

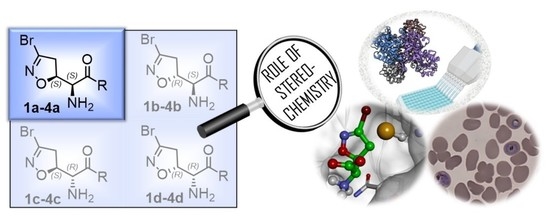
Role of Stereochemistry on the Biological Activity of Nature-Inspired 3-Br-Acivicin Isomers and Derivatives
 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Enantiomerically Pure Unnatural Isomers
2.2. Antimalarial Activity against Plasmodium falciparum
2.3. In Vitro Inhibitory Activity towards PfGAPDH
2.4. Molecular Modeling
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure A
3.1.3. General Procedure B
3.1.4. General Procedure C
3.1.5. General Procedure D
3.1.6. General Procedure E
3.1.7. General Procedure F
3.1.8. tert-Butyl ((R)-1-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-hydroxyethyl)carbamate (7a) and tert-butyl ((R)-1-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-hydroxyethyl)carbamate (7b)
3.1.9. tert-Butyl ((S)-1-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-hydroxyethyl)carbamate (7c) and tert-butyl ((S)-1-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-hydroxyethyl)carbamate (7d)
3.1.10. (S)-2-((S)-3-Bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetic acid (8a)
3.1.11. (S)-2-((R)-3-Bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetic acid (8b)
3.1.12. (R)-2-((S)-3-Bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetic acid (8c)
3.1.13. (R)-2-((R)-3-Bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetic acid (8d)
3.1.14. (S)-2-Amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetic acid (1b)
3.1.15. (R)-2-Amino-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)acetic acid (1c)
3.1.16. (R)-2-Amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetic acid (1d)
3.1.17. Methyl (S)-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (9b)
3.1.18. Methyl (R)-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (9c)
3.1.19. Methyl (R)-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (9d)
3.1.20. Benzyl (S)-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (10b)
3.1.21. Benzyl (R)-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (10c)
3.1.22. Benzyl (R)-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-((tert-butoxycarbonyl)amino)acetate (10d)
3.1.23. tert-Butyl ((S)-2-(benzylamino)-1-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-oxoethyl)carbamate (11b)
3.1.24. tert-Butyl ((R)-2-(benzylamino)-1-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-oxoethyl)carbamate (11c)
3.1.25. tert-Butyl ((R)-2-(benzylamino)-1-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)-2-oxoethyl)carbamate (11d)
3.1.26. Methyl (S)-2-amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (2b)
3.1.27. Methyl (R)-2-amino-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (2c)
3.1.28. Methyl (R)-2-amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (2d)
3.1.29. Benzyl (S)-2-amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (3b)
3.1.30. Benzyl (R)-2-amino-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (3c)
3.1.31. Benzyl (R)-2-amino-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetate (3d)
3.1.32. (S)-2-Amino-N-benzyl-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetamide (4b)
3.1.33. (R)-2-Amino-N-benzyl-2-((S)-3-bromo-4,5-dihydroisoxazol-5-yl)acetamide (4c)
3.1.34. (R)-2-Amino-N-benzyl-2-((R)-3-bromo-4,5-dihydroisoxazol-5-yl)acetamide (4d)
3.2. Molecular Modeling
3.2.1. Conformational Analysis
3.2.2. Docking Studies
3.3. Biological Assays
3.3.1. Expression and Purification of PfGAPDH
3.3.2. Enzyme Assays
3.3.3. Parasite Growth and Drug Susceptibility Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug. Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Aldas-Bulos, V.D.; Medina-Franco, J.L.; Plisson, F. Natural product drug discovery in the artificial intelligence era. Chem. Sci. 2022, 13, 1526–1546. [Google Scholar] [CrossRef]
- Nelson, A.; Karageorgis, G. Natural product-informed exploration of chemical space to enable bioactive molecular discovery. RSC Med. Chem. 2021, 12, 353–362. [Google Scholar] [CrossRef] [PubMed]
- A, N.L.B.; F, M.D.S.; Batista, J.M.; Cass, Q.B. Enantiomeric mixtures in natural product chemistry: Separation and absolute configuration assignment. Molecules 2018, 23, 492. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.A.; Ropek, N.; Melillo, B.; Schreiber, S.L.; Cravatt, B.F.; Vinogradova, E.V. Stereochemical diversity as a source of discovery in chemical biology. Curr. Res. Chem. Biol. 2022, 2, 100028. [Google Scholar] [CrossRef]
- Elder, F.C.T.; Feil, E.J.; Snape, J.; Gaze, W.H.; Kasprzyk-Hordern, B. The role of stereochemistry of antibiotic agents in the development of antibiotic resistance in the environment. Environ. Int. 2020, 139, 105681. [Google Scholar] [CrossRef]
- Miles, B.W.; Thoden, J.B.; Holden, H.M.; Raushel, F.M. Inactivation of the amidotransferase activity of carbamoyl phosphate synthetase by the antibiotic acivicin. J. Biol. Chem. 2002, 277, 4368–4373. [Google Scholar] [CrossRef] [Green Version]
- Denton, J.E.; Lui, M.S.; Aoki, T.; Sebolt, J.; Weber, G. Rapid in vivo inactivation by acivicin of CTP synthetase, carbamoyl-phosphate synthetase II, and amidophosphoribosyltransferase in hepatoma. Life Sci. 1982, 30, 1073–1080. [Google Scholar] [CrossRef]
- Earhart, R.H.; Neil, G.L. Acivicin in 1985. Adv. Enzym. Regul. 1985, 24, 179–205. [Google Scholar] [CrossRef]
- Williams, K.; Cullati, S.; Sand, A.; Biterova, E.I.; Barycki, J.J. Crystal structure of acivicin-inhibited gamma-glutamyltranspeptidase reveals critical roles for its C-terminus in autoprocessing and catalysis. Biochemistry 2009, 48, 2459–2467. [Google Scholar] [CrossRef] [Green Version]
- Chittur, S.V.; Klem, T.J.; Shafer, C.M.; Davisson, V.J. Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry 2001, 40, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Zana, A.; Conti, P. Covalent inhibitors of GAPDH: From unspecific warheads to selective compounds. Eur. J. Med. Chem. 2020, 207, 112740. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Pinto, A.; Wong, P.E.; Major, L.L.; Tamborini, L.; Iannuzzi, M.C.; De Micheli, C.; Barrett, M.P.; Smith, T.K. Synthesis and in vitro/in vivo evaluation of the antitrypanosomal activity of 3-bromoacivicin, a potent CTP synthetase inhibitor. ChemMedChem 2011, 6, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Pinto, A.; Paredi, G.; Tamborini, L.; De Micheli, C.; La Pietra, V.; Marinelli, L.; Novellino, E.; Conti, P.; Mozzarelli, A. Discovery of covalent inhibitors of glyceraldehyde-3-phosphate dehydrogenase, a target for the treatment of malaria. J. Med. Chem. 2014, 57, 7465–7471. [Google Scholar] [CrossRef]
- van Niekerk, D.D.; Penkler, G.P.; du Toit, F.; Snoep, J.L. Targeting glycolysis in the malaria parasite Plasmodium falciparum. Febs J 2016, 283, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Cullia, G.; Bruno, S.; Parapini, S.; Margiotta, M.; Tamborini, L.; Pinto, A.; Galbiati, A.; Mozzarelli, A.; Persico, M.; Paladino, A.; et al. Covalent inhibitors of Plasmodium falciparum glyceraldehyde 3-phosphate dehydrogenase with antimalarial activity in vitro. ACS Med. Chem. Lett. 2019, 10, 590–595. [Google Scholar] [CrossRef]
- Bruno, S.; Margiotta, M.; Pinto, A.; Cullia, G.; Conti, P.; De Micheli, C.; Mozzarelli, A. Selectivity of 3-bromo-isoxazoline inhibitors between human and Plasmodium falciparum glyceraldehyde-3-phosphate dehydrogenases. Bioorg Med. Chem. 2016, 24, 2654–2659. [Google Scholar] [CrossRef]
- Pacchiana, R.; Mullappilly, N.; Pinto, A.; Bova, S.; Forciniti, S.; Cullia, G.; Dalla Pozza, E.; Bottani, E.; Decimo, I.; Dando, I.; et al. 3-Bromo-isoxazoline derivatives inhibit GAPDH enzyme in PDAC cells triggering autophagy and apoptotic cell death. Cancers 2022, 14, 3153. [Google Scholar] [CrossRef]
- Pinto, A.; Conti, P.; De Amici, M.; Tamborini, L.; Madsen, U.; Nielsen, B.; Christesen, T.; Bräuner-Osborne, H.; De Micheli, C. Synthesis and pharmacological characterization at glutamate receptors of the four enantiopure isomers of tricholomic acid. J. Med. Chem. 2008, 51, 2311–2315. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, J.; Ma, S. Iron catalysis for room-temperature aerobic oxidation of alcohols to carboxylic acids. J. Am. Chem. Soc. 2016, 138, 8344–8347. [Google Scholar] [CrossRef]
- Huber, K.R.; Rosenfeld, H.; Roberts, J. Uptake of glutamine antimetabolites 6-diazo-5-oxo-L-norleucine (DON) and acivicin in sensitive and resistant tumor cell lines. Int. J. Cancer 1988, 41, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Nakajima, Y.; Motoyama, T.; Kitou, Y.; Kosaki, T.; Saito, T.; Nishiuchi, T.; Kanamaru, K.; Osada, H.; Kobayashi, T.; et al. Effects of acivicin on growth, mycotoxin production and virulence of phytopathogenic fungi. Lett. Appl. Microbiol. 2014, 59, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Elford, B.C. L-Glutamine influx in malaria-infected erythrocytes: A target for antimalarials? Parasitol. Today 1986, 2, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [CrossRef]
- Moniot, S.; Bruno, S.; Vonrhein, C.; Didierjean, C.; Boschi-Muller, S.; Vas, M.; Bricogne, G.; Branlant, G.; Mozzarelli, A.; Corbier, C. Trapping of the thioacylglyceraldehyde-3-phosphate dehydrogenase intermediate from Bacillus stearothermophilus. Direct evidence for a flip-flop mechanism. J. Biol. Chem. 2008, 283, 21693–21702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, W.J.; Senkovich, O.; Chattopadhyay, D. An unexpected phosphate binding site in glyceraldehyde 3-phosphate dehydrogenase: Crystal structures of apo, holo and ternary complex of Cryptosporidium parvum enzyme. BMC Struct. Biol. 2009, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Ewig, C.S.; Berry, R.; Dinur, U.; Hill, J.R.; Hwang, M.J.; Li, H.; Liang, C.; Maple, J.; Peng, Z.; Stockfisch, T.P.; et al. Derivation of class II force fields. VIII. Derivation of a general quantum mechanical force field for organic compounds. J. Comput. Chem. 2001, 22, 1782–1800. [Google Scholar] [CrossRef]
- Fletcher, R. Unconstrained optimization. In Practical Methods of Optimization; John Wiley & Sons Ltd.: New York, NY, USA, 1980; Volume 1, pp. 1–128. [Google Scholar]
- Senderowitz, H.; Guarnieri, F.; Still, W.C. A Smart Monte Carlo Technique for Free Energy Simulations of multiconformational molecules. Direct calculations of the conformational populations of organic molecules. J. Am. Chem. Soc. 1995, 117, 8211–8219. [Google Scholar] [CrossRef]
- Ding, H.Q.; Karasawa, N.; III, W.A.G. Atomic level simulations on a million particles: The cell multipole method for Coulomb and London nonbond interactions. J. Chem. Phys. 1992, 97, 4309–4315. [Google Scholar] [CrossRef] [Green Version]
- Lodola, A.; Branduardi, D.; De Vivo, M.; Capoferri, L.; Mor, M.; Piomelli, D.; Cavalli, A. A catalytic mechanism for cysteine N-terminal nucleophile hydrolases, as revealed by free energy simulations. PLoS ONE 2012, 7, e32397. [Google Scholar] [CrossRef] [Green Version]
- Arafet, K.; Ferrer, S.; Gonzalez, F.V.; Moliner, V. Quantum mechanics/molecular mechanics studies of the mechanism of cysteine protease inhibition by peptidyl-2,3-epoxyketones. Phys. Chem. Chem. Phys. 2017, 19, 12740–12748. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Baker, E.N.; Hubbard, R.E. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984, 44, 97–179. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Imperatore, C.; Persico, M.; Aiello, A.; Luciano, P.; Guiso, M.; Sanasi, M.F.; Taramelli, D.; Parapini, S.; Cebrián-Torrejón, G.; Doménech-Carbó, A.; et al. Marine inspired antiplasmodial thiazinoquinones: Synthesis, computational studies and electrochemical assays. RSC Adv. 2015, 5, 70689–70702. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Makler, M.T.; Hinrichs, D.J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 1993, 48, 205–210. [Google Scholar] [CrossRef]
- Byun, D.P.; Ritchie, J.; Holewinski, R.; Kim, H.-R.; Tagirasa, R.; Ivanic, J.; Weekley, C.M.; Parker, M.W.; Adresson, T.; Yoo, E. Covalent inhibition by a natural product-inspired latent electrophile. bioRxiv 2023. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Compound | clogD a | P. falciparum D10 IC50 (µM) b | P. falciparum W2 IC50 (µM) b |
|---|---|---|---|---|
 | (5S, αS)-1a | 0.02 | 0.35 ± 0.08 | 0.34 ± 0.12 |
| (5R, αS)-1b | 23.54 ± 0.31 | 24.75 ± 0.90 | ||
| (5S, αR)-1c | 7.49 ± 1.48 | 8.47 ± 2.06 | ||
| (5R, αR)-1d | 8.79 ± 1.12 | 10.18 ± 1.75 | ||
 | (5S, αS)-2a | 0.04 | 0.79 ± 0.21 | 0.88 ± 0.23 |
| (5R, αS)-2b | 17.14 ± 5.91 | 17.18 ± 7.39 | ||
| (5S, αR)-2c | 40.67 ± 16.20 | 48.18 ± 14.81 | ||
| (5R, αR)-2d | 8.27 ± 1.05 | 7.42 ± 3.54 | ||
 | (5S, αS)-3a | 1.41 | 0.37 ± 0.12 | 0.26 ± 0.05 |
| (5R, αS)-3b | 32.65 ± 18.10 | 34.85 ± 5.30 | ||
| (5S, αR)-3c | 19.35 ± 4.34 | 16.51 ± 8.11 | ||
| (5R, αR)-3d | 4.31 ± 0.51 | 4.50 ± 1.82 | ||
 | (5S, αS)-4a | 0.76 | 0.36 ± 0.11 | 0.48 ± 0.18 |
| (5R, αS)-4b | 18.20 ± 1.15 | 18.20 ± 0.93 | ||
| (5S, αR)-4c | 19.35 ± 2.15 | 19.35 ± 3.36 | ||
| (5R, αR)-4d | 8.55 ± 1.95 | 8.91 ± 3.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbiati, A.; Zana, A.; Borsari, C.; Persico, M.; Bova, S.; Tkachuk, O.; Corfu, A.I.; Tamborini, L.; Basilico, N.; Fattorusso, C.; et al. Role of Stereochemistry on the Biological Activity of Nature-Inspired 3-Br-Acivicin Isomers and Derivatives. Molecules 2023, 28, 3172. https://doi.org/10.3390/molecules28073172
Galbiati A, Zana A, Borsari C, Persico M, Bova S, Tkachuk O, Corfu AI, Tamborini L, Basilico N, Fattorusso C, et al. Role of Stereochemistry on the Biological Activity of Nature-Inspired 3-Br-Acivicin Isomers and Derivatives. Molecules. 2023; 28(7):3172. https://doi.org/10.3390/molecules28073172
Chicago/Turabian StyleGalbiati, Andrea, Aureliano Zana, Chiara Borsari, Marco Persico, Stefania Bova, Oleh Tkachuk, Alexandra Ioana Corfu, Lucia Tamborini, Nicoletta Basilico, Caterina Fattorusso, and et al. 2023. "Role of Stereochemistry on the Biological Activity of Nature-Inspired 3-Br-Acivicin Isomers and Derivatives" Molecules 28, no. 7: 3172. https://doi.org/10.3390/molecules28073172