) | 53 | n.d. c |
| 3 | 1a | 2 (1.2 equiv.) | Na2S2O3 (0.25 equiv.), K2CO3 (2.0 equiv.) | 70 (50) | n.d. c |
| 4 | 1a | 2 (1.2 equiv.) | K2CO3 (2.0 equiv.) | 20 | n.d. c |
| 5 | 1a | 2 (3.0 equiv.) | Na2S2O3 (0.25 equiv.), K2CO3 (2.0 equiv.) | 66 | n.d. c |
| 6 | 1b | 3 (3.0 equiv.) | DIPEA (2.0 equiv.) | n.d. | 70 (72) |
a Reaction conditions: The mixture of 1 (0.25 mmol), Eosin Y (10 mol%), styrene (2 or 3), and additives in CH3CN/H2O (7.5 mL/1 mL) under an argon atmosphere was irradiated for 3 h with white LED light at 25 °C. b Yields were determined by 19F NMR using PhCF3 as the internal standard. Yields in parentheses are isolated yields. c n.d.; Not detected.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shibata, H.; Nakayama, M.; Tagami, K.; Kanbara, T.; Yajima, T. Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y. Molecules 2023, 28, 7577. https://doi.org/10.3390/molecules28227577
AMA Style
Shibata H, Nakayama M, Tagami K, Kanbara T, Yajima T. Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y. Molecules. 2023; 28(22):7577. https://doi.org/10.3390/molecules28227577
Chicago/Turabian StyleShibata, Haruko, Moeko Nakayama, Koto Tagami, Tadashi Kanbara, and Tomoko Yajima. 2023. "Hydroxy- and Hydro-Perfluoroalkylation of Styrenes by Controlling the Quenching Cycle of Eosin Y" Molecules 28, no. 22: 7577. https://doi.org/10.3390/molecules28227577