
Synthesis of a 3,7-Disubstituted Isothiazolo[4,3-b]pyridine as a Potential Inhibitor of Cyclin G-Associated Kinase

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
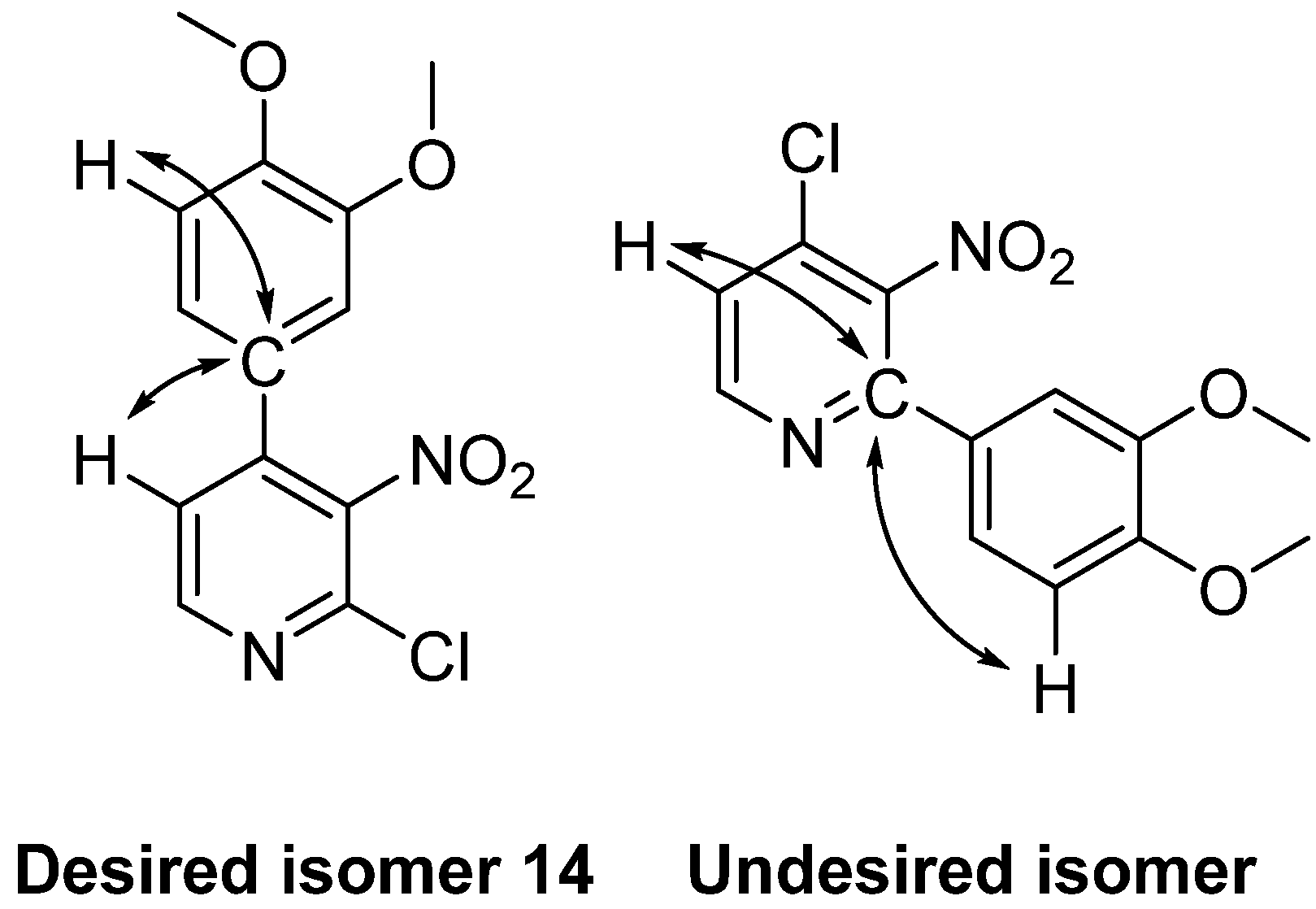
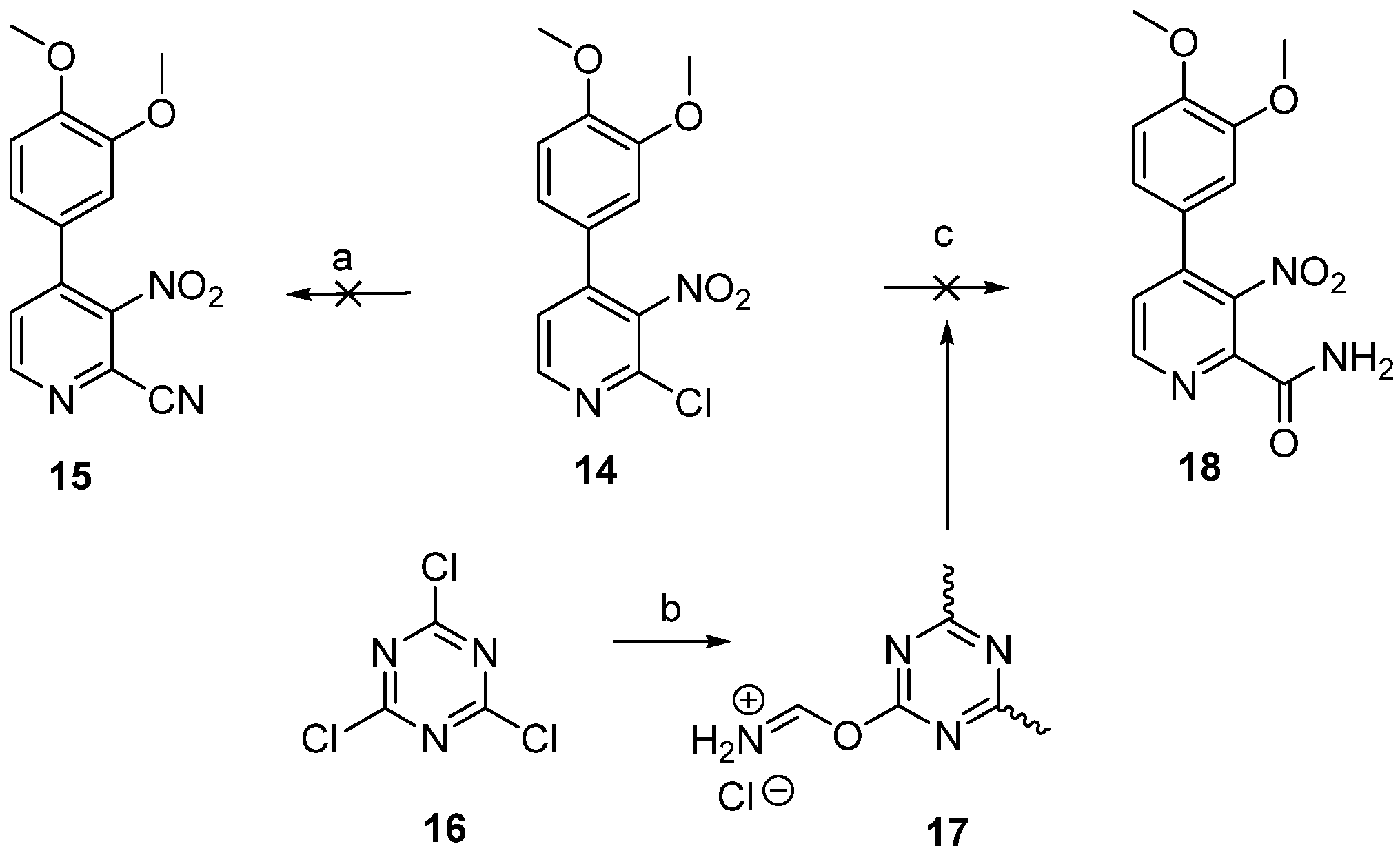
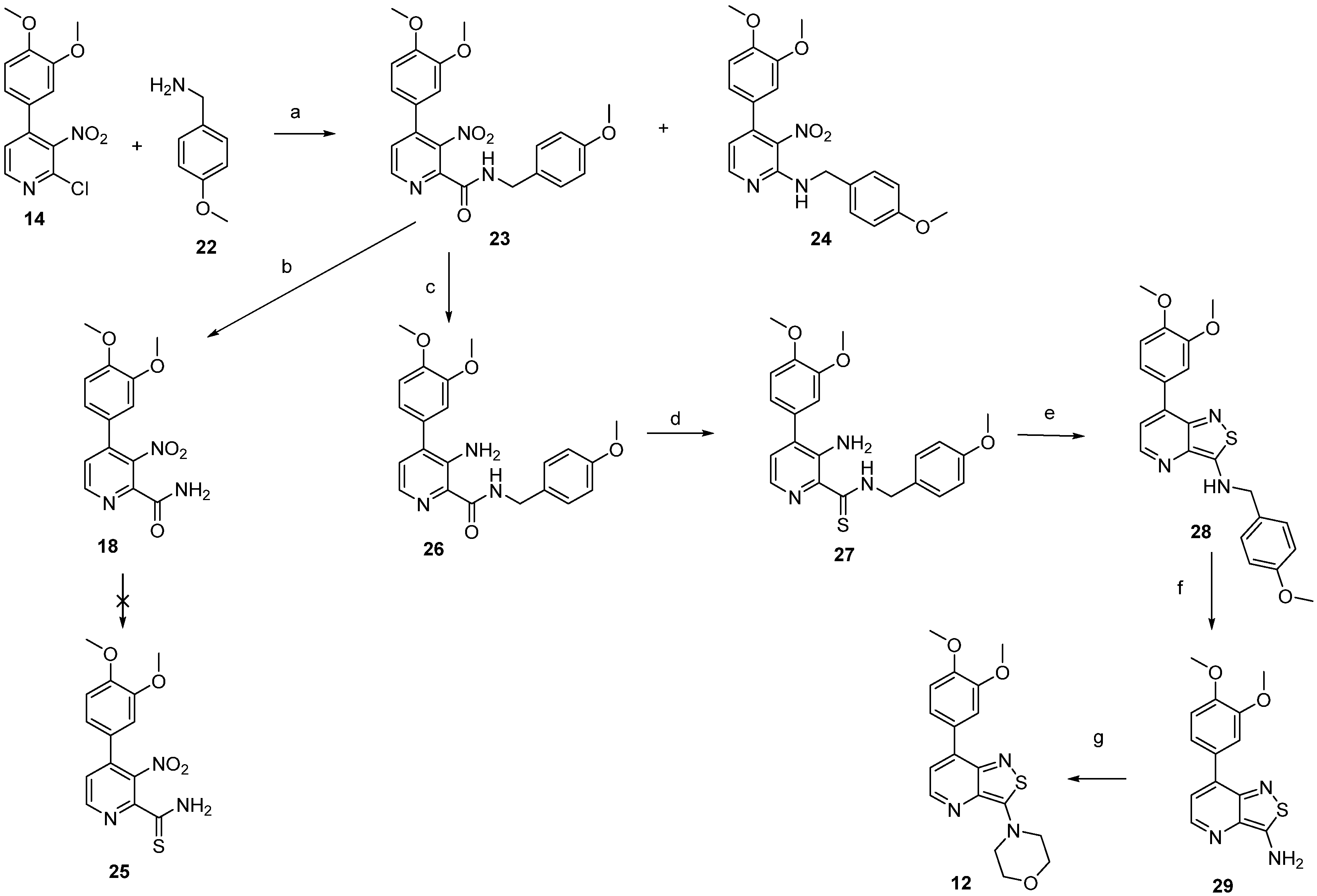
2.1. Chemistry
2.2. GAK Affinity Studies
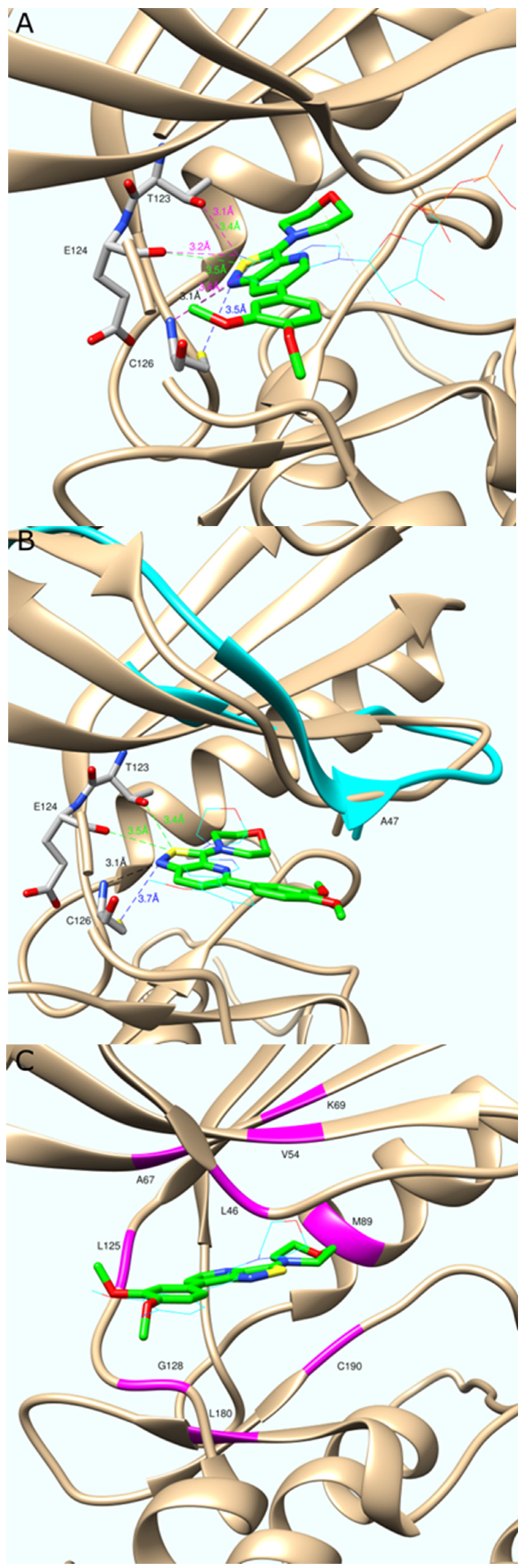
2.3. Molecular Modeling
3. Materials and Methods
3.1. Chemistry
3.1.1. 2-Chloro-4-(3,4-dimethoxyphenyl)-3-nitropyridine (14)
3.1.2. 4-(3,4-Dimethoxyphenyl)-3-nitropicolinamide (18)
3.1.3. N-Benzyl-4-(3,4-dimethoxyphenyl)-3-nitropicolinamide (20)
3.1.4. N-Benzyl-4-(3,4-dimethoxyphenyl)-3-nitropyridin-2-amine (21)
3.1.5. 4-(3,4-Dimethoxyphenyl)-N-(4-methoxybenzyl)-3-nitropicolinamide (23)
3.1.6. 4-(3,4-Dimethoxyphenyl)-N-(4-methoxybenzyl)-3-nitropyridin-2-amine (24)
3.1.7. 3-Amino-4-(3,4-dimethoxyphenyl)-N-(4-methoxybenzyl)picolinamide (26)
3.1.8. 3-Amino-4-(3,4-dimethoxyphenyl)-N-(4-methoxybenzyl)pyridine-2-carbothioamide (27)
3.1.9. 7-(3,4-Dimethoxyphenyl)-N-(4-methoxybenzyl)isothiazolo[4,3-b]pyridin-3-amine (28)
3.1.10. 7-(3,4-Dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-amine (29)
3.1.11. 4-(7-(3,4-Dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12)
3.2. GAK Affinity Studies
3.3. X-ray Crystallography
3.4. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorrell, F.J.; Szklarz, M.; Abdul Azeez, K.R.; Elkins, J.M.; Knapp, S. Family-Wide Structural Analysis of Human Numb-Associated Protein Kinases. Structure 2016, 24, 401–411. [Google Scholar] [CrossRef]
- Ricotta, D.; Conner, S.D.; Schmid, S.L.; von Figura, K.; Höning, S. Phosphorylation of the AP2 μ Subunit by AAK1 Mediates High Affinity Binding to Membrane Protein Sorting Signals. J. Cell Biol. 2002, 156, 791–795. [Google Scholar] [CrossRef]
- Borner, G.H.H.; Antrobus, R.; Hirst, J.; Bhumbra, G.S.; Kozik, P.; Jackson, L.P.; Sahlender, D.A.; Robinson, M.S. Multivariate Proteomic Profiling Identifies Novel Accessory Proteins of Coated Vesicles. J. Cell Biol. 2012, 197, 141–160. [Google Scholar] [CrossRef]
- Chaikuad, A.; Keates, T.; Vincke, C.; Kaufholz, M.; Zenn, M.; Zimmermann, B.; Gutiérrez, C.; Zhang, R.; Hatzos-Skintges, C.; Joachimiak, A.; et al. Structure of Cyclin G-Associated Kinase (GAK) Trapped in Different Conformations Using Nanobodies. Biochem. J. 2014, 459, 59–69. [Google Scholar] [CrossRef]
- Kearns, A.E.; Donohue, M.M.; Sanyal, B.; Demay, M.B. Cloning and Characterization of a Novel Protein Kinase That Impairs Osteoblast Differentiation In Vitro *. J. Biol. Chem. 2001, 276, 42213–42218. [Google Scholar] [CrossRef]
- Shimizu, H.; Nagamori, I.; Yabuta, N.; Nojima, H. GAK, a Regulator of Clathrin-Mediated Membrane Traffic, Also Controls Centrosome Integrity and Chromosome Congression. J. Cell Sci. 2009, 122 Pt 17, 3145–3152. [Google Scholar] [CrossRef]
- Zhang, C.X.; Engqvist-Goldstein, Å.E.Y.; Carreno, S.; Owen, D.J.; Smythe, E.; Drubin, D.G. Multiple Roles for Cyclin G-Associated Kinase in Clathrin-Mediated Sorting Events. Traffic 2005, 6, 1103–1113. [Google Scholar] [CrossRef]
- Neveu, G.; Barouch-Bentov, R.; Ziv-Av, A.; Gerber, D.; Jacob, Y.; Einav, S. Identification and Targeting of an Interaction between a Tyrosine Motif within Hepatitis C Virus Core Protein and AP2M1 Essential for Viral Assembly. PLoS Pathog. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Aguilar, R.C.; Ohno, H.; Roche, K.W.; Bonifacino, J.S. Functional Domain Mapping of the Clathrin-Associated Adaptor Medium Chains Mu1 and Mu2. J. Biol. Chem. 1997, 272, 27160–27166. [Google Scholar] [CrossRef]
- Owen, D.J.; Evans, P.R. A Structural Explanation for the Recognition of Tyrosine-Based Endocytotic Signals. Science 1998, 282, 1327–1332. [Google Scholar] [CrossRef]
- Olusanya, O.; Andrews, P.D.; Swedlow, J.R.; Smythe, E. Phosphorylation of Threonine 156 of the Μ2 Subunit of the AP2 Complex Is Essential for Endocytosis In Vitro and In Vivo. Curr. Biol. 2001, 11, 896–900. [Google Scholar] [CrossRef]
- Ghosh, P.; Kornfeld, S. AP-1 Binding to Sorting Signals and Release from Clathrin-Coated Vesicles Is Regulated by Phosphorylation. J. Cell Biol. 2003, 160, 699–708. [Google Scholar] [CrossRef]
- Collins, B.M.; McCoy, A.J.; Kent, H.M.; Evans, P.R.; Owen, D.J. Molecular Architecture and Functional Model of the Endocytic AP2 Complex. Cell 2002, 109, 523–535. [Google Scholar] [CrossRef]
- Lee, D.; Zhao, X.; Zhang, F.; Eisenberg, E.; Greene, L.E. Depletion of GAK/Auxilin 2 Inhibits Receptor-Mediated Endocytosis and Recruitment of Both Clathrin and Clathrin Adaptors. J. Cell Sci. 2005, 118, 4311–4321. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Banting, G. CK2 and GAK/Auxilin2 Are Major Protein Kinases in Clathrin-Coated Vesicles. Traffic 2002, 3, 428–439. [Google Scholar] [CrossRef]
- Hinshaw, J.E. Dynamin and Its Role in Membrane Fission. Annu. Rev. Cell Dev. Biol. 2000, 16, 483–519. [Google Scholar] [CrossRef]
- Xing, Y.; Böcking, T.; Wolf, M.; Grigorieff, N.; Kirchhausen, T.; Harrison, S.C. Structure of Clathrin Coat with Bound Hsc70 and Auxilin: Mechanism of Hsc70-Facilitated Disassembly. EMBO J. 2010, 29, 655–665. [Google Scholar] [CrossRef]
- Neveu, G.; Ziv-Av, A.; Barouch-Bentov, R.; Berkerman, E.; Mulholland, J.; Einav, S. AP-2-Associated Protein Kinase 1 and Cyclin G-Associated Kinase Regulate Hepatitis C Virus Entry and Are Potential Drug Targets. J. Virol. 2015, 89, 4387–4404. [Google Scholar] [CrossRef]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.-Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer Kinase Inhibitors Impair Intracellular Viral Trafficking and Exert Broad-Spectrum Antiviral Effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 Are Host Factors for Hepatitis C Virus Entry and Possible Targets for Antiviral Therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Godoi, P.H.; East, M.P.; Tizzard, G.J.; Graves, L.M.; Johnson, G.L.; Dornsife, R.E.; Wells, C.I.; et al. Identification and Optimization of 4-Anilinoquinolines as Inhibitors of Cyclin G Associated Kinase. ChemMedChem 2018, 13, 48–66. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Wells, C.I.; Elkins, J.M.; Zuercher, W.J.; Tizzard, G.J.; Poso, A. Design and Analysis of the 4-Anilinoquin(Az)Oline Kinase Inhibition Profiles of GAK/SLK/STK10 Using Quantitative Structure-Activity Relationships. ChemMedChem 2020, 15, 26–49. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Berger, B.-T.; Wan, J.; Bennett, J.M.; Capuzzi, S.J.; Crona, D.J.; Drewry, D.H.; East, M.P.; Elkins, J.M.; Fedorov, O.; et al. SGC-GAK-1: A Chemical Probe for Cyclin G Associated Kinase (GAK). J. Med. Chem. 2019, 62, 2830–2836. [Google Scholar] [CrossRef]
- Saul, S.; Pu, S.-Y.; Zuercher, W.J.; Einav, S.; Asquith, C.R.M. Potent Antiviral Activity of Novel Multi-Substituted 4-Anilinoquin(Az)Olines. Bioorg. Med. Chem. Lett. 2020, 30, 127284. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Huang, P.-T.; Einav, S.; Asquith, C.R.M. Identification and Evaluation of 4-Anilinoquin(Az)Olines as Potent Inhibitors of Both Dengue Virus (DENV) and Venezuelan Equine Encephalitis Virus (VEEV). Bioorg. Med. Chem. Lett. 2021, 52, 128407. [Google Scholar] [CrossRef] [PubMed]
- Kovackova, S.; Chang, L.; Bekerman, E.; Neveu, G.; Barouch-Bentov, R.; Chaikuad, A.; Heroven, C.; Šála, M.; De Jonghe, S.; Knapp, S.; et al. Selective Inhibitors of Cyclin G Associated Kinase (GAK) as Anti-Hepatitis C Agents. J. Med. Chem. 2015, 58, 3393–3410. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gualda, B.; Saul, S.; Froeyen, M.; Schols, D.; Herdewijn, P.; Einav, S.; De Jonghe, S. Discovery of 3-Phenyl- and 3-N-Piperidinyl-Isothiazolo[4,3-b]Pyridines as Highly Potent Inhibitors of Cyclin G-Associated Kinase. Eur. J. Med. Chem. 2021, 213, 113158. [Google Scholar] [CrossRef]
- Martinez-Gualda, B.; Pu, S.-Y.; Froeyen, M.; Herdewijn, P.; Einav, S.; De Jonghe, S. Structure-Activity Relationship Study of the Pyridine Moiety of Isothiazolo[4,3-b]Pyridines as Antiviral Agents Targeting Cyclin G-Associated Kinase. Bioorg. Med. Chem. 2020, 28, 115188. [Google Scholar] [CrossRef]
- Norman, J.P.; Larson, N.G.; Entz, E.D.; Neufeldt, S.R. Unconventional Site Selectivity in Palladium-Catalyzed Cross-Couplings of Dichloroheteroarenes under Ligand-Controlled and Ligand-Free Systems. J. Org. Chem. 2022, 87, 7414–7421. [Google Scholar] [CrossRef]
- Piersanti, G.; Giorgi, L.; Bartoccini, F.; Tarzia, G.; Minetti, P.; Gallo, G.; Giorgi, F.; Castorina, M.; Ghirardi, O.; Carminati, P. Synthesis of Benzo[1,2-d;3,4-D′]Diimidazole and 1H-Pyrazolo[4,3-b]Pyridine as Putative A2A Receptor Antagonists. Org. Biomol. Chem. 2007, 5, 2567–2571. [Google Scholar] [CrossRef]
- Niknam, E.; Panahi, F.; Khalafi-Nezhad, A. Palladium-Catalyzed Cyanation of Aryl Halides Using Formamide and Cyanuric Chloride as a New “CN” Source. Eur. J. Org. Chem. 2020, 2020, 2699–2707. [Google Scholar] [CrossRef]
- Iranpoor, N.; Panahi, F.; Roozbin, F.; Erfan, S.; Rahimi, S. Palladium-Catalyzed Aminocarbonylation of Aryl Halides with 2,4,6-Trichloro-1,3,5-Triazine/Formamide Mixed Reagent. Eur. J. Org. Chem. 2016, 2016, 1781–1787. [Google Scholar] [CrossRef]
- Veryser, C.; Mileghem, S.V.; Egle, B.; Gilles, P.; Borggraeve, W.M.D. Low-Cost Instant CO Generation at Room Temperature Using Formic Acid, Mesyl Chloride and Triethylamine. React. Chem. Eng. 2016, 1, 142–146. [Google Scholar] [CrossRef]
- Gadge, S.T.; Bhanage, B.M. A Phosphine-Free Approach to Primary Amides by Palladium-Catalyzed Aminocarbonylation of Aryl and Heteroaryl Iodides Using Methoxylamine Hydrochloride as an Ammonia Equivalent. Synlett 2014, 25, 85–88. [Google Scholar] [CrossRef]
- Schnyder, A.; Beller, M.; Mehltretter, G.; Nsenda, T.; Studer, M.; Indolese, A.F. Synthesis of Primary Aromatic Amides by Aminocarbonylation of Aryl Halides Using Formamide as an Ammonia Synthon. J. Org. Chem. 2001, 66, 4311–4315. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Zhou, J.; Chen, S.; Ding, K. Room-Temperature Debenzylation of N-Benzylcarboxamides by N-Bromosuccinimide. Synthesis 2007, 20, 3129–3134. [Google Scholar] [CrossRef]
- Lebakken, C.S.; Riddle, S.M.; Singh, U.; Frazee, W.J.; Eliason, H.C.; Gao, Y.; Reichling, L.J.; Marks, B.D.; Vogel, K.W. Development and Applications of a Broad-Coverage, TR-FRET-Based Kinase Binding Assay Platform. J. Biomol. Screen. 2009, 14, 924–935. [Google Scholar] [CrossRef]
- Zhou, P.; Tian, F.; Lv, F.; Shang, Z. Geometric Characteristics of Hydrogen Bonds Involving Sulfur Atoms in Proteins. Proteins Struct. Funct. Bioinforma. 2009, 76, 151–163. [Google Scholar] [CrossRef]
- Kříž, K.; Fanfrlík, J.; Lepšík, M. Chalcogen Bonding in Protein−Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef]
- Hermange, P.; Lindhardt, A.T.; Taaning, R.H.; Bjerglund, K.; Lupp, D.; Skrydstrup, T. Ex Situ Generation of Stoichiometric and Substoichiometric 12CO and 13CO and Its Efficient Incorporation in Palladium Catalyzed Aminocarbonylations. J. Am. Chem. Soc. 2011, 133, 6061–6071. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Equivalents of CO | Catalyst Loading | Yield of 20 | Yield of 21 |
| 1 | 1.3 | 1 mol% | 23% | 15% |
| 2 | 2.6 | 1 mol% | 42% | 50% |
| 3 | 2.6 | 2.5 mol% | 78% | 15% |
| Compound | 3,5-disubstituted isothiazolo[4,3-b]pyridine 10 | 3,6-disubstituted isothiazolo[4,3-b]pyridine 8 | 3,7-disubstituted isothiazolo[4,3-b]pyridine 12 |
| Structure | 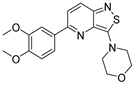 | 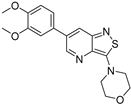 | 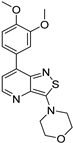 |
| GAK IC50 (µM) | 0.124 [28] | 0.442 | >10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grisez, T.; Ravi, N.P.; Froeyen, M.; Schols, D.; Van Meervelt, L.; De Jonghe, S.; Dehaen, W. Synthesis of a 3,7-Disubstituted Isothiazolo[4,3-b]pyridine as a Potential Inhibitor of Cyclin G-Associated Kinase. Molecules 2024, 29, 954. https://doi.org/10.3390/molecules29050954
Grisez T, Ravi NP, Froeyen M, Schols D, Van Meervelt L, De Jonghe S, Dehaen W. Synthesis of a 3,7-Disubstituted Isothiazolo[4,3-b]pyridine as a Potential Inhibitor of Cyclin G-Associated Kinase. Molecules. 2024; 29(5):954. https://doi.org/10.3390/molecules29050954
Chicago/Turabian StyleGrisez, Tom, Nitha Panikkassery Ravi, Mathy Froeyen, Dominique Schols, Luc Van Meervelt, Steven De Jonghe, and Wim Dehaen. 2024. "Synthesis of a 3,7-Disubstituted Isothiazolo[4,3-b]pyridine as a Potential Inhibitor of Cyclin G-Associated Kinase" Molecules 29, no. 5: 954. https://doi.org/10.3390/molecules29050954