


Evaluation of an Oral Fluid Collection Device and a Solid-Phase Extraction Method for the Determination of Coca Leaf Alkaloids by Gas Chromatography–Mass Spectrometry
 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
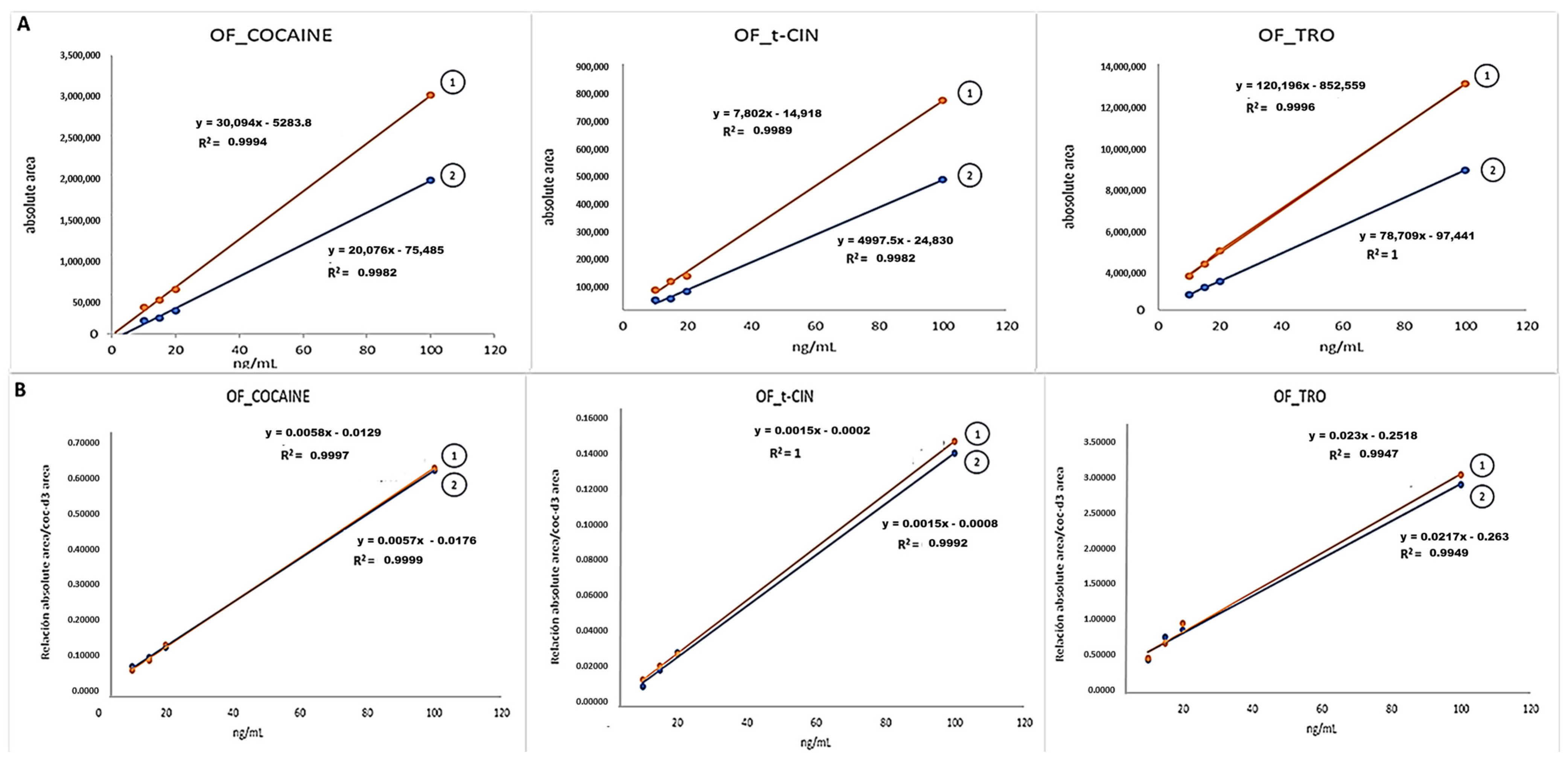
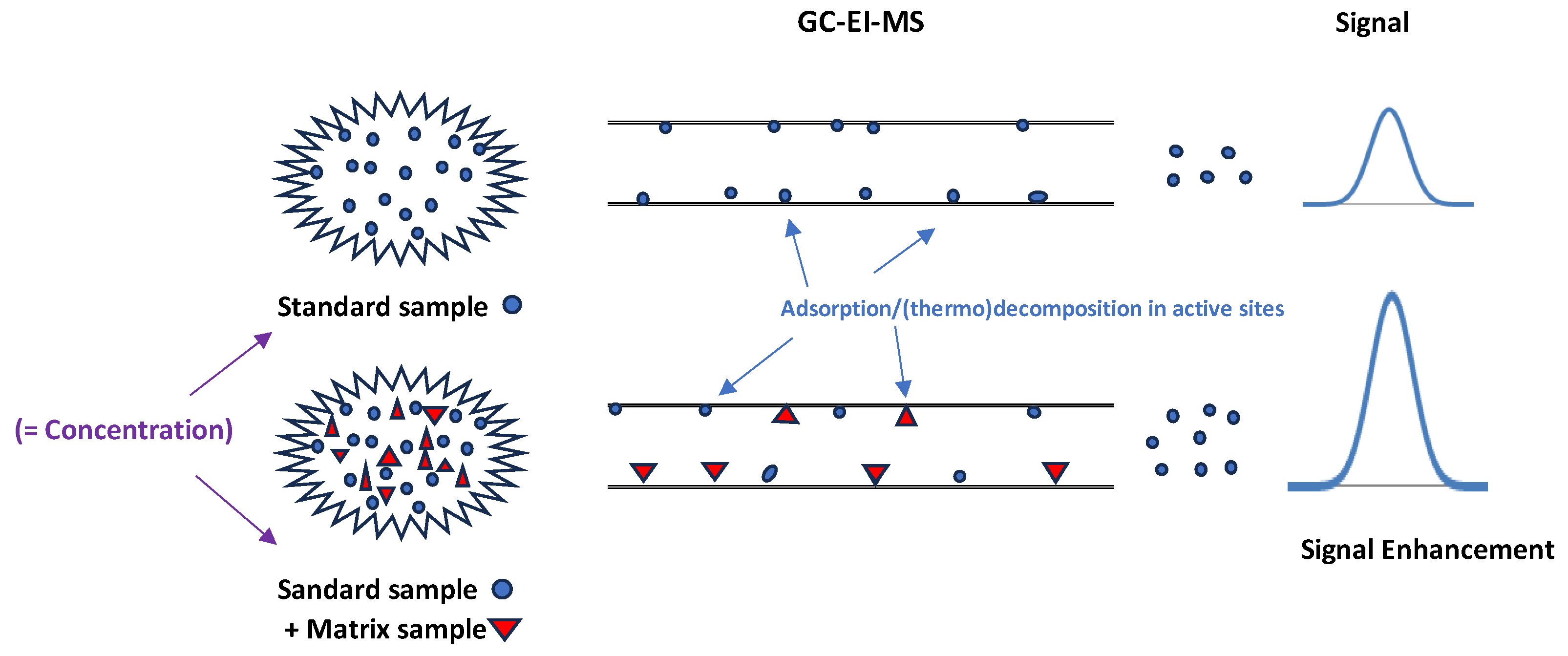
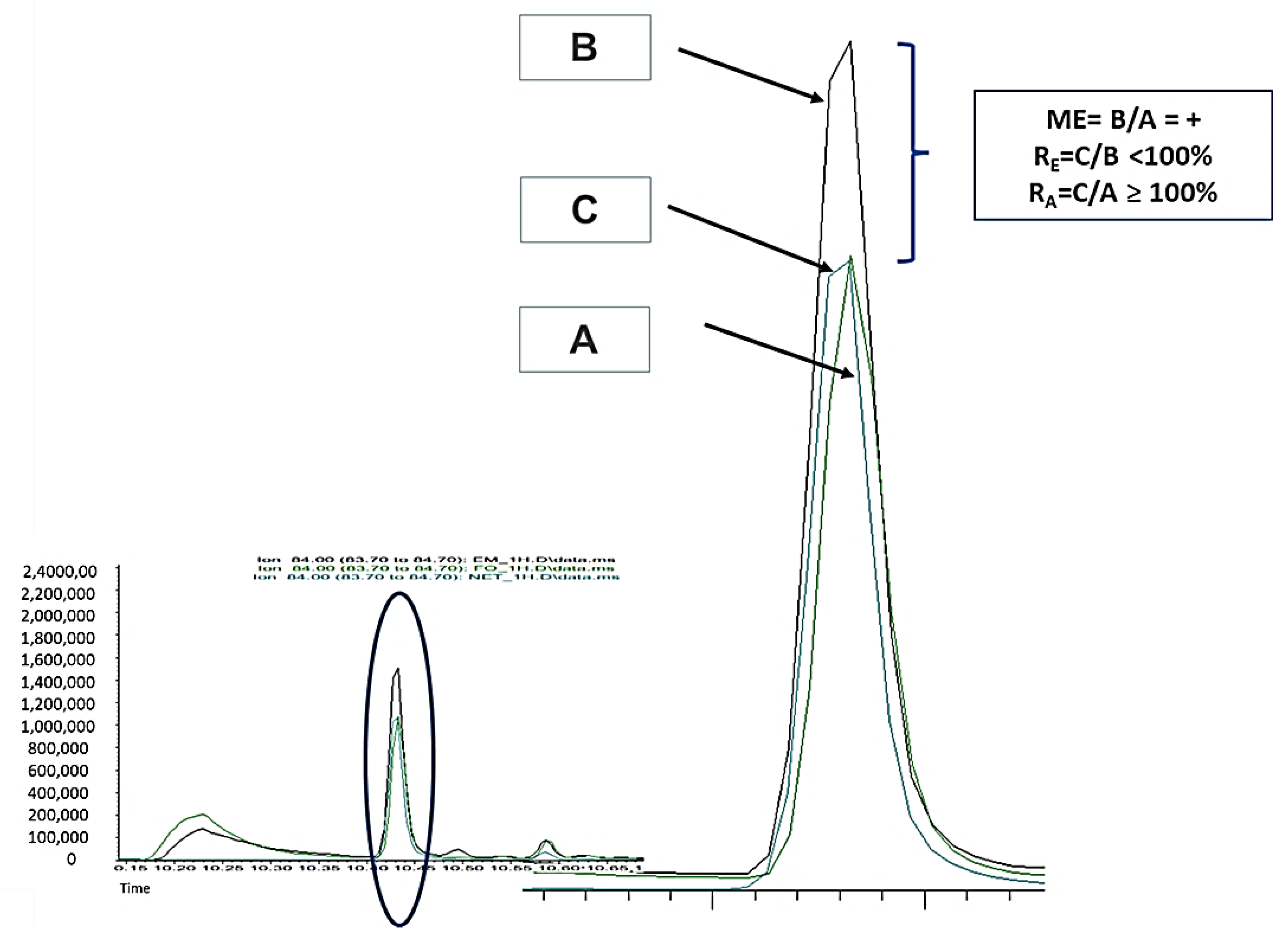
2.1. Experimental Part 1: Matrix Effect (ME)
2.2. Experimental Part 2: Recovery of Alkaloids from Quantisal® Pad
2.3. Experimental Part 3: Process Efficiency or Apparent Recovery (RA) and Extraction Recovery (RE) or Extraction Efficiency of Alkaloids from Oral Fluid and Quantisal®
2.4. Experimental Part 4: Limit of Detection and Limit of Quantification (LOD and LOQ)
2.5. Experimental Part 5: Application to Real Samples
3. Discussion
3.1. Experimental Part 1: Matrix Effect (ME)
3.2. Experimental Part 2: Recovery of Alkaloids from Quantisal® Pad
3.3. Experimental Part 3: Process Efficiency or Apparent Recovery (RA) and Extraction Recovery (RE) or Extraction Efficiency of Alkaloids from Oral Fluid and Quantisal®
3.4. Experimental Part 4: Limit of Detection and Limit of Quantification (LOD and LOQ)
3.5. Experimental Part 5: Application to Real Samples
4. Materials and Methods
4.1. Reagents and Standards
4.2. Working Solutions
4.3. Instrumental Analysis (GC-EI/MS)
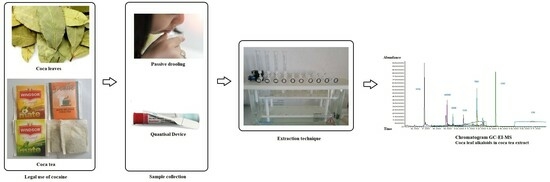
4.4. Sample Collection
4.5. Sample Preparation
- a.
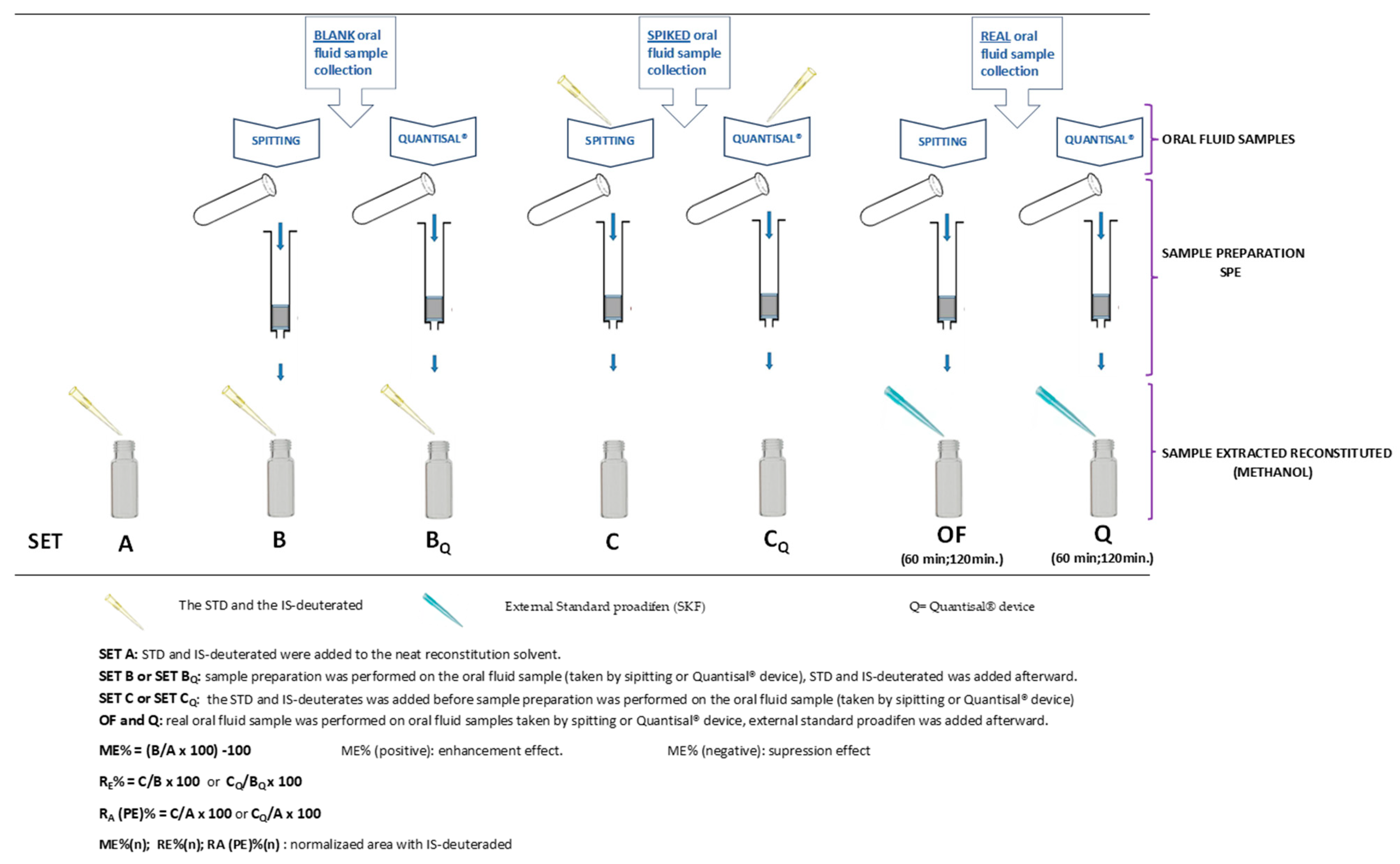
- One mL of oral fluid sample was spiked on the Quantisal® device pad at different concentrations of CUS, EME, TRO, COC, t-CIN, and the internal standard (IS: mix of COC-d3 and EME-d3; 10 µL Sol. 1.5 µg/mL). The protocol we followed adhered to the manufacturer’s specifications and was as described by Cohier et al. [24], except for the fact that we used 1 mL of buffer instead of 3 mL. In addition, blank OF samples were processed in the same way.
- b.
- One mL of oral fluid sample containing different concentrations of CUS, EME, TRO, COC, t-CIN, and IS (mix of COC-d3 and EME-d3; 10 µL Sol. 1.5 µg/mL) was added directly to one mL of Quantisal® preservative buffer (without pad) over 24 h at 4 °C. In addition, blank oral fluid samples were processed in the same way.
- c.
- One mL of oral fluid sample was taken from volunteers who had previously drunk coca tea using the Quantisal® device. Then, the sample was transferred to a 10 mL glass tube and mixed with 10 µL of 1.5 µg/mL internal standard (IS) (mix of COC-d3 and EME-d3).
4.6. Extraction via SPE
4.7. Experimental Study
4.7.1. Experimental Part 1: Matrix Effect (ME)
4.7.2. Experimental Part 2: Recovery of Alkaloids from the Quantisal® Device (Using Pad)
4.7.3. Experimental Part 3: Process Efficiency or Apparent Recovery (RA) and Extraction Recovery (RE) or Extraction Efficiency of Alkaloids from Oral Fluid and the Quantisal® Device
4.7.4. Experimental Part 4: Determination of Limit of Detection and Limit of Quantification (LOD and LOQ)
4.7.5. Experimental Part 5: Application to Real Samples
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Argentine Federal Law 23737, Art.15. O.B. October 10, 1989. Ley 23737/1989. Available online: https://www.argentina.gob.ar/ (accessed on 9 January 2023).
- Rubio, N.C.; Strano-Rossi, S.; Tabernero, M.J.; Gonzalez, J.L.; Anzillotti, L.; Chiarotti, M.; Bermejo, A.M. Application of hygrine and cuscohygrine as possible markers to distinguish coca chewing from cocaine abuse on WDT and forensic cases. Forensic Sci. Int. 2014, 243, 30–34. [Google Scholar] [CrossRef]
- Rubio, N.C.; Hastedt, M.; Gonzalez, J.; Pragst, F. Possibilities for discrimination between chewing of coca leaves and abuse of cocaine by hair analysis including hygrine, cuscohygrine, cinnamoylcocaine and cocaine metabolite/cocaine ratios. Int. J. Leg. Med. 2014, 129, 69–84. [Google Scholar] [CrossRef]
- Rubio, N.C.; Bermejo-Barrera, P.; Bermejo, A.M.; Moreda-Piñeiro, A. Development of a Reliable Method for Assessing Coca Alkaloids in Oral Fluid by HPLC–MS-MS. J. Anal. Toxicol. 2019, 43, 196–202. [Google Scholar] [CrossRef]
- Rubio, N.C.; Denise Thurmann, D.; Krumbiegel, F.; Pragst, F. Behaviour of hygrine and cuscohygrine in illicit cocaine production establishes their use as markers for chewing coca leaves in contrast with cocaine abuse. Drug Test Anal. 2017, 9, 323–326. [Google Scholar] [CrossRef]
- Bosker, W.M.; Huesti, M.A. Oral fluid testing for drugs of abuse. Clin. Chem. 2009, 55, 1910–1931. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sparve, E.; Sparring, S.; Bergström, M. Detection of drugs in oral fluid samples using a commercially available collection device: Agreement with urine testing and evaluation of a and b samples obtained from employees at different workplace settings with uncontrolled sampling procedures. J. Anal. Toxicol. 2020, 44, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Quintela, O.; Crouch, D.J.; Andrenyak, D.M. Recovery of Drugs of Abuse from the Immunalysis Quantisal Oral Fluid Collection Device. J. Anal. Toxicol. 2006, 30, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, N.A.; Huestis, M.A. Oral fluid drug testing: Analytical approaches, issues and interpretation of results. J. Anal. Toxicol. 2019, 43, 415–443. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for Workplace Drug Testing in Oral Fluid 2022-11-01 Version 3.0 FINAL. EWDTS. Available online: www.ewdts.org (accessed on 9 January 2023).
- Proposed Revisions to Mandatory Guidelines for Federal Workplace Drug Testing Programs-Oral Fluid. Federal Register, 80 FR 28053-2015 Version. Department of Health and Human Services, SAMHSA. Available online: https://www.federalregister.gov/documents/2015/05/15/2015-11523/mandatory-guidelines-for-federal-workplace-drug-testing-programs (accessed on 30 March 2023).
- Logan, B.K.; D’Orazio, A.L.; Mohr, A.L.; Limoges, J.F.; Miles, A.K.; Scarneo, C.E.; Kerrigan, S.; Liddicoat, L.J.; Scott, K.S.; Huestis, M.A. Recommendations for Toxicological Investigation of Drug-Impaired Driving and Motor Vehicle Fatalities—2017 Update. J. Anal. Toxicol. 2018, 42, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.T.; Wissenbach, D.K.; Busardò, F.P.; Marchei, E.; Pichini, S. Method Development in Forensic Toxicology. Curr. Pharm. Des. 2017, 23, 5455–5467. [Google Scholar] [CrossRef]
- GTFCh Guidelines for Quality Assurance in Forensic-Toxicological Analyses-Requirements for the Validation of Analytical Methods Änderungshinweise. APPENDIX B. Available online: gtfch.org (accessed on 1 April 2023).
- Scientific Working Group for Forensic Toxicology. Scientific working groups for forensic toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J. Anal. Toxicol. 2013, 37, 452–474. [Google Scholar] [CrossRef]
- Bienvenu, J.F.; Provencher, G.; Bélanger, P.; Bérubé, R.; Dumas, P.; Gagné, S.; Gaudreau, É.; Fleury, N. Standardized Procedure for the Simultaneous Determination of the Matrix Effect, Recovery, Process Efficiency, and Internal Standard Association. Anal. Chem. 2017, 89, 7560–7568. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Raposo, F.; Barceló, D. Challenges and strategies of matrix effects using chromatography-mass spectrometry: An overview from research versus regulatory viewpoints. Anal. Chem. 2021, 134, 116068. [Google Scholar] [CrossRef]
- Hayama, T. Matrix effects in mass spectrometry analysis. Anal. Sci. 2020, 36, 1151. [Google Scholar] [CrossRef] [PubMed]
- Hajslová, J.; Zrostlíková, J. Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. J. Chromatogr. A 2003, 1000, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.C.; Moreda-Piñeiro, A.; Bermejo-Barrera, P.; Bermejo, A.M. Coca alkaloids profile in oral fluid from people chewing coca leaves and drinking coca tea. Preliminary study. Acta Toxicol. Argent. 2019, 27, 72–80. [Google Scholar]
- Álvarez-Freire, I.; Cabarcos-Fernández, P.; Rubio, N.C.; Moreda-Piñeiro, A.; Tabernero-Duque, M.J.; Sánchez-Sellero, I.; Bermejo-Barrera, P.; Bermejo-Barrera, A.M. Detection of coca alkaloids in oral fluid from coca leaf (tea) consumers: Using solid phase extraction to improve validation parameters and widen the detection window. Anal. Methods 2023, 15, 6177. [Google Scholar] [CrossRef]
- FDA. FDA Guide Bioanalytical Method Validation Guidance for Industry; U.S. Department of Health and Human Services; Food and Drug Administration (FDA); Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM): Silver Spring, MD, USA, 2018.
- Cohier, C.; Mégarbane, B.; Roussel, O. Illicit Drugs in Oral Fluid: Evaluation of Two Collection Devices. J. Anal. Toxicol. 2017, 41, 71–76. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis; European Medicines Agency: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Raposo, F.; Ibelli-Bianco, C. Performance parameters for analytical method validation: Controversies and discrepancies among numerous guidelines. TrAC Trends Anal. Chem. 2020, 129, 115913. [Google Scholar] [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. For. Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.T.; Maurer, H.H. Review: Bioanalytical method validation-How, how much and why? Toxichem. Krimtech. 2010, 68, 1–11. Available online: https://gtfch.org/cms/images/stories/media/tk/tk68_3/Peters.pdf (accessed on 22 January 2024).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ME (%) | ME (n) (%) | ME (%) | ME (n) (%) | ME (%) | ME (n) (%) | ME (%) | ME (n) (%) | ME (%) | ME (n) (%) | ME (%) | ME (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (EME_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 2000 | - | - | 21 | - | - | - | - | - | - | - | - | - |
| CV% (n = 3) | - | - | 5.8% | - | - | - | - | - | - | - | - | - |
| 1000 | - | - | 64 | - | - | - | - | - | - | - | - | - |
| CV% (n = 3) | - | - | 13.0% | - | - | - | - | - | - | - | - | - |
| 500 | 9; 21 | −8; 14 | 19; 68 | 22; 36 | 13; 17 | −7; 2 | 0–9 | 0 | 3; 47; 80 | −5; −4; 21 | 9 | 21–34 |
| CV% (n = 6) | 2.5% | 4.7% | 12.7% | 7.5 | 2.3% | 3.4% | 13.9% | 1.4% | 19.6% | 17.9% | 14.7% | 10.3% |
| 20 | 21; 26 | 15; −3 | 0 | 21; 385 | 4; 17 | −5; −6 | 5–15 | −1; 0 | 41; 31 | 21; 18 | 20 | 30 |
| CV% (n = 6) | 6.8% | 2.3% | 9.3% | 10.6% | 5.6% | 7.8% | 18.7% | 12.6% | 18.6% | 15.4% | 2.3% | 4.9% |
| MEQ (%) | MEQ (n) (%) | MEQ (%) | MEQ (n) (%) | MEQ (%) | MEQ (n) (%) | MEQ (%) | MEQ (n) (%) | MEQ (%) | MEQ (n) (%) | MEQ (%) | MEQ (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (COC_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 10 | 13 | nd | 130 | - | 2 | −9 | 16 | 3 | 46 | 30 | 12 | nd |
| CV% (n = 3) | 3.7% | nd | 16.6% | - | 2.1% | 3.3% | 4.3% | 2.5% | 6.4% | 4.2% | 5.3% | nd |
| 20 | 2 | nd | 75 | - | −4 | −9 | 7 | 2 | 36 | 30 | 5 | nd |
| CV% (n = 3) | 5.5% | nd | 6.8% | - | 2.6% | 1.5% | 2.7% | 0.5% | 4.6% | 2.2% | 3.7% | nd |
| 50 | 8 | 11 | 35 | 13 | - | - | 16 | 0 | 18 | 2 | 16 | −3 |
| CV% (n = 3) | 6.0% | 1.5% | 26.7% | 27.0% | - | - | 3.1% | 0.4% | 2.6% | 5.0% | 2.7% | 7.5% |
| Nominal Conc. | R (EME) | Rn (EME/EME-d3) | R (CUS) | R (TRO) | Rn (TRO/COC-d3) | R (COC) | Rn (COC/COC-d3) | R (t-CIN) | Rn (t_CIN/COC-d3) | R (COC-d3) | R (EME-d3) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 ng/mL | nd | nd | nd | 45% | 95% | 52% | 111% | 51% | 76% | 47% | nd |
| 15 ng/mL | nd | nd | nd | 50% | 112% | 47% | 107% | 40% | 91% | 44% | nd |
| 20ng/mL | nd | nd | nd | 49% | 90% | 52% | 96% | 55% | 102% | 54% | nd |
| 100 ng/mL | nd | nd | nd | 62% | 95% | 64% | 99% | 62% | 96% | 64% | nd |
| 500 ng/mL | 93 | 91 | 90 | 69 | 54 | 77 | 93 | 83 | 101 | 83 | 110 |
| CV% (n = 3) | 11.6% | 9.9% | 52.2% | 12.5% | 10.8% | 4.5% | 3.9% | 5.6% | 2.9% | 5.1% | 8.9% |
| RA (%) | RA (n) (%) | RA (%) | RA (n) (%) | RA (%) | RA (n) (%) | RA (n) (%) | RA (%) | RA (n) (%) | RA (%) | RA (n) (%) | RA (%) | RA (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (EME_d3) | TROn (COC_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 5 | 67.5% | 99.4% | nd | - | 40.2% | 58.7% | 72.1% | 61.5% | 110.8% | 65.8% | 117.3% | 55.4% | 68.1% |
| CV% (n = 3) | 9.3% | 4.6% | - | - | 11.6% | 8.2% | 8.0% | 9.4% | 2.4% | 12.2% | 6.7% | 7.0% | 7.4% |
| 10 | 79% | 94% | nd | - | 48.9% | 58.2% | 65.7% | 80.9% | 111.1% | 99.0% | 138.2% | 73.2% | 83.9% |
| CV% (n = 3) | 5.0% | 6.2% | - | - | 4.2% | 2.7% | 9.6% | 10.9% | 5.0% | 25.3% | 13.0% | 13.0% | 6.0% |
| 20 | 53% | 104% | nd | - | 40.1% | 78.6% | 71.9% | 47.4% | 84.5% | 64.2% | 113.8 | 55.7 | 51.0% |
| CV% (n = 3) | 5.6 | 2.8% | - | - | 3.2% | 4.3% | 7.3% | 12.7% | 13.6% | 22.4% | 23.8% | 4.8% | 7.5% |
| 50 | 70.6% | 89.1% | 73.0% | 141.0% | 45.5% | 57.5% | 55.1% | 78.0% | 95.6% | 126.5% | 157.8% | 81.6% | 79.2% |
| CV% (n = 3) | 5.5% | 1.5% | 14.0% | 6.2% | 3.1% | 3.2% | 5.5% | 9.5% | 7.0% | 15.6% | 12.9% | 5.8% | 6.3% |
| 500 | 41% | 66% | 60–104% (▪) | 66–95% (▪) | - | - | - | - | - | - | - | - | - |
| CV% (n = 6) | 6.9% | 4.5% | 16.8–22.3% | 14.8–19.8% | - | - | - | - | - | - | - | - | - |
| RA(Q) (%) | RA(Q)(n) (%) | RA(Q) (%) | RA(Q)(n) (%) | RA(Q) (%) | RA(Q)(n) (%) | RA(Q) (%) | RA(Q)(n) (%) | RA(Q) (%) | RA(Q)(n) (%) | RA(Q) (%) | RA(Q) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (COC_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 10 | nd | nd | nd | nd | 25% | 64% | 41% | 107% | 48% | 127% | 38% | nd |
| CV% (n = 3) | - | - | - | - | 12.7% | 3.8% | 11.0% | 0.7% | 5.0% | 7.3% | 10.7% | - |
| 20 | nd | nd | nd | nd | 19–20.2% | 63–64.3% (▪) | 29.3–31% (▪) | 93.7–100% (▪) | 29.6–34% (▪) | 94.7–112% (▪) | 31–31.3% (▪) | nd |
| CV% (n = 6) | - | - | - | - | 5.3 | 3.5 | 6.5% | 3.4% | 8.4% | 6.5% | 8.6% | - |
| 100 | nd | nd | nd | nd | 34% | 73% | 44% | 94% | 49% | 104.4% | 47% | nd |
| CV% (n = 3) | - | - | - | - | - | - | - | - | - | - | - | - |
| 500 | 12.2% | 48% | 44.1% | - | - | - | 105 | 114% | 190% | 159% | 90% | 28.9% |
| CV% (n = 3) | 8.5% | 7.2% | 13.3% | - | - | - | 14% | 3.8% | 16.0% | 8.4% | 17% | 6.4% |
| RE(Q) (%) | RE(Q)(n) (%) | RE(Q) (%) | RE(Q)(n) (%) | RE(Q) (%) | RE(Q)(n) (%) | RE(Q) (%) | RE(Q)(n) (%) | RE(Q) (%) | RE(Q)(n) (%) | RE(Q) (%) | RE(Q) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (COC_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 10 | nd | nd | nd | nd | 24% | 70% | 35% | 104% | 33% | 98% | 34% | nd |
| CV% (n = 3) | - | - | - | - | 11.1% | 2.7% | 9.3% | 0.6% | 4.4% | 9.5% | 11% | - |
| 20 | nd | nd | nd | nd | 18–20.1% (▪) | 69% | 25–29% (▪) | 94–98% (▪) | 20–25% (▪) | 78–86% (▪) | 26–29% (▪) | nd |
| CV% (n = 3) | - | - | - | - | 3.9% | 3.4% | 5.22% | 2.6% | 4.2% | 4.5% | 5.5% | - |
| 100 | nd | nd | nd | nd | 32.0% | 77.0% | 40% | 95% | 35% | 77% | 42% | nd |
| n = 1 | - | - | - | - | - | - | - | - | - | - | - | - |
| 500 | 9.6% | 57.3% | 33.5% | - | - | - | 104% | 106% | 129 | 132 | 98.9% | 19.6% |
| CV% (n = 3) | 6.0% | 5.8% | 46.2% | - | - | - | 4.0% | 2.5% | 8.6% | 6.7% | 5.28 | 7.5% |
| RE(%) | RE(n) (%) | RE(%) | RE(n) (%) | RE(%) | RE(n) (%) | RE(n) (%) | RE(%) | RE(n) (%) | RE(%) | RE(n) (%) | RE(%) | RE(%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | EME | EMEn (EME_d3) | CUS | CUSn (EME_d3) | TRO | TROn (EME_d3) | TROn (COC_d3) | COC | COCn (COC_d3) | t-CIN | t-CINn (COC_d3) | COC-d3 | EME-d3 |
| 50 | 60.0% | 108.0% | 53.0% | 92.0% | - | - | - | 46.0% | 119.0% | 26.2% | 68.0% | 39% | 56.0% |
| CV% (n = 3) | 6.5% | 5.7% | 34.0% | 39.0% | - | - | - | 2.5% | 2.0% | 4.8% | 3.5% | 4.0% | 7.0% |
| 500 | 37 | 74% | 36% | 70% | 47% | 93% | 54% | 63% | 73% | 64% | 74% | 87% | 50% |
| CV% (n = 3) | 7.1% | 5.0%% | 14.3% | 7.1% | 15.0% | 10.8% | 12.0% | 4.0% | 0.3% | 2.9% | 1.5% | 4.1% | 7.3% |
| Nominal Conc. | EME | CUS | TRO | COC | t-CIN | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| OF | Q® | OF | Q® | OF | Q® | OF | Q® | OF | Q® | |
| 2.5 ng/mL | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| 5 ng/mL | + | nd | nd | nd | + | + | + | + | + | nd |
| 10 ng/mL | ++ | nd | nd | nd | ++ | ++ | ++ | ++ | ++ | + |
| 20 ng/mL | - | nd | + | nd | - | - | - | - | - | ++ |
| 50 ng/mL | - | nd | ++ | nd | - | - | - | - | - | - |
| 100 ng/mL | - | nd | - | nd | - | - | - | - | - | - |
| 500 ng/mL | - | ++ | - | ++ | - | - | - | - | - | - |
| Volunteer 1 | ||||||
| HYG | CUS | EME | TRO | COC | t-CIN | |
| OF-60 min | 0.28 | 0.43 | 0.85 | nd | 11.94 | 0.95 |
| Q-60 min | nd | 0.08 | 0.08 | nd | 1.14 | 0.07 |
| OF-120 min | 0.11 | 0.05 | 0.40 | nd | 1.11 | 0.13 |
| Q-120 min | nd | nd | nd | nd | 0.28 | nd |
| Volunteer 2 | ||||||
| HYG | CUS | EME | TRO | COC | t-CIN | |
| OF-60 min | 0.49 | 0.08 | 1.66 | nd | 5.20 | 0.34 |
| Q-60 min | 0.13 | 0.04 | 0.27 | nd | 1.93 | 0.06 |
| OF-120 min | 0.40 | 0.04 | 1.73 | nd | 0.24 | 0.04 |
| Q-120 min | nd | nd | 0.11 | nd | 0.08 | nd |
| Group Name | Start Time (min) | Ion Mass-To Charge |
|---|---|---|
| HYG | 5.00 | 42; 84; 141 |
| EME; EME-d3; CUS; TRO | 7.50 | (82; 94; 96); (99; 85); (84; 42; 209); (124; 82; 245) |
| COC; COC-d3; SKF | 12.50 | (182; 198; 303); (185; 306); (86; 99) |
| t-CIN | 15.10 | 96; 103; 182; 329 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabarcos-Fernández, P.; Álvarez-Freire, I.; Rubio, N.C.; Bermejo-Barrera, A.M.; Moreda-Piñeiro, A.; Sánchez-Sellero, I.; Tabernero-Duque, M.J. Evaluation of an Oral Fluid Collection Device and a Solid-Phase Extraction Method for the Determination of Coca Leaf Alkaloids by Gas Chromatography–Mass Spectrometry. Molecules 2024, 29, 592. https://doi.org/10.3390/molecules29030592
Cabarcos-Fernández P, Álvarez-Freire I, Rubio NC, Bermejo-Barrera AM, Moreda-Piñeiro A, Sánchez-Sellero I, Tabernero-Duque MJ. Evaluation of an Oral Fluid Collection Device and a Solid-Phase Extraction Method for the Determination of Coca Leaf Alkaloids by Gas Chromatography–Mass Spectrometry. Molecules. 2024; 29(3):592. https://doi.org/10.3390/molecules29030592
Chicago/Turabian StyleCabarcos-Fernández, Pamela, Ivan Álvarez-Freire, Nelida Cristina Rubio, Ana Maria Bermejo-Barrera, Antonio Moreda-Piñeiro, Ines Sánchez-Sellero, and Maria Jesus Tabernero-Duque. 2024. "Evaluation of an Oral Fluid Collection Device and a Solid-Phase Extraction Method for the Determination of Coca Leaf Alkaloids by Gas Chromatography–Mass Spectrometry" Molecules 29, no. 3: 592. https://doi.org/10.3390/molecules29030592