

Design, Synthesis, and Evaluation of B-(Trifluoromethyl)phenyl Phosphine–Borane Derivatives as Novel Progesterone Receptor Antagonists


Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(7), 1587; https://doi.org/10.3390/molecules29071587
Submission received: 16 February 2024
/
Revised: 26 March 2024
/
Accepted: 27 March 2024
/
Published: 2 April 2024
(This article belongs to the Special Issue Recent Development of Organophosphorus Chemistry)
Abstract
:We previously revealed that phosphine–boranes can function as molecular frameworks for biofunctional molecules. In the present study, we exploited the diversity of available phosphines to design and synthesize a series of B-(trifluoromethyl)phenyl phosphine–borane derivatives as novel progesterone receptor (PR) antagonists. We revealed that the synthesized phosphine–borane derivatives exhibited LogP values in a predictable manner and that the P–H group in the phosphine–borane was almost nonpolar. Among the synthesized phosphine–boranes, which exhibited PR antagonistic activity, B-(4-trifluoromethyl)phenyl tricyclopropylphosphine–borane was the most potent with an IC50 value of 0.54 μM. A docking simulation indicated that the tricyclopropylphosphine moiety plays an important role in ligand–receptor interactions. These results support the idea that phosphine–boranes are versatile structural options in drug discovery, and the developed compounds are promising lead compounds for further structural development of next-generation PR antagonists.
1. Introduction
Phosphine–boranes are complexes of phosphines and boranes bearing P–B bonds [1,2,3]. Owing to the stability of the P–B bond in the adducts, phosphine–boranes are utilized as synthetic intermediates of phosphines: the borane moiety functions as a protective group for the oxidation-susceptible lone pair of trivalent phosphorus [4,5,6]. P–B adducts have also been investigated for biomedical applications such as boranophosphate-type nucleic acids (1), in which the P=O double bond of the phosphate moiety is replaced by a P–B bond [7,8]. Levin et al. developed phosphine–boranes bearing a P–BH3 moiety, such as compound 2, as neuroprotective agents (Figure 1) [9,10]. From a structural arrangement perspective, the P–B substructures in phosphine–boranes can be regarded as isosteric with alkanes because of their tetrahedral sp3–sp3 character. Based on this consideration, we recently developed phosphine–borane-containing estrogen receptor (ER) modulators such as 3 and 4 [11,12]. Subsequently, we had revealed that the P–BH2–Ph moiety as well as P–BH3 moieties are useful structural options for the development of biologically active compounds, particularly for the optimization of hydrophobic substructures by controlling the hydrophobicity of the compounds. In the current study, to investigate the general usefulness of phosphine–boranes for other biological targets, we aimed to investigate the development of novel progesterone receptor (PR) antagonists based on a phosphine–borane framework.
The progesterone receptor (PR) is a member of the nuclear receptor superfamily of ligand-dependent transcription factors and plays important roles in multiple physiological processes including the female reproductive system, such as in uterine cell proliferation and differentiation, ovulation cycle, and mammary gland growth and differentiation [13,14]. The PR is regulated by the endogenous steroidal agonist progesterone (P4, 5, Figure 2). Various steroidal PR agonists have been developed and are clinically used for the treatment of gynecological disorders, contraception, and hormone replacement therapy [15,16]. In addition to PR agonists, PR antagonists have attracted considerable attention as drug candidates. Although the representative, approved PR antagonist mifepristone (6) is currently in limited clinical use as an abortifacient, ulipristal acetate (7) is used not only as a contraceptive agent but also as a treatment for uterine fibroids [17,18,19,20]. Studies using PR antagonists, such as 6 and the investigational drug onapristone (8), indicated that PR antagonists might be effective not only for contraception and uterine fibroids but also for the treatment of endometriosis, breast cancer, ovarian cancer, uterine cancer, and some psychiatric disorders [21,22,23,24,25,26,27]. In contrast, all PR antagonists used clinically, including 6 and 7, are steroidal compounds. Therefore, the development of nonsteroidal PR antagonists is preferred to avoid side effects related to target selectivity and metabolic pathways. Indeed, a few steroidal PR antagonists, including 6, demonstrate potent activities against other steroid receptors, such as the glucocorticoid receptor (GR) [28,29,30]. Thus, various nonsteroidal PR antagonists such as 10–13 [31,32,33,34,35,36,37] have been synthesized based on the structure of the nonsteroidal PR agonist tanaproget (9) [38]. These compounds have a common cyanoaryl moiety as the pharmacophore motif, and minor structural modifications of PR ligands bearing this motif can cause agonist/antagonist activity switching [32,33,38,39]. We have been investigating the development of new-generation PR antagonists and have reported that PR antagonists such as 14 and 15 are structurally distinct from conventional nonsteroidal PR antagonists (Figure 2) [40,41]. Regarding PR antagonists and ligands of other nuclear receptors, hydrophobic interactions are essential for ligand activity. Thus, we hypothesized that the application of the phosphine–borane moiety is advantageous for the structural development of the hydrophobic moiety in PR antagonists. Consequently, we developed novel phosphine–borane framework-based PR antagonists.
2. Results and Discussion
2.1. Molecular Design
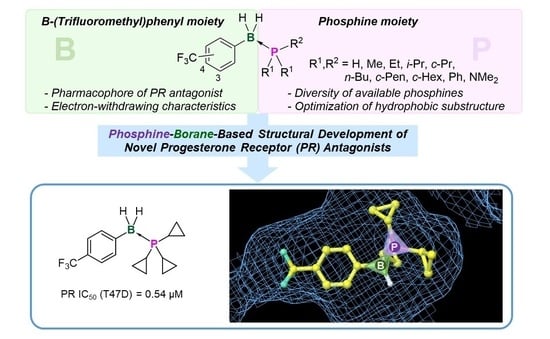
To develop a novel nonsteroidal PR antagonist, we adopted the (trifluoromethyl)phenyl group as the key structural motif, which has been found to function as a pharmacophore for PR antagonists such as 14. Our recent results indicated that the electronic profile of the B-substituents of phosphine–boranes partly influences the stability of the compounds, and an increase in the electrophilicity of the boron atom can increase the stability of the phosphine–borane adducts [12]. Thus, the introduction of an electron-withdrawing trifluoromethyl group onto the B-phenyl moiety is a reasonable approach for designing novel biologically active phosphine–borane derivatives. In addition, phosphine–borane frameworks are less hydrophobic than the corresponding carbon-based frameworks [11,12]; therefore, the introduction of a highly hydrophobic trifluoromethyl group on the phosphine–borane framework can provide appropriate hydrophobicity to the compounds. Regarding the phosphine moiety, various phosphines with diverse shapes and bulkiness are available because of their utility as phosphine ligands in catalysts. We assumed that the structural diversity of phosphines is particularly useful for the optimization of a hydrophobic motif in PR antagonists. Hence, we designed a series of B-(trifluoromethyl)phenyl phosphine–borane derivatives bearing a wide variety of phosphine moieties (Figure 3).
2.2. Synthesis
The designed B-(trifluoromethyl)phenyl phosphine–borane derivatives 16–39 were synthesized from the corresponding phosphines and 3- or 4-(trifluoromethyl)phenyl boronic acid under reductive conditions (Scheme 1) [12,42,43]. Interestingly, we could obtain phosphine–borane derivatives 26 and 38 containing the P–H moiety under normal preparation conditions using an aqueous workup. We also obtained tris(dimethylamino)phosphine-borane derivatives 27 and 39.
2.3. Hydrophobicity
Hydrophobicity is a key determinant of the activity and pharmacokinetics of biologically active compounds, and the octanol–water partition coefficient (P) and logarithm value (LogP) are widely used hydrophobicity parameters [44]. The LogP values of the synthesized compounds were determined using HPLC [45,46]. Table 1 summarizes the experimentally determined LogP values and calculated values. The trimethylphosphine–borane derivatives 16 and 28 exhibited a LogP value of 3.65. In our previous study, the corresponding phenol derivative B-4-hydroxyphenyl trimethylphosphine–borane exhibited a LogP value of 2.44 [12]. The reported substituent constants of hydrophobicity of the CF3 and phenolic OH groups were 0.88 and −0.67, respectively [47], and therefore, the difference between the LogP values of trifluoride and the corresponding phenol derivative moderately agreed with the reported difference. From the viewpoint of the phosphine structure, an increase in the number of carbon atoms increased hydrophobicity in a predictable manner. Compounds 26 and 38 bearing a P–H group and two cyclohexyl groups exhibited LogP values of 7.28 and 7.34, respectively. These LogP values were similar to those of the tri-n-butyl derivatives 23 (LogP = 7.34) and 35 (LogP = 7.50) possessing the same number of carbon atoms (C12), indicating that the P–H group in the phosphine–borane derivative was almost nonpolar and did not significantly reduce the hydrophobicity of the compounds.
Our previous results for B-hydroxyphenyl phosphine–borane derivatives indicated that some of these compounds showed LogP values with non-negligible differences from the calculated values. Therefore, we calculated the hydrophobicity parameters using SwissADME [48,49]. MLOGP is one of the most representative calculation methods of LogP defined by Moriguchi et al. [50,51], which is referred to as Lipinski’s Rule of Five [52,53]. Figure 4A shows the correlation between the measured LogP and MLOGP values, with the exception of amide compounds 27 and 39, which is expressed by the following equation: LogP = 1.735MOLGP − 4.093 (R2 = 0.911). The correlation coefficient was above 0.9 but not sufficiently large, and moreover, the slope and intercept were significantly large. The difference between the calculated and measured values of each compound ranged from –0.77 (17) to +1.60 (37). MLOGP is a calculation method based on the topology of chemical structures using 13 molecular descriptors that are correlated with LogP values. Thereafter, we used the WLOGP calculation method, defined by Wildman and Crippen based on the classification of constituent atoms using 68 atomic descriptors [54]. Figure 4B shows the correlation between the measured LogP and WLOGP values, which was expressed by the following equation: LogP = 1.016WLOGP + 0.271 (R2 = 0.980). The correlation coefficient was close to 1.0, and the slope and intercept were smaller than those of MLOGP. These results suggest that WLOGP was more suitable for predicting the hydrophobicity of phosphine–borane derivatives.
2.4. PR Antagonistic Activity
The PR agonistic and antagonistic activities of the synthesized phosphine–borane derivatives were evaluated using an alkaline phosphatase assay with the T47D human breast cancer cell line [55]. None of the test compounds induced alkaline phosphatase activity alone, indicating that they do not act as PR agonists. The basal alkaline phosphatase activity was also not affected by the tested compounds alone. The results also indicated that these compounds exhibited no significant cytotoxicity (Figure S1). The PR antagonistic activities of the compounds were assessed in the presence of 1 nM of P4 (5). We also calculated the molecular volume of each compound to investigate the structure–activity relationship (SAR). Table 2 summarizes the PR antagonistic activity and calculated volumes of the synthesized compounds. All tested compounds exhibited PR antagonistic activity. Among the 3-CF3 derivatives 16–27, the trimethylphosphine derivative 16, tri-n-butylphosphine derivative 23, and tricyclohexylphosphine derivative 25 exhibited only weak potency, and the triethylphosphine derivative 18, diisopropyl(phenyl)phosphine derivative 21, and tricyclopentylphosphine derivative 24 exhibited moderate PR antagonistic activity. The dimethyl(phenyl)phosphine derivative 17, diethyl(phenyl)phosphine derivative 19, triisopropylphosphine derivative 20, and tricyclopropylphosphine derivative 22, as well as the amide derivative 27, exhibited potent activities. These results indicate that the trimethylphosphine moiety cannot sufficiently fill the hydrophobic cavity of the ligand-binding pocket of PR and that the bulky phosphine moiety is too large to enter the ligand-binding pocket. The findings also indicated that phosphines bearing 8–10 carbon atoms are suitable for the hydrophobic substructure of the designed PR antagonists, with the tricyclopropylphosphine substructure as the most suitable hydrophobic motif. The SAR of the 4-CF3 derivatives 28–39 were similar to those of 3-CF3 derivatives, and among the synthesized compounds, B-(4-trifluoromethyl)phenyl tricyclopropylphosphine–borane (34) was the most potent with an IC50 value of 0.54 μM. Interestingly, tricyclopropylphosphine derivative 34 was approximately three times more potent than triisopropylphosphine derivative 32 (IC50 = 1.51 μM). The differences in the molecular volumes of these compounds resulted in large differences in potency. We investigated the binding affinity of the compounds toward the PR ligand-binding domain (LBD) using a fluorescence polarization assay system. Compound 34 exhibited significant affinity toward PR LBD, whereas the less potent compound 28 did not show the affinity, indicating that the potent antagonistic activity of 34 in the alkaline phosphatase assay was mediated by the binding to PR (Figure 5). We also investigated the activity of compounds 28 and 34 toward the androgen receptor (AR) by means of cell-proliferation promoting/inhibitory activity toward androgen-dependent SC-3 cells [56]. These compounds did not affect cell growth both in the presence or absence of dihydrotestosterone, indicating that these compounds did not have AR agonistic or antagonistic activity (Tabel S1).
2.5. Docking Simulations
To estimate the binding mode of the developed phosphine–borane-based PR antagonists, we conducted docking simulations of tricyclopropylphosphine derivatives 22 and 34 with the X-ray crystal structure of the nonsteroidal ligand-bound form of the hPR LBD (PDB ID: 3G8O) using AutoDock 4.2 [57,58]. Figure 6 shows the docking models of phosphine–boranes 22 and 34 in hPR LBD. In the docked structure of B-(4-trifluoromethyl)phenyl tricyclopropylphosphine–borane (34), the tricyclopropylphosphine moiety occupied the hydrophobic cavity of the ligand-binding pocket, and the 4-(trifluoromethyl)phenyl group was located in the hydrophobic cavity on the contralateral side of the phosphine moiety. In the docked structure of the 3-trifluoromethyl isomer 22, the tricyclopropylphosphine moiety occupied the hydrophobic cavity in the same manner as that in 34, and the 3-(trifluoromethyl)phenyl group was located near the center of the pocket. These results indicated that the tricyclopropylphosphine moiety played an important role in ligand–receptor interactions and that the optimization of the phenyl moiety could lead to the development of novel PR antagonists with improved potency. Overall, a wide variety of phosphine structures, which are difficult to construct using carbon-based functionalities, could enable fine tuning of the hydrophobic interactions, and phosphine–boranes could be a versatile option for the structural optimization of diverse drug candidates.
3. Materials and Methods
3.1. Chemistry
All the reagents were purchased from Sigma-Aldrich Inc., St. Louis, MO, USA, Tokyo Chemical Industry Co., Ltd., Tokyo, Japan, Fujifilm Wako Pure Chemical Corporation, Osaka, Japan, or Kanto Chemical Co., Inc., Tokyo, Japan, and used without further purification. Thin-layer chromatography (TLC) was performed using a silica gel coated with the fluorescent indicator F254 (Merck, Boston, MA, USA, #1.05715.0001). Silica gel column chromatography was performed using a neutral silica gel (60 Å, 40–50 μm) purchased from Kanto Chemical Co., Inc. NMR spectra were recorded using Bruker Avance 400 (1H: 400 MHz, 13C: 100 MHz, 11B: 128 MHz, 19F: 376 MHz, and 31P: 161 MHz) and Bruker Avance 500 (1H: 500 MHz and 13C: 125 MHz) spectrometers. High-resolution mass (HRMS) spectra were recorded on a Bruker Daltonics micrOTOF-2 focus using electron spray ionization time-of-flight (ESI-TOF).
3.2. General Procedure for the Synthesis of Phosphine–Borane Derivatives 16–39
A dry round-bottomed flask, equipped with a magnetic stirring bar, sealed with a septum, and protected with an Ar balloon, was charged with 4- or 3-(trifluoromethyl)phenyl boronic acid (190 mg, 1.0 mmol or 570 mg, 3.0 mmol) in 1.0 mL of THF. A solution of 1.0 M diisobutyl aluminium hydride (DIBAL) in toluene (3.0 or 5.0 mmol) was added to the mixture, and the corresponding phosphine was added at 0 °C. The mixture was stirred at room temperature and monitored using TLC. Once completed, the reaction was quenched by the addition of a saturated aqueous potassium sodium tartrate solution and extracted with AcOEt. The organic layer was washed with brine and dried over Na2SO4. The solvent was evaporated, and the residue was purified using silica gel column chromatography (eluent = hexane/AcOEt) to generate the desired phosphine–borane derivatives. For compounds 20–22 and 32–34, the corresponding phosphines were prepared immediately before addition, without purification. The details of the characterization and spectra of the compounds are presented in the Supporting Information.
3.2.1. B-3-(Trifluoromethyl)phenyl Trimethylphosphine–Borane (16)
Colorless oil, Yield 67%, 154 mg; 1H NMR (400 MHz, DMSO-d6): δ 7.49–7.46 (m, 2H), 7.37–7.31 (m, 2H), 2.23–1.41 (br, 2H), 1.23 (d, J = 11.0 Hz, 9H); 11B NMR (128 MHz, DMSO-d6): δ −23.7 (d, JBP = 59.3 Hz); 13C NMR (125 MHz, CDCl3): δ 138.9 (d, JCP = 6.8 Hz), 131.74–131.66 (m), 129.1 (qd, JCF = 30.8 Hz, JCP = 3.3 Hz), 127.2 (d, JCP = 3.4 Hz), 125.0 (q, JCF = 272.2 Hz), 121.5–121.4 (m), 10.2 (d, JCP = 37.5 Hz); 19F NMR (376 MHz, DMSO-d6): δ −61.1 (s); 31P NMR (161 MHz, DMSO-d6): δ −6.1 (s); HRMS (ESI) m/z calcd. for C10H15BF3NaP [M + Na]+ 257.0849, found 257.0852.
3.2.2. B-(3-Trifluoromethyl)phenyl Dimethyl(phenyl)phosphine–Borane (17)
White solid; Yield 68%, 177 mg; MP 75.0–75.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.65 (t, J = 8.7 Hz, 2H), 7.57–7.43 (m, 4H), 7.36–7.26 (m, 3H), 2.35–1.72 (br, 2H), 1.53 (d, J = 10.7 Hz, 6H); 11B NMR (128 MHz, DMSO-d6) δ −23.8 (s); 13C NMR (125 MHz, CDCl3) δ 139.2 (d, JCP = 7.0 Hz), 132.1–132.0 (m), 131.3 (d, JCP = 2.4 Hz), 130.8 (d, JCP = 8.4 Hz), 129.3 (d, JCP = 53.5 Hz), 129.1 (qd, JCF = 31.0 Hz, JCP = 3.3 Hz), 128.9 (d, JCP = 10 Hz), 127.2 (d, JCP = 3.4 Hz), 124.9 (q, JCF = 272.1 Hz), 121.6–121.5 (m), 10.0 (d, JCP = 38.2 Hz; 19F NMR (376 MHz, DMSO-d6) δ −61.1 (s); 31P NMR (161 MHz, DMSO-d6) δ −1.2 (s); HRMS (ESI) m/z calcd. for C15H17BF3NaP [M + Na]+ 319.1005, found 319.0998.
3.2.3. B-3-(Trifluoromethyl)phenyl Triethylphosphine–Borane (18)
Colorless oil; Yield 26%, 71 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.50–7.48 (m, 2H), 7.35–7.29 (m, 2H), 1.63–1.54 (m, 6H), 1.04–0.96 (m, 9H); 11B NMR (128 MHz, DMSO-d6) δ −27.4 (d, JBP = 50.1 Hz); 13C NMR (125 MHz, CDCl3) δ 139.1 (d, JCP = 6.4 Hz), 132.0–131.9 (m), 129.0 (qd, JCF = 30.9 Hz, JCP = 3.0 Hz), 127.1 (d, JCP = 2.9 Hz), 125.0 (q, JCF = 272.3 Hz), 121.3–121.2 (m), 12.8 (d, JCP = 34.1 Hz), 6.4 (d, JCP = 3.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.1 (s); 31P NMR (161 MHz, DMSO-d6) δ 12.5 (s); HRMS (ESI) m/z calcd. for C13H21BF3NaP [M + Na]+ 299.1318, found 299.1325.
3.2.4. B-3-(Trifluoromethyl)phenyl Diethyl(phenyl)phosphine–Borane (19)
White solid; Yield 68%, 215 mg; MP 40.0–40.5 °C; 1H NMR (400 MHz, CD2Cl2) δ 7.62–7.58 (m, 2H), 7.56–7.46 (m, 5H), 7.30 (d, J = 7.5 Hz, 1H), 7.23 (t, J = 7.5 Hz, 1H), 1.89–1.76 (m, 4H), 1.09–1.01 (m, 6H); 11B NMR (128 MHz, DMSO-d6) δ −27.1 (s); 13C NMR (100 MHz, DMSO-d6): δ 139.8 (d, JCP = 6.4 Hz), 132.5 (d, JCP = 7.6 Hz), 131.9–131.7 (m), 131.7 (d, JCP = 2.2 Hz), 129.3 (d, JCP = 9.3 Hz), 128.2 (qd, JCF = 30.3 Hz, JCP = 3.1 Hz), 127.9 (d, JCP = 3.0 Hz), 126.4 (d, JCP = 51.1 Hz), 125.3 (q, JCF = 272.2 Hz), 121.5–121.4 (m), 14.5 (d, JCP = 36.0 Hz), 6.9 (d, JCP = 3.0 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 13.1 (s); ESMS (ESI) m/z calcd. for C17H21BF3NaP [M + Na]+ 347.1318, found 347.1310.
3.2.5. B-3-(Trifluoromethyl)phenyl Triisopropylphosphine–Borane (20)
Colorless oil; Yield 54%, 514 mg (2 steps); 1H NMR (400 MHz, DMSO-d6) δ= 7.57–7.55 (m, 2H), 7.34–7.28 (m, 2H), 2.28–1.50 (br, 2H), 2.26–2.16 (m, 3H), 1.17–1.12 (m, 18H); 11B NMR (128 MHz, DMSO-d6) δ −28.8 (s); 13C NMR (125 MHz, DMSO-d6) δ 140.2 (d, JCP = 5.5 Hz), 132.3–132.2 (m), 128.0 (qd, JCF = 30.2 Hz, JCP = 3.4 Hz), 127.9 (d, JCP = 3.0 Hz), 125.4 (q, JCF = 272.2 Hz), 121.31–121.25 (m), 20.6 (d, JCP = 29.5 Hz), 18.1 (d, JCP = 1.5 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 24.6 (s); HRMS (ESI) m/z calcd. for C16H27BF3NaP [M + Na]+ 341.1788, found 341.1800.
3.2.6. B-3-(Trifluoromethyl)phenyl Diisopropyl(phenyl)phosphine–Borane (21)
White solid; Yield 25%, 263 mg (2 step); MP 118.7–119.1 °C; 1H NMR (400 MHz, CD2Cl2) δ 7.80–7.76 (m, 2H), 7.69 (s, 1H), 7.62 (d, J = 6.9 Hz, 1H), 7.59–7.50 (m, 3H), 7.29 (d, J = 7.3 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 2.72–1.90 (br, 2H), 2.49–2.39 (m, 2H), 1.08–0.95 (m, 12H); 11B NMR (128 MHz, DMSO-d6) δ −28.5 (s); 13C NMR (100 MHz, DMSO-d6) δ 140.2 (d, JCP = 6.8 Hz), 134.1 (d, JCP = 6.7 Hz), 132.3–132.2 (m), 132.0 (d, JCP = 2.2 Hz), 129.2 (d, JCP = 8.9 Hz), 128.2 (qd, JCF = 27.6 Hz, JCP = 2.5 Hz), 127.9 (d, JCP = 2.2 Hz), 125.4 (q, JCF = 272.2 Hz), 123.5 (d, JCP = 46.9 Hz), 121.53–121.46 (m), 20.7 (d, JCP = 32.7 Hz), 16.8–16.7 (m); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 22.1 (s); HRMS (ESI) m/z calcd. for C19H25BF3NaP [M + Na]+ 375.1631, found 375.1653.
3.2.7. B-3-(Trifluoromethyl)phenyl Tricyclopropylphosphine–Borane (22)
Colorless oil; Yield 26%, 80 mg (2 steps); 1H NMR (400 MHz, DMSO-d6) δ 7.54–7.52 (m, 2H), 7.34–7.26 (m, 2H), 2.00–1.05 (br, 2H), 0.80–0.62 (m, 15H); 11B NMR (128 MHz, DMSO-d6) δ −29.1 (s); 13C NMR (125 MHz, CDCl3) δ 139.5 (d, JCP = 6.9 Hz), 132.5–132.4 (m), 128.7 (q, JCF = 31.4 Hz), 126.9 (d, JCP = 2.5 Hz), 125.0 (q, JCF = 272.3 Hz), 121.1–121.0 (m), 2.1 (d, JCP = 3.0 Hz), 1.6 (d, JCP = 58.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.1 (s); 31P NMR (161 MHz, DMSO-d6) δ 17.3 (s); HRMS (ESI) m/z calcd. for C16H21BF3NaP [M + Na]+ 335.1318, found 335.1323.
3.2.8. B-3-(Trifluoromethyl)phenyl Tri-n-Butylphosphine–Borane (23)
Colorless oil; Yield 66%, 238 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.47 (s, 2H), 7.36–7.30 (m, 2H), 1.57–1.51 (m, 6H), 1.38–1.29 (m, 12H), 0.85 (t, J = 7.0 Hz, 9H); 11B NMR (128 MHz, DMSO-d6) δ −26.8 (s); 13C NMR (125 MHz, CDCl3) δ 139.0 (d, JCP = 6.4 Hz), 132.0–131.9 (m), 128.9 (qd, JCF = 30.8 Hz, JCP = 3.4 Hz), 127.1 (d, JCP = 3.4 Hz), 125.0 (q, JCF = 272.2 Hz), 121.2–121.1 (m), 24.4 (d, JCP = 2.8 Hz), 24.3 (d, JCP = 12.5 Hz), 20.2 (d, JCP = 33.0 Hz), 13.4 (s); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 7.03 (s); ESMS (ESI) m/z calcd. for C19H33BF3NaP [M + Na]+ 383.2257, found 383.2276.
3.2.9. B-3-(Trifluoromethyl)phenyl Tricyclopentylphosphine–Borane (24)
Colorless oil; Yield 60%, 239 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.56–7.53 (m, 2H), 7.32–7.26 (m, 2H), 2.23–1.69 (br, 2H), 2.19–2.10 (m, 3H), 1.81–1.79 (m, 6H), 1.58–1.50 (m, 18H); 11B NMR (128 MHz, DMSO-d6) δ −28.4 (s); 13C NMR (125 MHz, CDCl3) δ 139.6 (d, JCP = 6.0 Hz), 132.8–132.7 (m), 128.7 (qd, JCF = 30.7 Hz, JCP = 2.9 Hz), 126.8 (d, JCP = 2.8 Hz), 125.0 (q, JCF = 272.3 Hz), 121.0–120.9 (m), 32.4 (d, JCP = 31.9 Hz), 28.3(s), 26.0 (d, JCP = 8.8 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 19.1 (s); HRMS (ESI) m/z calcd. for C22H33BF3NaP [M + Na]+ 419.2257, found 419.2265.
3.2.10. B-3-(Trifluoromethyl)phenyl Tricyclohexylphosphine–Borane (25)
White solid; Yield:45%, 213 mg; MP 100.0–100.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.53 (s, 2H), 7.35–7.30 (m, 2H), 2.08–1.10 (br, 2H),1.94–1.65 (m, 18H), 1.32–1.14 (m, 15H); 11B NMR (128 MHz, DMSO-d6) δ −28.8 (s); 13C NMR (125 MHz, DMSO-d6) δ 140.2 (d, JCP = 5.2 Hz), 132.4–132.3 (m), 128.0 (qd, JCF = 30.1 Hz, JCP = 2.6 Hz), 128.0 (d, JCP = 2.3 Hz), 125.5 (q, JCF = 272.6 Hz), 121.33–121.27 (m), 30.3 (d, JCP = 28.7 Hz), 27.8 (d, JCP = 1.4 Hz), 27.2 (d, JCP = 10.0 Hz), 26.1 (s); 19F NMR (376 MHz, DMSO-d6) δ −61.2 (s); 31P NMR (161 MHz, DMSO-d6) δ 15.7 (s); HRMS (ESI) m/z calcd. for C25H39BF3NaP [M + Na]+ 461.2727, found 461.2739.
3.2.11. B-3-(Trifluoromethyl)phenyl Dicyclohexylphosphine–Borane (26)
Colorless oil; Yield 31%, 109 mg; 1H NMR (400 MHz, acetone-d6) δ 7.65–7.60 (m, 2H), 7.35–7.28 (m, 2H), 4.26 (d, J = 357.0 Hz, 1H), 2.82–1.66 (br, 2H), 2.12–2.01 (m, 2H), 1.85–1.66 (m, 10H), 1.47–1.15 (m, 10H); 11B NMR (128 MHz, DMSO-d6) δ −29.18 (s); 13C NMR (100 MHz, acetone-d6) δ 140.4 (d, JCP = 7.0 Hz), 132.8–132.7 (m), 129.5 (qd, JCF = 30.3 Hz, JCP = 2.9 Hz), 128.3 (d, JCP = 2.6 Hz), 126.2 (q, JCF = 271.5 Hz), 121.9–121.8 (m), 30.7 (s), 29.8 (d, JCP = 31.1 Hz), 29.1 (s), 27.4 (d, JCP = 10.3 Hz), 27.2 (d, JCP = 11.5 Hz), 26.5 (s); 19F NMR (376 MHz, DMSO-d6): δ −61.12 (s); 31P NMR (161 MHz, DMSO-d6) δ 10.0 (d, J = 353.3 Hz); HRMS (ESI) m/z calcd. for C19H29BF3NaP [M + Na]+ 379.1944, found 379.1929.
3.2.12. B-3-(Trifluoromethyl)phenyl Tris(dimethylamino)phosphine–Borane (27)
Colorless oil; Yield:23%, 75 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.57–7.53 (m, 2H), 7.33–7.527 (m, 2H), 2.52 (d, J = 9.1 Hz, 18H), 2.30–1.56 (br, 2H); 11B NMR (128 MHz, DMSO-d6) δ −27.1 (d, J = 104.8 Hz); 13C NMR (125 MHz, DMSO-d6): δ 140.3 (d, JCP = 8.1 Hz), 132.3–132.2 (m), 127.8 (qd, JCF = 30.3 Hz, JCP = 2.9 Hz), 127.7 (s), 125.5 (q, JCF = 272.2 Hz), 121.18–121.12 (m), 37.3 (d, JCP = 3.4 Hz); 19F NMR (376 MHz, DMSO-d6) δ −61.1 (s); 31P NMR (161 MHz, DMSO-d6) δ 92.3 (s); HRMS (ESI) m/z calcd. for C13H24BF3N3NaP [M + Na]+ 344.1645, found 344.1640.
3.2.13. B-4-(Trifluoromethyl)phenyl Trimethylphosphine–Borane (28)
Colorless oil; Yield: 24%, 55 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.41 (s, 4H), 2.29–1.42 (br, 2H), 1.23 (d, J = 10.9 Hz, 9H); 11B NMR (128 MHz, DMSO-d6): δ −23.8 (d, JBP = 63.1 Hz); 13C NMR (125 MHz, CDCl3) δ 135.6 (d, JCP = 7.0 Hz), 126.8 (qd, JCF = 31.5 Hz, JCP = 4.7 Hz), 125.0 (qd, JCF = 271.3 Hz, JCP = 1.7 Hz), 123.7 (s), 10.3 (d, JCP = 37.5 Hz); 19F NMR (376 MHz, DMSO-d6) δ −55.9 (s); 31P NMR (161 MHz, DMSO-d6) δ −6.2 (s); HRMS (ESI) m/z calcd. for C10H15BF3NaP [M + Na]+ 257.0849, found 257.0842.
3.2.14. B-4-(Trifluoromethyl)phenyl Dimethyl(phenyl)phosphine–Borane (29)
White solid; Yield:28%, 73 mg; MP 78.0–78.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.69–7.66 (m, 2H), 7.58–7.49 (m, 3H), 7.38 (s, 4H), 2.30–1.72 (br, 2H), 1.54 (d, J = 10.7 Hz, 6H); 11B NMR (128 MHz, DMSO-d6) δ −23.8 (s); 13C NMR (100 MHz, DMSO-d6) δ 136.3 (d, JCP = 7.2 Hz), 131.6 (d, JCP = 2.1 Hz), 131.4 (d, JCP = 8.8 Hz), 130.1 (d, JCP = 54.4 Hz), 129.3 (d, JCP = 9.6 Hz), 125.7 (qd, JCF = 31.2 Hz, JCP = 4.3 Hz), 125.5 (qd, JCF = 270.9 Hz, JCP = 1.3 Hz), 123.67–13.60 (m), 9.7 (d, JCP = 38.8 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.8 (d, J = 4.1 Hz); 31P NMR (161 MHz, DMSO-d6) δ −1.4 (s); HRMS (ESI) m/z calcd. for C15H17BF3NaP [M + Na]+ 319.1005, found 319.1013.
3.2.15. B-4-(Trifluoromethyl)phenyl Triethylphosphine–Borane (30)
Colorless oil; Yield:49%, 135 mg; 1H NMR (400 MHz, DMSO-d6) δ 7.41 (s, 4H), 2.24–1.43 (br, 2H), 1.63–1.55 (m, 6H), 1.05–0.97 (m, 9H); 11B NMR (128 MHz, DMSO-d6) δ −27.6 (d, JBP = 53.0 Hz); 13C NMR (125 MHz, CDCl3) δ 135.8 (d, JCP = 6.6 Hz), 126.5 (qd, JCF = 31.7 Hz, JCP = 4.2 Hz), 125.0 (qd, JCF = 271.6 Hz), 123.6–123.5 (m), 12.8 (d, JCP = 34.1 Hz), 6.4 (d, JCP = 3.8 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.7 (d, JFP = 3.0 Hz); 31P NMR (161 MHz, DMSO-d6) δ 12.8 (s); HRMS (ESI) m/z calcd. for C13H21BF3NaP [M + Na]+ 299.1318, found 299.1319.
3.2.16. B-4-(Trifluoromethyl)phenyl Diethyl(phenyl)phosphine–Borane (31)
White solid; Yield:38%, 121 mg; MP 62.0–62.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.75–7.70 (m, 2H), 7.61–7.52 (m, 3H), 7.39 (s,4H), 2.45–1.76 (br, 2H), 2.02–1.77 (m, 4H), 0.98–0.90 (m, 6H); 11B NMR (128 MHz, DMSO-d6) δ −27.3 (s); 13C NMR (125 MHz, CDCl3) δ 135.9 (d, JCP = 7.1 Hz), 132.1 (d, JCP = 7.5 Hz), 131.3 (d, JCP = 2.5 Hz), 128.9 (d, JCP = 9.4 Hz), 126.8 (qd, JCF = 31.7 Hz, JCP = 4.2 Hz), 126.1 (d, JCP = 50.3 Hz), 125.0 (q, JCF = 271.5 Hz), 123.61–123.56 (m), 15.1 (d, JCP = 35.6 Hz), 6.7 (d, JCP = 3.0 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.7 (d, J = 3.8 Hz); 31P NMR (161 MHz, DMSO-d6) δ 13.3 (s); ESMS (ESI) m/z calcd. for C17H21BF3PNa [M + Na]+ 347.1318, found 347.1317.
3.2.17. B-4-(Trifluoromethyl)phenyl Triisopropylphosphine–Borane (32)
White solid; Yield:13%, 123 mg (2 steps); MP 59.0–59.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.49 (d, J = 7.4 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 2.30–1.52 (br, 2H), 2.28–2.18 (m, 3H), 1.17–1.12 (m, 18H); 11B NMR (128 MHz, DMSO-d6) δ −28.7 (d, J = 37.6 Hz,); 13C NMR (125 MHz, CDCl3) δ 136.3 (d, JCP = 6.2 Hz), 126.5 (q, JCF = 30.0 Hz), 125.1 (q, JCF = 271.4 Hz), 123.4 (s), 20.8 (d, JCP = 29.2 Hz), 18.0 (s); 19F NMR (376 MHz, DMSO-d6) δ −60.6 (d, J = 3.5 Hz); 31P NMR (161 MHz, DMSO-d6) δ 24.9 (s); HRMS (ESI) m/z calcd. for C16H27BF3NaP [M + Na]+ 341.1788, found 341.1791.
3.2.18. B-4-(Trifluoromethyl)phenyl Diisopropyl(phenyl)phosphine–Borane (33)
White solid; Yield 18%, 186 mg (2 steps); MP 94.8–95.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.87–7.65 (m, 2H), 7.65–7.57 (m, 3H), 7.52 (d, J = 7.1 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 2.65–2.00 (br, 2H), 2.63–2.53 (m, 2H), 1.01–0.86 (m, 12H); 11B NMR (128 MHz, DMSO-d6) δ −28.9 (s); 13C NMR (125 MHz, CDCl3) δ 136.3 (d, JCP = 7.0 Hz), 133.6 (d, JCP = 6.5 Hz), 131.3 (d, JCP = 2.4 Hz), 128.6 (d, JCP = 8.7 Hz), 126.7 (qd, JCF = 31.8 Hz, JCP = 3.8 Hz), 125.0 (q, JCF = 271.2 Hz), 123.9 (s), 123.53–123.48 (m), 21.2 (d, JCP = 31.8 Hz), 16.7–16.5 (m); 19F NMR (376 MHz, DMSO-d6) δ −60.7 (d, J = 2.8 Hz); 31P NMR (161 MHz, DMSO-d6) δ 22.5 (s); HRMS (ESI) m/z calcd. for C19H25BF3NaP [M + Na]+ 375.1631, found 375.1632.
3.2.19. B-4-(Trifluoromethyl)phenyl Tricyclopropylphosphine–Borane (34)
White solid; Yield:22%, 68 mg (2 steps); MP 72.6–72.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.46 (d, J = 6.9 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 1.99–1.12 (br, 2H), 0.81–0.66 (m, 15H); 11B NMR (128 MHz, DMSO-d6) δ −29.4 (s); 13C NMR (125 MHz, DMSO-d6) δ 136.6 (d, JCP = 6.8 Hz), 125.5 (qd, JCF = 271.4 Hz), 125.4 (qd, JCF = 31.1 Hz, JCP = 4.1 Hz), 123.42–123.39 (m), 2.37 (d, JCP = 3.3 Hz), 1.65 (d, JCP = 59.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.6 (d, J = 4.0 Hz); 31P NMR (161 MHz, DMSO-d6) δ 17.7 (s); HRMS (ESI) m/z calcd. for C16H21BF3NaP [M + Na]+ 335.1318, found 335.1321.
3.2.20. B-4-(Trifluoromethyl)phenyl Tri-n-butylphosphine–Borane (35)
White solid; Yield:15%, 54 mg; MP 33.2–33.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.41 (s, 4H), 1.59–1.52 (m, 6H), 2.10–1.48 (br, 2H), 1.38–1.29 (m, 12H), 0.85 (t, J = 7.0 Hz, 9H); 11B NMR (128 MHz, DMSO-d6) δ −26.8 (s); 13C NMR (125 MHz, DMSO-d6) δ 136.2 (d, JCP = 6.6 Hz), 125.54 (q, JCF = 270.9 Hz), 125.48 (qd, JCF = 31.1 Hz, JCP = 4.0 Hz), 123.7–123.6 (m), 24.43 (d, JCP = 2.7 Hz), 24.37 (d, JCP = 12.5 Hz), 20.1 (d, JCP = 33.8 Hz), 13.9 (s); 19F NMR (376 MHz, DMSO-d6) δ −60.7 (s); 31P NMR (161 MHz, DMSO-d6) δ 7.4 (s); ESMS (ESI) calcd. for C19H33BF3PNa [M + Na]+ 383.2257, found 383.2270.
3.2.21. B-4-(Trifluoromethyl)phenyl Tricyclopentylphosphine–Borane (36)
White solid; Yield:32%, 126 mg; MP 88.0–88.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.48 (d, J = 7.0 Hz, 2H), 7.38 (d, J = 7.8 Hz, 2H), 2.24–1.73 (br, 2H), 2.20–2.12 (m, 3H), 1.82–1.80 (m, 6H), 1.58–1.50 (m, 18H); 11B NMR (128 MHz, DMSO-d6) δ −28.9 (s); 13C NMR (125 MHz, CDCl3) δ 136.4 (d, JCP = 6.0 Hz), 126.4 (qd, JCF = 31.6 Hz, JCP = 4.0 Hz), 125.1 (q, JCF = 271.1 Hz), 123.31–123.26 (m), 34.3 (d, JCP = 32.0 Hz), 28.4 (s), 26.1 (d, JCP = 8.4 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.9 (t, J = 2.3 Hz); 31P NMR (161 MHz, DMSO-d6) δ 19.4 (s); HRMS (ESI) m/z calcd. for C22H33BF3NaP [M + Na]+ 419.2257, found 419.2247.
3.2.22. B-4-(Trifluoromethyl)phenyl Tricyclohexylphosphine–Borane (37)
White solid; Yield:29%, 140 mg; MP 126.7–127.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.47–7.40 (m, 4H), 1.98–1.10 (br, 2H), 1.93–1.65 (m, 18H), 1.32–1.20 (m, 15H); 11B NMR (128 MHz, DMSO-d6) δ −28.8 (s); 13C NMR (125 MHz, DMSO-d6): δ 136.8 (d, JCP = 6.3 Hz), 125.5 (d, JCF = 271.7 Hz), 125.4 (qd, JCF = 31.1 Hz, JCP = 3.6 Hz), 123.6 (s), 30.4 (d, JCP = 28.7 Hz), 27.8 (s), 27.2 (d, JCP = 10.0 Hz), 26.2 (s); 19F NMR (376 MHz, DMSO-d6) δ −69.6 (d, J = 2.8 Hz); 31P NMR (161 MHz, DMSO-d6) δ 16.2 (s); HRMS (ESI) m/z calcd. for C25H39BF3ClP [M + Cl]− 473.2529, found 473.2529.
3.2.23. B-4-(Trifluoromethyl)phenyl Dicyclohexylphosphine–Borane (38)
White solid; Yield 37%, 132 mg, MP 52.7–53.7 °C; 1H NMR (400 MHz, acetone-d6) δ 7.54 (d, J = 7.1 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 4.27 (d, J = 357.0 Hz, 1H), 2.45–1.63 (br, 2H), 2.13–2.03 (m, 2H), 1.86–1.66 (m, 10H), 1.46–1.25 (m, 10H); 11B NMR (128 MHz, DMSO-d6) δ −29.4 (s); 13C NMR (100 MHz, acetone-d6) δ 137.0 (d, JCP = 7.7 Hz), 126.9 (qd, JCF = 31.4 Hz, JCP = 4.1 Hz), 126.3 (qd, JCF = 270.8 Hz, JCP = 1.3 Hz), 124.3–124.2 (m), 30.1 (s), 29.8 (d, JCP = 31.3 Hz), 29.1 (s), 27.4 (d, JCP = 10.4 Hz), 27.2 (d, JCP = 11.7 Hz), 26.5 (d, JCP = 1.1 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.7 (d, JFP = 3.4 Hz); 31P NMR (161 MHz, DMSO-d6) δ 10.5 (d, J = 368.2 Hz); HRMS (ESI) m/z calcd. for C19H29BF3NaP [M + Na]+ 379.1944, found 379.1964.
3.2.24. B-4-(Trifluoromethyl)phenyl Tris(dimethylamino)phosphine–Borane (39)
White solid; Yield:23%, 73 mg; MP 41.1–41.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.48 (d, J = 7.1 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 2.53 (d, J = 9.1 Hz, 18H), 2.30–1.56 (br, 2H); 11B NMR (128 MHz, DMSO-d6) δ −27.2 (d, J = 102.7 Hz); 13C NMR (125 MHz, DMSO-d6): δ 136.8 (d, JCP = 7.8 Hz), 125.6 (q, JCF = 271.7 Hz), 125.3 (qd, JCF = 31.2 Hz, JCP = 3.9 Hz), 123.42–123.37 (m), 37.4 (d, JCP = 3.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ −60.6 (d, J = 3.9 Hz); 31P NMR (161 MHz, DMSO-d6) δ 92.5 (s); HRMS (ESI) m/z calcd. for C13H24BF3N3KP [M + K]+ 360.1385, found 360.1398.
3.3. Determination of Hydrophobicity (LogP)
The 1-octanol/water partition coefficient P was determined using HPLC based on the OECD Guideline for Testing Chemicals. A COSMOSIL Packed Column 5C18-MS-II (5 μm, 150 mm × 4.6 mm id, Nacalai Tesque, Inc., Kyoto, Japan) was fitted on an HPLC instrument (Multiwavelength Detector, MD-2010 Plus. JASCO, Tokyo, Japan) equipped with a pump (PU-2080, JASCO) and an oven (SSC-2120, Senshu Scientific Co., Ltd., Tokyo, Japan). The injection volume was 20 µL; the mobile phase was methanol–water 75% (v/v); and the flow rate was 1.0 mL/min in all cases. Compounds were detected by measuring the UV absorption at 240 nm. The temperature of the column was kept at 40.0 (± 0.1) °C during the measurement. The measurement was performed in triplicate, and the mean value was calculated. The dead time t0 was measured with thiourea as the unretained compound, and the capacity factor k was calculated using the equation logk = log (tr − t0/t0), where tr represents the retention time of the compound. A calibration graph was determined experimentally using reference compounds (4-methylphenol, 4-chlorophenol, 4-phenylphenol, diphenylether, fluoranthene, and dichlorodiphenyltrichloroethane) with known logP values. The LogP values of phosphine–boranes were calculated based on a calibration graph (LogP = 2.6269 logkw + 3.059, R2 = 0.9915).
3.4. T47D Alkaline Phosphatase Assay for the Evaluation of PR Antagonistic Activity
T47D alkaline phosphatase assays were performed as previously described, with minor modifications [40]. Briefly, the human breast cancer cell line T47D (HMS LINCS database Accession Numbers: 50541) was routinely cultivated in RPMI 1640 medium with 10% FBS at 37 °C in a 5% CO2 humidified incubator. The cells were plated in 96-well plates and incubated overnight in a 5% CO2 humidified incubator at 37 °C. The next day, the cells were treated with a fresh medium containing the test compound in the presence of 1 nM of progesterone and further incubated for 48 h. The medium was aspirated, and the cells were fixed with 100 μL of 1.8% formalin–PBS. The fixed cells were washed with PBS, and 100 μL of an assay buffer (1 mg/mL p-nitrophenol phosphate in diethanolamine water solution, pH 9.0) was added. The mixture was incubated at room temperature for 2 h under light-shielded conditions. The absorbance was measured at 405 nm using a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA). All data points were measured in triplicate, and IC50 values were calculated from three independent experiments.
3.5. PR LBD Competitive Binding Assay
PR binding affinity was assessed using a PolarScreen Progesterone Receptor Competitor Assay Kit, Green (Invitrogen, Waltham, MA, USA, A15905) based on the manufacturer’s instruction. Briefly, PR-LBD(GST)/Fluormone PR Green Complex (final concentration: 6.5 nM) and test compounds (final concentration: 100 nM to 32 μM for the phosphine-boranes and 1 nM–320 nM for P4) in the assay buffer (final volume: 32 μM) were mixed on a 384-well plate (black, polypropylene). Then, the mixture was incubated at room temperature for 1 h under shade. The fluorescence polarization value (mP) was measured at 485 nm/535 nm (excitation/emission) on a DTX 880 Multimode Detector (Beckman Coulter).
3.6. Docking Simulation
The structure of the LBD of hPR was prepared from the Protein Data Bank accession number 3G8O [56]. Polar hydrogen atoms and partial atomic charges were assigned using AutoDockTools (ADT). Molecular docking was performed using AutoDock 4.2 with the genetic algorithm. The AutoDock parameters for boron atoms were Rii = 4.08 and eii = 0.180.
4. Conclusions
To expand the utility of phosphine–boranes in medicinal chemistry, we designed and synthesized a series of B-(trifluoromethyl)phenyl phosphine–borane derivatives and investigated their hydrophobic characteristics and biological activity toward the PR. The synthesized B-(trifluoromethyl)phenyl phosphine–borane derivatives exhibited predictable LogP values. We also demonstrated that the P–H group in phosphine–borane was nonpolar. Biological evaluation revealed that all synthesized phosphine–boranes, except for the secondary phosphine derivatives, exhibited PR antagonistic activity. The potency of the compounds depended on the bulkiness of the phosphine moiety, and the tricyclopropylphosphine substructure was found to be the most suitable for the designed PR antagonists. Docking simulations suggested that the tricyclopropylphosphine moiety plays an important role in ligand–receptor interactions and that optimization of the phenyl moiety could lead to the development of novel PR antagonists with improved potency. These results support the idea that phosphine–boranes are versatile structural options in medicinal chemistry for a wide variety of drug candidates, and the developed compounds, including 22 and 34, are promising lead compounds for further structural development of next-generation PR antagonists.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29071587/s1, 1H, 13C, 11B, 19F and 31P NMR Spectra of synthesized compounds.
Author Contributions
Conceptualization, S.F.; formal analysis, Y.M., funding acquisition, S.F. investigation, Y.M., K.O. and S.F.; project administration, S.F.; writing—original draft preparation, Y.M.; writing—review and editing, S.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by Grants-in-Aid for Scientific Research from JSPS (KAKENHI Grant Nos. 23K06046 (S.F.)), the Takeda Science Foundation (S.F.), the Hoansha Fundation (S.F.), and the Research Center for Biomedical Engineering (S.F.). This research was also partially supported by AMED under Grant Number JP23ama121043 (Research Support Project for Life Science and Drug Discovery, BINDS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article and Supplementary Materials.
Acknowledgments
We thank Yosuke Demizu for kind assistance with the PR competitive binding assay.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Staubitz, A.; Robertson, A.P.; Sloan, M.E.; Manners, I. Amine− and phosphine− borane adducts: New interest in old molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef]
- Brunel, J.M.; Faure, B.; Maffei, M. Phosphane-boranes: Synthesis, characterization and synthetic applications. Coord. Chem. Rev. 1998, 178, 665–698. [Google Scholar] [CrossRef]
- Paine, R.T.; Noth, H. Recent advances in phosphinoborane chemistry. Chem. Rev. 1995, 95, 343–379. [Google Scholar] [CrossRef]
- Imamoto, T.; Kusumoto, T.; Suzuki, N.; Sato, K. Phosphine oxides and LiAlH4–NaBH4–CeCl3; synthesis and reactions of phosphine-boranes. J. Am. Chem. Soc. 1985, 107, 5301–5302. [Google Scholar] [CrossRef]
- Imamoto, T.; Oshiki, T. Synthesis of Organic phosphorus compounds containing a linear P–B bond chain. Tetrahedron Lett. 1989, 30, 383–384. [Google Scholar] [CrossRef]
- Imamoto, T.; Oshiki, T.; Onozawa, T.; Kusumoto, T.; Sato, K. Synthesis and reactions of phosphine–boranes. Synthesis of new bidentate ligands with homochiral phosphine centers via optically pure phosphine–boranes. J. Am. Chem. Soc. 1990, 112, 5244–5252. [Google Scholar] [CrossRef]
- Li, P.; Sergueeva, Z.A.; Dobrikov, M.; Shaw, B.R. Nucleoside and oligonucleoside boranophosphates: Chemistry and properties. Chem. Rev. 2007, 107, 4746–4796. [Google Scholar] [CrossRef]
- Summers, J.S.; Shaw, B. Boranophosphates as mimics of natural phosphodiesters in DNA. Curr. Med. Chem. 2001, 8, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Schlieve, C.R.; Tam, A.; Nilsson, B.L.; Lieven, C.J.; Raines, R.T.; Levin, L.A. Synthesis and characterization of a novel class of reducing agents that are highly neuroprotective for retinal ganglion cells. Exp. Eye Res. 2006, 83, 1252–1259. [Google Scholar] [CrossRef]
- Remtulla, R.; Das, S.K.; Levin, L.A. Predicting absorption-distribution properties of neuroprotective phosphine-borane compounds using in silico modeling and machine learning. Molecules 2021, 26, 2505. [Google Scholar] [CrossRef]
- Saito, H.; Matsumoto, Y.; Hashimoto, Y.; Fujii, S. Phosphine boranes as less hydrophobic building blocks than alkanes and silanes: Structure-property relationship and estrogen-receptor-modulating potency of 4-phosphinophenol derivatives. Bioorg. Med. Chem. 2020, 28, 115310. [Google Scholar] [CrossRef]
- Miyajima, Y.; Noguchi-Yachide, T.; Ochiai, K.; Fujii, S. Physicochemical characterization of B-hydroxyphenyl phosphine borane derivatives and their evaluation as nuclear estrogen receptor ligands. RSC Med. Chem. 2024, 15, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Aupperlee, M.D.; Smith, K.T.; Kariagina, A.; Haslam, S.Z. Progesterone receptor isoforms A and B: Temporal and spatial differences in expression during murine mammary gland development. Endocrinology 2005, 146, 3577–3588. [Google Scholar] [CrossRef]
- Kolatorova, L.; Vitku, J.; Suchopar, J.; Hill, M.; Parizek, A. Progesterone: A steroid with wide range of effects in physiology as well as human medicine. Int. J. Mol. Sci. 2022, 23, 7989. [Google Scholar] [CrossRef]
- Africander, D.; Verhoog, N.; Hapgood, J.P. Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception. Steroids 2011, 76, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Spitz, I.M. Progesterone antagonists and progesterone receptor modulators: An overview. Steroids 2003, 68, 981–993. [Google Scholar] [CrossRef]
- Islam, M.S.; Afrin, S.; Jones, S.I.; Segars, J. Selective progesterone receptor modulators-mechanisms and therapeutic utility. Endocr. Rev. 2020, 41, bnaa012. [Google Scholar] [CrossRef] [PubMed]
- Critchley, H.O.D.; Chodankar, R.R. Selective progesterone receptor modulators in gynaecological therapies. J. Mol. Endocrinol. 2020, 65, T15–T33. [Google Scholar] [CrossRef]
- Singh, S.S.; Belland, L. Contemporary management of uterine fibroids: Focus on emerging medical treatments. Curr. Med. Res. Opin. 2015, 31, 1–12. [Google Scholar] [CrossRef]
- Proietti, C.J.; Cenciarini, M.E.; Elizalde, P.V. Revisiting progesterone receptor (PR) actions in breast cancer: Insights into PR repressive functions. Steroids 2018, 133, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Soria, V.; Gonzalez-Rodriguez, A.; Huerta-Ramos, E.; Usall, J.; Cobo, J.; Bioque, M.; Barbero, J.D.; Garcia-Rizo, C.; Tost, M.; Monreal, J.A.; et al. Targeting hypothalamic-pituitary-adrenal axis hormones and sex steroids for improving cognition in major mood disorders and schizophrenia: A systematic review and narrative synthesis. Psychoneuroendocrinology 2018, 93, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C. Progesterone signalling in breast cancer: A neglected hormone coming into the limelight. Nat. Rev. Cancer 2013, 13, 385–396. [Google Scholar] [CrossRef]
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef]
- Goyeneche, A.A.; Carón, R.W.; Telleria, C.M. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin. Cancer Res. 2007, 13, 3370–3379. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Shi, P.; Nie, Z.; Liang, H.; Zhou, Z.; Chen, W.; Chen, H.; Dong, C.; Yang, R.; Liu, S.; et al. Mifepristone suppresses basal triple-negative breast cancer stem cells by down-regulating KLF5 expression. Theranostics 2016, 6, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Choi, M.R.; Christov, K.; Ivancic, D.; Khan, S.A. Progesterone receptor antagonism inhibits progestogen-related carcinogenesis and suppresses tumor cell proliferation. Cancer Lett. 2016, 376, 310–317. [Google Scholar]
- Sitruk-Ware, R. Reprint of pharmacological profile of progestins. Maturitas 2008, 61, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Moguilewsky, M.; Philibert, D. RU 38486: Potent antiglucocorticoid activity correlated with strong binding to the cytosolic glucocorticoid receptor followed by an impaired activation. J. Steroid Biochem. 1984, 20, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.H.; Langan-Fahey, S.M.; Jordan, V.C. Estrogenic actions of RU486 in hormone-responsive MCF-7 human breast cancer cells. Endocrinology 1993, 6, 2622–2630. [Google Scholar] [CrossRef]
- Winneker, R.C.; Fensome, A.; Zhang, P.; Yudt, M.R.; McComas, C.C.; Unwalla, R.J. A new generation of progesterone receptor modulators. Steroids 2008, 73, 689–701. [Google Scholar] [CrossRef]
- Zhang, P.; Terefenko, E.A.; Fensome, A.; Wrobel, J.; Winneker, R.; Lundeen, S.; Marschke, K.B.; Zhang, Z. 6-Aryl-1,4-dihydro-benzo[d][1,3]oxazin-2-ones: A novel class of potent, selective, and orally active nonsteroidal progesterone receptor antagonists. J. Med. Chem. 2002, 45, 4379–4382. [Google Scholar] [CrossRef]
- Fensome, A.; Adams, W.R.; Adams, A.L.; Berrodin, T.J.; Cohen, J.; Huselton, C.; Illenberger, A.; Kern, J.C.; Hudak, V.A.; Marella, M.A.; et al. Design, synthesis, and SAR of new pyrrole-oxindole progesterone receptor modulators leading to 5-(7-fluoro-3,3-dimethyl-2-oxo-2,3-dihydro-1H-indol-5-yl)-1-methyl-1H-pyrrole-2-carbonitrile (WAY-255348). J. Med. Chem. 2008, 51, 1861–1873. [Google Scholar] [CrossRef]
- Sakai, H.; Hirano, T.; Mori, S.; Fujii, S.; Masuno, H.; Kinoshita, M.; Kagechika, H.; Tanatani, A. 6-Arylcoumarins as novel nonsteroidal type progesterone antagonists: An example with receptor-binding-dependent fluorescence. J. Med. Chem. 2011, 54, 7055–7065. [Google Scholar] [CrossRef]
- Fujii, S.; Yamada, A.; Nakano, E.; Takeuchi, Y.; Mori, S.; Masuno, H.; Kagechika, H. Design and synthesis of nonsteroidal progesterone receptor antagonists based on C,C′-diphenylcarborane scaffold as a hydrophobic pharmacophore. Eur. J. Med. Chem. 2014, 84, 264–277. [Google Scholar] [CrossRef]
- Fujii, S.; Nakano, E.; Yanagida, N.; Mori, S.; Masuno, H.; Kagechika, H. Development of p-carborane-based nonsteroidal progesterone receptor antagonists. Bioorg. Med. Chem. 2014, 22, 5329–5337. [Google Scholar] [CrossRef]
- Mori, S.; Takeuchi, Y.; Tanatani, A.; Kagechika, H.; Fujii, S. Development of 1, 3-diphenyladamantane derivatives as nonsteroidal progesterone receptor antagonists. Bioorg. Med. Chem. 2015, 23, 803–809. [Google Scholar] [CrossRef]
- Fensome, A.; Bender, R.; Chopra, R.; Cohen, J.; Collins, M.A.; Hudak, V.; Malakian, K.; Lockhead, S.; Olland, A.; Svenson, K.; et al. Synthesis and structure-activity relationship of novel 6-aryl-1,4-dihydrobenzo[d]-[1,3]oxazine-2-thiones as progesterone receptor modulators leading to the potent and selective nonsteroidal progesterone receptor agonist tanaproget. J. Med. Chem. 2005, 48, 5092–5095. [Google Scholar] [CrossRef]
- Collins, M.A.; Hudak, V.; Bender, R.; Fensome, A.; Zhang, P.; Miller, L.; Winneker, R.C.; Zhang, Z.; Zhu, Y.; Cohen, J.; et al. Novel pyrrole-containing progesterone receptor modulators. Bioorg. Med. Chem. Lett. 2004, 14, 2185–2189. [Google Scholar] [CrossRef]
- Yamada, A.; Kazui, Y.; Yoshioka, H.; Tanatani, A.; Mori, S.; Kagechika, H.; Fujii, S. Development of N-(4-phenoxyphenyl) benzenesulfonamide derivatives as novel nonsteroidal progesterone receptor antagonists. ACS Med. Chem. Lett. 2016, 7, 1028–1033. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Mori, S.; Makishima, M.; Fujii, S.; Kagechika, H.; Hashimoto, Y.; Ishikawa, M. Novel nonsteroidal progesterone receptor (PR) antagonists with a phenanthridinone skeleton. ACS Med. Chem. Lett. 2018, 9, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Baban, J.A.; Roberts, B.P. Homolytic reactions of ligated boranes. Part 9. Overall addition of alkanes to electron-deficient alkenes by a radical chain mechanism. J. Chem. Soc. Perkin Trans. 1988, 7, 1195–1200. [Google Scholar] [CrossRef]
- Kawano, Y.; Yamaguchi, K.; Miyake, S.Y.; Kakizawa, T.; Shimoi, M. Investigation of the stability of the M-H-B bond in borane σ complexes [M(CO)5(η1-BH2R⋅L)] and [CpMn(CO)2(η1-BH2R⋅L)](M= Cr, W; L= tertiary amine or phosphine): Substituent and Lewis base effects. Chem. Eur. J. 2007, 13, 6920–6931. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- OECD. Guideline for Testing of Chemicals 117, Partition Coefficient (n-octanol/Water), High Performance Liquid Chromatography (HPLC) Method; OECD Publishing: Paris, France, 2022. [Google Scholar]
- Finizio, A.; Vighi, M.; Sandroni, D. Determination of n-octanol/water partition coefficient (Kow) of pesticide critical review and comparison of methods. Chemosphere 1997, 34, 131–161. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995; Volume 2, ISBN 978-084-122-991-4. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- SwissADME. Available online: http://www.swissadme.ch (accessed on 27 September 2023).
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Di Lorenzo, D.; Albertini, A.; Zava, D. Progestin regulation of alkaline phosphatase in the human breast cancer cell line T47D. Cancer Res. 1991, 51, 4470–4475. [Google Scholar] [PubMed]
- Hiraoka, D.; Nakamura, N.; Nishizawa, Y.; Uchida, N.; Noguchi, S.; Matsumoto, K.; Sato, B. Inhibitory and stimulatory effects of glucocorticoid on androgen-induced growth of murine Shionogi carcinoma 115 in vivo and in cell culture. Cancer Res. 1987, 47, 6560–6564. [Google Scholar] [PubMed]
- Thompson, S.K.; Washburn, D.G.; Frazee, J.S.; Madauss, K.P.; Hoang, T.H.; Lapinski, L.; Grygielko, E.T.; Glace, L.E.; Trizna, W.; Williams, S.P.; et al. Rational design of orally-active, pyrrolidine-based progesterone receptor partial agonists. Bioorg. Med. Chem. Lett. 2009, 19, 4777–4780. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of P–B bond-containing biofunctional molecules.

Figure 2.
Structures of the endogenous PR agonist 5 and representative synthetic PR agonists and antagonists.
Figure 2.
Structures of the endogenous PR agonist 5 and representative synthetic PR agonists and antagonists.
Figure 3.
Design scheme of the B-(trifluoromethyl)phenyl phosphine–borane derivatives as PR antagonists.
Figure 3.
Design scheme of the B-(trifluoromethyl)phenyl phosphine–borane derivatives as PR antagonists.

Scheme 1.
Synthesis of the designed B-(trifluoromethyl)phenyl phosphine–borane derivatives 16–39. a The yield was calculated in two steps: (1) synthesis of phosphines without purification, (2) reductive P–B bond formation using DIBAL-H.
Scheme 1.
Synthesis of the designed B-(trifluoromethyl)phenyl phosphine–borane derivatives 16–39. a The yield was calculated in two steps: (1) synthesis of phosphines without purification, (2) reductive P–B bond formation using DIBAL-H.
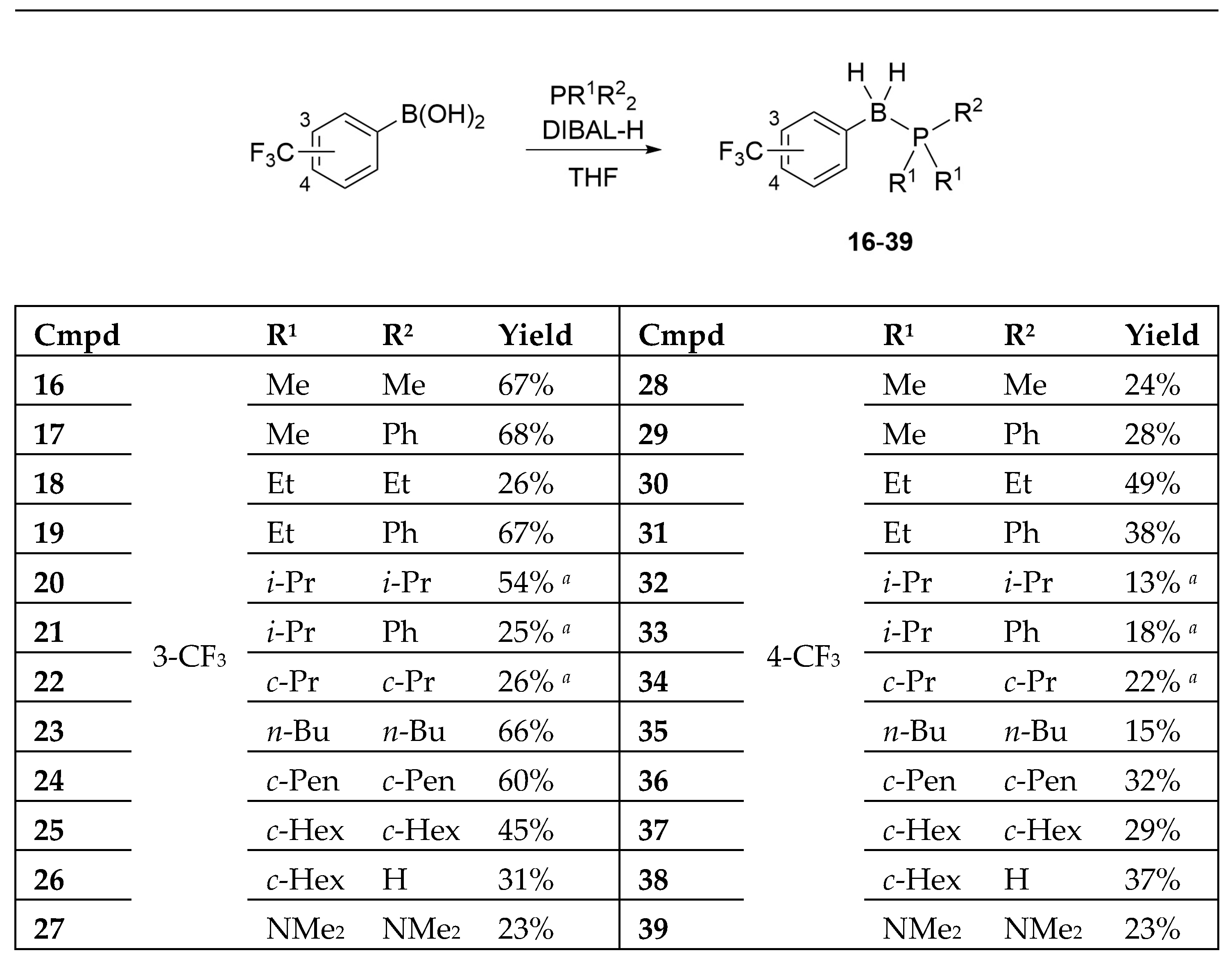
Figure 4.
Correlation (A) between experimentally measured LogP and calculated MLOGP values and (B) between experimentally measured LogP and calculated WLOGP values with the exception of amide derivatives 27 and 39.
Figure 4.
Correlation (A) between experimentally measured LogP and calculated MLOGP values and (B) between experimentally measured LogP and calculated WLOGP values with the exception of amide derivatives 27 and 39.
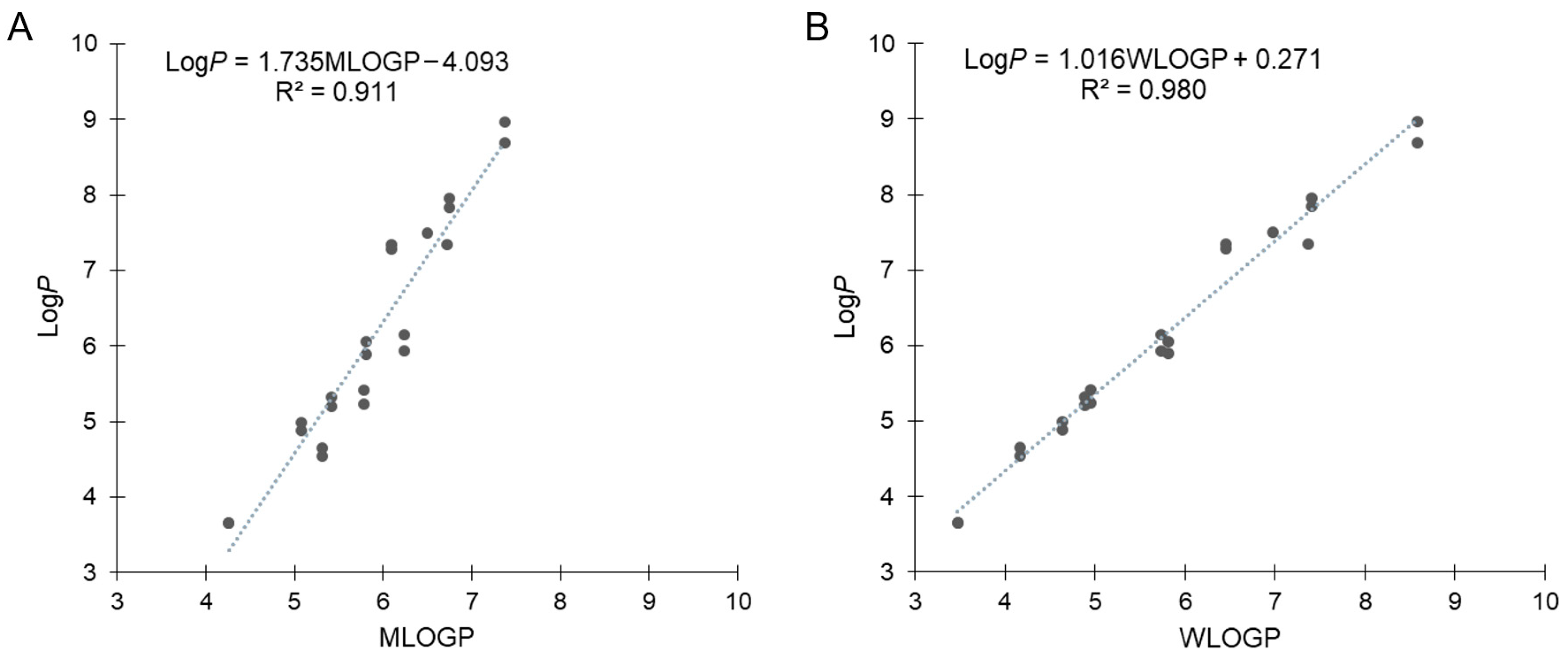
Figure 5.
Competitive binding assay using a fluorescence polarization system.

Figure 6.
Superimposition of the docked structures of B-(3-trifluoromethyl)phenyl tricyclopropylphosphine–borane (22: carbon in green) and B-(4-trifluoromethyl)phenyl tricyclopropylphosphine–borane (34: carbon in yellow), with hPR LBD, calculated using the docking program AutoDock. The protein surface is displayed as a blue mesh.
Figure 6.
Superimposition of the docked structures of B-(3-trifluoromethyl)phenyl tricyclopropylphosphine–borane (22: carbon in green) and B-(4-trifluoromethyl)phenyl tricyclopropylphosphine–borane (34: carbon in yellow), with hPR LBD, calculated using the docking program AutoDock. The protein surface is displayed as a blue mesh.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Determined LogP values and calculated hydrophobicity parameters MLOGP and WLOGP of the synthesized phosphine–borane derivatives 16–39.
Table 1.
Determined LogP values and calculated hydrophobicity parameters MLOGP and WLOGP of the synthesized phosphine–borane derivatives 16–39.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | LogP | MLOGP | Δ(LogP−MLOGP) | WLOGP | Δ(LogP−WLOGP) | |
| 16 | 3-CF3 | Me | Me | 3.65 | 4.26 | −0.61 | 3.47 | 0.18 |
| 17 | Me | Ph | 4.54 | 5.31 | −0.77 | 4.17 | 0.37 | |
| 18 | Et | Et | 4.88 | 5.07 | −0.19 | 4.64 | 0.24 | |
| 19 | Et | Ph | 5.24 | 5.78 | −0.54 | 4.95 | 0.29 | |
| 20 | i-Pr | i-Pr | 5.89 | 5.81 | 0.08 | 5.81 | 0.08 | |
| 21 | i-Pr | Ph | 5.93 | 6.24 | −0.31 | 5.73 | 0.20 | |
| 22 | c-Pr | c-Pr | 5.21 | 5.41 | −0.20 | 4.88 | 0.33 | |
| 23 | n-Bu | n-Bu | 7.34 a | 6.72 | 0.62 | 7.37 | −0.03 | |
| 24 | c-Pen | c-Pen | 7.84 a | 6.75 | 1.09 | 7.41 | 0.43 | |
| 25 | c-Hex | c-Hex | 8.69 a | 7.37 | 1.32 | 8.58 | 0.11 | |
| 26 | c-Hex | H | 7.28 a | 6.10 | 1.18 | 6.45 | 0.83 | |
| 27 | NMe2 | NMe2 | 5.39 | 3.00 | 2.39 | 3.01 | 2.38 | |
| 28 | 4-CF3 | Me | Me | 3.65 | 4.26 | −0.61 | 3.47 | 0.18 |
| 29 | Me | Ph | 4.65 | 5.31 | −0.66 | 4.17 | 0.48 | |
| 30 | Et | Et | 4.99 | 5.07 | −0.08 | 4.64 | 0.35 | |
| 31 | Et | Ph | 5.41 | 5.78 | −0.37 | 4.95 | 0.46 | |
| 32 | i-Pr | i-Pr | 6.06 | 5.81 | 0.25 | 5.81 | 0.25 | |
| 33 | i-Pr | Ph | 6.15 | 6.24 | −0.09 | 5.73 | 0.42 | |
| 34 | c-Pr | c-Pr | 5.32 | 5.41 | −0.09 | 4.88 | 0.44 | |
| 35 | n-Bu | n-Bu | 7.50 a | 6.50 | 1.00 | 6.98 | 0.52 | |
| 36 | c-Pen | c-Pen | 7.95 a | 6.75 | 1.20 | 7.41 | 0.54 | |
| 37 | c-Hex | c-Hex | 8.97 a | 7.37 | 1.60 | 8.58 | 0.39 | |
| 38 | c-Hex | H | 7.34 a | 6.10 | 1.24 | 6.45 | 0.89 | |
| 39 | NMe2 | NMe2 | 5.64 | 3.00 | 2.64 | 3.01 | 2.63 | |
a These LogP values are tentative because the hydrophobicity was beyond the range of the reference compounds (1.9 < LogP < 6.5).
Table 2.
PR antagonistic activity assessed using a T47D alkaline phosphatase assay and the calculated molecular volumes of synthesized phosphine–borane derivatives 16–39.
Table 2.
PR antagonistic activity assessed using a T47D alkaline phosphatase assay and the calculated molecular volumes of synthesized phosphine–borane derivatives 16–39.
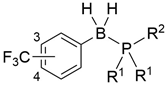 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | LogP | Volume (Å3) a | IC50 (μM) b | |
| 16 | 3-CF3 | Me | Me | 3.65 | 238 | >10 |
| 17 | Me | Ph | 4.54 | 304 | 1.16 ± 0.60 | |
| 18 | Et | Et | 4.88 | 294 | 3.67 ± 3.34 | |
| 19 | Et | Ph | 5.24 | 341 | 1.12 ± 0.22 | |
| 20 | i-Pr | i-Pr | 5.89 | 348 | 1.35 ± 0.47 | |
| 21 | i-Pr | Ph | 5.93 | 389 | 3.35 ± 1.49 | |
| 22 | c-Pr | c-Pr | 5.21 | 328 | 0.65 ± 0.17 | |
| 23 | n-Bu | n-Bu | 7.34 | 404 | >10 | |
| 24 | c-Pen | c-Pen | 7.84 | 424 | 6.64 ± 0.87 | |
| 25 | c-Hex | c-Hex | 8.69 | 474 | >10 | |
| 26 | c-Hex | H | 7.28 | 376 | N.D. c | |
| 27 | NMe2 | NMe2 | 5.39 | 332 | 1.20 ± 0.63 | |
| 28 | 4-CF3 | Me | Me | 3.65 | 238 | >10 |
| 29 | Me | Ph | 4.65 | 304 | 1.61 ± 0.87 | |
| 30 | Et | Et | 4.99 | 294 | 1.76 ± 1.44 | |
| 31 | Et | Ph | 5.41 | 341 | 1.78 ± 0.97 | |
| 32 | i-Pr | i-Pr | 6.06 | 347 | 1.51 ± 0.53 | |
| 33 | i-Pr | Ph | 6.15 | 377 | 2.01 ± 0.58 | |
| 34 | c-Pr | c-Pr | 5.32 | 328 | 0.54 ± 0.09 | |
| 35 | n-Bu | n-Bu | 7.50 | 404 | >10 | |
| 36 | c-Pen | c-Pen | 7.95 | 424 | 2.63 ± 0.72 | |
| 37 | c-Hex | c-Hex | 8.97 | 474 | >10 | |
| 38 | c-Hex | H | 7.34 | 376 | N.D. c | |
| 39 | NMe2 | NMe2 | 5.64 | 332 | 1.40 ± 0.88 | |
a These values were calculated for structures optimized at the B3LYP/6-311 + G** level using the Spartan’ 18 program (Wavefunction, Inc.). b The IC50 values are shown as the mean ± SD from three independent experiments. The P4 concentration was 1.0 nM. c Not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Miyajima, Y.; Ochiai, K.; Fujii, S. Design, Synthesis, and Evaluation of B-(Trifluoromethyl)phenyl Phosphine–Borane Derivatives as Novel Progesterone Receptor Antagonists. Molecules 2024, 29, 1587. https://doi.org/10.3390/molecules29071587
AMA Style
Miyajima Y, Ochiai K, Fujii S. Design, Synthesis, and Evaluation of B-(Trifluoromethyl)phenyl Phosphine–Borane Derivatives as Novel Progesterone Receptor Antagonists. Molecules. 2024; 29(7):1587. https://doi.org/10.3390/molecules29071587
Chicago/Turabian StyleMiyajima, Yu, Kotaro Ochiai, and Shinya Fujii. 2024. "Design, Synthesis, and Evaluation of B-(Trifluoromethyl)phenyl Phosphine–Borane Derivatives as Novel Progesterone Receptor Antagonists" Molecules 29, no. 7: 1587. https://doi.org/10.3390/molecules29071587