
Organophosphates as Versatile Substrates in Organic Synthesis

Department of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(7), 1593; https://doi.org/10.3390/molecules29071593
Submission received: 1 February 2024
/
Revised: 25 March 2024
/
Accepted: 26 March 2024
/
Published: 2 April 2024
(This article belongs to the Section Organic Chemistry)
Abstract
:This review summarizes the applications of organophosphates in organic synthesis. After a brief introduction, it discusses cross-coupling reactions, including both transition-metal-catalyzed and transition-metal-free substitution reactions. Subsequently, oxidation and reduction reactions are described. In addition, this review highlights the applications of organophosphates in the synthesis of natural compounds, demonstrating their versatility and importance in modern synthetic chemistry.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Organophosphates are recognized as organic esters derived from phosphoric acid and are of significant interest because of their biological activities. A prominent example is adenosine triphosphate (ATP), which serves as a crucial energy carrier within the human body (Scheme 1). Compounds, such as dicrotophos and monocrotophos, function as acetylcholinesterase inhibitors [1] and are used as insecticides [2]. Paraoxon, a metabolic derivative of the pesticide parathion, is acknowledged as a potent cholinesterase inhibitor [3]. In addition, tochlophos-methyl is used as a fungicide [4]. Fenitrothion, used as an insecticide and known commercially as Sumithion, is an essential inclusion in the list of significant organophosphates.
Organic synthesis has experienced significant advancements in recent decades. Beyond the realm of traditional methods, techniques involving transition-metal complexes [5,6,7], electrosynthesis [8,9,10,11,12], and photochemical transformations [13,14,15] are widely used. These developments have allowed for the preparation of a wide range of compounds as well as transformations that include the formation of C–C bonds [16,17,18,19,20,21], the synthesis of heterocyclic compounds [22,23,24,25,26,27,28], and the preparation of tetrasubstituted alkenes [29,30,31,32,33,34,35,36]. In addition to conventional starting compounds, organophosphates are increasingly utilized in organic synthesis, likely due to their availability. The phosphorylation of alcohols, phenols [37], and enolates [38,39,40] represents the methods that are frequently used. Furthermore, specific reactions have been developed for the synthesis of compounds such as vinyl phosphates. The Perkow reaction [41] serves as an example, and is widely adopted for the production of vinyl phosphates.
Significant interest in the chemistry of organophosphorus compounds is evident from several recent books and Special Issues in prominent journals, including Molecules [42,43], Beilstein Journal of Organic Chemistry [44], and the Journal of Organic Chemistry [45]. However, a comprehensive review that systematically summarizes the reactivity of organophosphates is currently missing. Therefore, the objective of this review is to summarize the main applications of organophosphates in organic synthesis over the past decade (Scheme 2). This review is structured into distinct sections for thorough coverage. The first part begins with an in-depth examination of cross-coupling reactions involving organophosphates. This section includes detailed discussion of C–H bond activations and various substitution reactions. The final part concludes with a comprehensive summary of oxidations and reductions, primarily focusing on vinyl and alkyl phosphates.
2. Organophosphates as Electrophiles in Transition-Metal-Catalyzed Reactions
Cross-coupling reactions frequently make use of a wide range of electrophiles. This holds true for organophosphates, which are often used due to their availability. Recent applications of organophosphates in cross-coupling reactions include the Kumada, Negishi, and Suzuki reactions. Alternatively, vinyl phosphates are employed as a directing group for the activation of the C–H bond (Scheme 3).
2.1. Kumada (Kumada–Tamao–Corriu) Reaction
Mazet developed a simple procedure for the preparation of conjugated dienes (Scheme 4) [46]. The process begins with diethyl vinyl phosphates 4–1, which are transformed into dienes 4–2 through a reaction with vinylmagnesium bromide, facilitated by a catalytic amount of nickel complex. Typically conducted at room temperature, the versatility of this method is evident in a wide range of synthesized dienes. It allows for the preparation of monosubstituted dienes featuring phenyl or ferrocenyl substituents 4–2a and 4–2b, as well as dienes with a steroid skeleton 4–2c. Although the scope has been predominantly explored using vinylmagnesium bromide, the authors also demonstrated its efficacy in generating simple disubstituted dienes, such as diene 4–2d. The stereoselectivity of this cross-coupling reaction is highly influenced by the structure of the initial phosphate. For example, the reaction of phosphates 4–1e and 4–1f with vinylmagnesium bromide predominantly produces E-alkenes 4–2e and 4–2f. In contrast, phosphate with a trimethylsilyl group tends to undergo isomerization at the double bond, resulting in Z-diene 4–2g. The simplicity and effectiveness of this methodology are further underscored by its widespread adoption in numerous publications [47,48,49,50,51,52,53].
Mazet and his team conducted an in-depth study of the mechanism underlying the Kumada reaction (Scheme 5) [54]. In this Ni(dppe)Br2-catalyzed reaction, active catalyst B is generated through a process of double transmetalation, followed by reductive elimination. Subsequently, the Ni(0) complex undergoes oxidative addition, leading to the formation of bromide complex D by ligand substitution. The rate-determining step of this reaction is transmetalation with vinylmagnesium bromide, which culminates in the creation of the reaction-product complex F via reductive elimination. A comparable mechanism is hypothesized for reactions catalyzed by the Ni(dmpe)Br2 complex.
The sensitivity of the Kumada reaction of phosphate 6–1 to the amount of vinylmagnesium bromide is reported (Scheme 6) [55]. When the amount of vinylmagnesium bromide exceeds 1.05 equivalence, the resultant diene 6–2 undergoes polymerization to produce a 1,4-cis polymer 6–3. This polymerization process of diene 6–2 is facilitated by magnesium salts, as illustrated by the fact that the average molecular weight of the resultant polymer is influenced by the ratio of the starting phosphate 6–1 to the magnesium salt.
The same research group adopted an identical methodology to synthesize [n]dendralenes by the Kumada reaction of vinyl phosphate 7–2 (Scheme 7) [56]. In this process, readily available phosphate 7–2 was subjected to a reaction with related Grignard reagents under different conditions. In particular, [3]dendralene 7–3a was efficiently synthesized using vinylmagnesium, achieving a high yield in just one hour. In contrast, the synthesis of [4]- and [6]dendralenes required significantly longer reaction times. The low yield of [6]dendralene 7–3c might be attributed to the reduced stability of [n]dendralenes with an increasing number of vinyl units [57].
In some cases, the Kumada reaction of organophosphates can be catalyzed by iron complexes. For example, Wang described a cross-coupling reaction of pyrimidyl diethyl phosphates with Grignard reagents that yields substituted pyrimidines 8–2 (Scheme 8) [58]. Although this method was successfully extended to the substituted pyridine derivative 8–3, it is important to note that the reaction conditions are limited to the preparation of substituted pyrimidines 8–2.
Sun and co-workers developed a novel class of ionic iron complexes. One of the prepared complexes successfully catalyzed the Kumada reaction of aryl or heteroaryl diethyl phosphates with alkylmagnesium bromides (Scheme 9a) [59]. The reaction conditions in this study were predominantly used to prepare naphthalene decorated with alkyl substituents. Additionally, alkylated pyridine and 1,1’-biphenyl derivatives were also successfully prepared, as evidenced from selected examples 9–2b and 9–2c. In addition to the aforementioned protocol, Shen [60] and Sun [61] demonstrated that the cross-coupling reaction of organophosphates 9–3a and 9–3b with organobromides 9–4 can be conducted under reductive cross-coupling conditions (Scheme 9b).
The Kumada reaction has also been used as a complementary method in some studies to broaden the scope of the research topic. Selected examples of this application are illustrated in Scheme 10. A substituted cyclobutene 10–2 was synthesized using a variation of the Mazet methodology (Scheme 10a) [62]. The Kumada reaction can be catalyzed via palladium complexes, as evidenced by the synthesis of cyclic alkenes 10–4 [63] and 10–6 [64] (Scheme 10b,c). Furthermore, Kotek published a stereoselective synthesis of tetrasubstituted alkenes from triply electrophilic templates, employing the Kumada reaction as the final step (Scheme 10d). Under optimized conditions, this method yielded tetrasubstituted alkene 10–8 as a single stereoisomer [65]. Brown [66] and Yoshikai [67] used similar conditions for the cross-coupling reaction of trisubstituted vinyl phosphates. In particular, the cross-coupling reaction of vinyl phosphates with Grignard reagents catalyzed by precatalysts, such as Ni(acac)2 [68], Fe(acac)3 [69,70], and Pd(PPh3)4 [71], has been used to synthesize a diverse range of alkenes.
The Kumada reaction has been extensively used in the total syntheses of natural substances. An illustrative example is the nickel-catalyzed Kumada reaction, where naphthyl phosphate 11–1 reacts with an alkyl Grignard reagent (Scheme 11) [72]. Additionally, the Kumada reaction catalyzed through iron and nickel complexes was used to prepare a sex pheromone of Lobesia botrana [73], (–)-piperitylmagnolol [74], and indole alkaloids [75].
2.2. Negishi Reaction
The efficiency of the cross-coupling reaction involving organophosphates and organozinc, as well as trialkylaluminum reagents, is significantly influenced by the molecular structures of both organophosphate and organometallic reagents. For example, trialkyl aluminums can react with aryl diethyl phosphate at room temperature in the presence of a nickel complex [76]. In contrast, trisubstituted vinyl phosphates 12–1 undergo a reaction with organozinc reagents in refluxing THF or acetonitrile (Scheme 12) [77]. Under these conditions, the successful incorporation of heterocyclic and arylzinc halides, both with electron-donating and electron-withdrawing groups, was achieved. Similarly demanding conditions for the Negishi reaction of vinyl diphenyl phosphate 12–3 have been published by Shi and Xiao [78].
Recently, a novel approach for the stereoselective synthesis of tetrasubstituted alkenes was introduced (Scheme 13a) [79]. Starting phosphate 13–1 was synthesized via the double Suzuki coupling of dibromovinyl phosphates. Subsequently, the phosphate moiety was replaced via Negishi coupling in the presence of aluminum chloride. The developed reaction conditions tolerate various functional groups, including ester and nitrile groups. The authors proposed that the coordination of aluminum chloride with the phosphate group facilitates the oxidative addition of Pd(0) to the C–O bond. However, they noted that vinyl phosphate undergoes simultaneous decomposition in the presence of aluminum chloride. This methodology offers a straightforward and stereoselective route to (Z)-tamoxifen and its derivatives from easily available phosphate 13–3 (Scheme 13b).
Dibromovinyl or bromovinyl phosphates have been proven to be effective for the synthesis of substituted indoles and [n]dendralenes (Scheme 14). The synthesis of substituted indoles starts with Negishi coupling using 14–1, which results in an indole derivative 14–2. Subsequently, the synthesis of biologically active indole 14–3 was achieved by removing the Boc group using additional aluminum chloride (Scheme 14a) [80]. Furthermore, Scheme 14b illustrates the modular synthesis of [4]dendralenes by the Negishi reaction of cyclobutenyl phosphate 14–5 [81]. The starting cyclobutenyl phosphate 14–5 is synthesized from 2-bromocyclobutenyl phosphate 14–4. Subsequent Negishi coupling yields cyclobutenyl derivatives 14–6. An electrocyclic ring opening of this compound produced [4]dendralene 14–7. A similar approach has been employed for the preparation of 2-substituted cyclobutanones [82] and cyclobutenones [83]. Further studies have shown that aluminum chloride facilitates the Negishi reaction of cycloalkenyl diphenyl phosphates at room temperature [84,85].
2.3. Suzuki Reaction
The Suzuki reaction has become a widely used tool for the formation of C–C bonds. The use of organophosphates in the Suzuki reaction has received significant attention in recent studies [86,87,88,89,90]. A recent example is the nickel-catalyzed transformation of aryl (diethyl) phosphates 14–1 in the presence of bis(tricyclohexylphosphine)nickel(II) dichloride (Scheme 15) [91]. This reaction is characterized by high yields of substituted biaryls. However, the scope of this reaction is limited to arylboronic acids. Vinyl and alkyl boronic acids were not used. The proposed mechanism for the published transformation involves the oxidative addition of a Ni(0)L2 complex, followed by transmetalation. The reaction product 15–2 is then produced by reductive elimination, and the nickel complex is regenerated by ligand substitution.
In contrast to diethyl phosphates, diphenyl phosphates are more common in the Suzuki reaction due to the higher reactivity of diphenyl phosphates. This is exemplified in a recent study presented in Scheme 16 [92]. Gigant and co-workers used dioxazaborocanes in a palladium-catalyzed reaction with phosphate enamides. The versatility of the reaction extends beyond the synthesis of azepine derivatives, since it accommodates a range of aromatic heterocyclic compounds, including substances 16–2b and 16–2c. However, the reaction conditions were specifically suited for vinyl phophates, and attempts to use aryl phosphates 16–1a were unsuccessful. In addition, Senra and colleagues introduced an innovative palladium-supported layered double-hydroxide catalytic system for the Suzuki reaction of vinyl phosphates. Their research focused primarily on exploring the properties of this catalytic system; therefore, the scope of their reported reaction remains limited [93].
Recently, the Suzuki reaction between dihydropyranylphosphates 17–2 and 4-methoxyphenylboronic acid pinacol ester has been published (Scheme 17) [94]. Following careful optimization, it was determined that the most effective reaction conditions involved the use of Pd(PPh3)4 in THF under reflux. The optimized reaction conditions were used for the preparation of C-arylglycal 17–3. The starting lactone 17–1 was synthesized from 4-di-O-acetyl-L-rhamnal and the phosphorylation itself was mediated by LiHMDS. However, the Suzuki reaction required 20 mol% of the catalyst to suppress the undesired chelation of the palladium atom to pyran oxygen. The final product 17–3 was obtained with a yield of 54% in two steps.
A popular variation of the Suzuki reaction is Miyaura borylation. This process typically involves the reaction of an electrophile with bis(pinacolato)diboron (Bpin-Bpin) in the presence of a catalytic amount of diverse transition-metal complexes [95,96]. Although organohalogens are the electrophiles of choice, organophosphates are also amenable to borylation. A recent example is the borylation of arylphosphates under photocatalytic conditions (Scheme 18) [97]. In this methodology, an aryl phosphate with a high negative reduction potential is activated via a phenothiazine photocatalyst, which exhibits enhanced reduction potential due to a proton-coupled electron transfer (PCET). Under optimized reaction conditions, a variety of aryl diethyl phosphates can be borylated, including esters and amides 18–2b and 18–2c. The synthesis of organotrifluoroborates is accessible; however, the crude reaction mixture after borylation requires treatment with KHF2 or KF. Furthermore, Li recently reported a metal-free borylation of aryl halides under continuous-flow conditions, although this method has a limited scope with respect to aryl diethylphosphates [98].
The Suzuki reaction of organophosphates has been widely used for the preparation of various substances. Selected examples 19–2a [99], 19–2b [100], 19–2c [101], and 19–2d [102] are given in Scheme 19. In most cases, diphenyl phosphates were treated with aromatic and heteroaromatic boronic acids or their pinacol esters in the presence of a catalytic amount of palladium or nickel complexes. In addition, Asano and co-workers used the double Suzuki reaction for the synthesis of the heterocyclic compound 19–2e [103].
The application of the Suzuki reaction of organophosphates includes the synthesis of natural substances. Fuwa and Sasaki published a palladium-catalyzed Suzuki reaction of borane 20–1 with diphenyl phosphate 20–2 for the preparation of the dihydropyran derivative 20–3 en route to didemnaketal B (Scheme 20) [104]. The same authors used a similar Suzuki reaction to synthesize the C15–C38 fragment of akadaic acid [105].
In contrast, the published preparation of (–)-anabasine (21–5) took advantage of the palladium-catalyzed Suzuki–Miyaura borylation. This process was applied to crudely isolated phosphate 21–2 in the presence of barium hydroxide (Scheme 21) [106]. The prepared pinacol boronic acid ester 21–3 was treated with 3-bromopyridine to give alkene 21–4 with a 56% isolated yield. Finally, (–)-anabasine was obtained by reducing cycloalkene 21–4.
2.4. C–H Activation
Organophosphates are also widely used in the transition-metal-catalyzed activation of the C–H bond. The phosphate group can serve as a directing group to facilitate the activation of the C–H bond. Alternatively, organophosphates are used as electrophiles for the C–H bond activation (Scheme 22).
An example of C–H-bond activation involves the rhodium-catalyzed vinylation of vinyl phosphates 23–2 (Scheme 23) [107]. A comprehensive series of dienyl diethyl phosphates was prepared under optimized reaction conditions. Although the scope of the reaction was extensively explored with methyl acrylate, styrene derivatives also performed similarly, as illustrated in example 23–2b. However, the reaction with acrylonitrile resulted in the vinylation product 23–2c, albeit with a reduced ZE/ZZ ratio. Adjusting the amount of copper acetate to 60 mol% and the use of enones led to the formation of hydroalkylation product 23–2d.
The phosphate-directing group has been applied to the synthesis of biaryls 24–2 from aryl diethyl phosphates 24–1 (Scheme 24) [108]. However, the scope of this reaction is limited to the preparation of simple biaryls and cyclic lactone 24–2c. The proposed mechanism involves the formation of Pd(IV) complex B, which then undergoes reductive elimination to yield the coupling product 24–2.
In addition to the dialkyl phosphate group, a monophosphoric acid-directing group can also facilitate the activation of the C–H bond (Scheme 25) [109]. The reaction scope was assessed using diphenyliodonium triflate in the presence of a catalytic amount of palladium trifluoroacetate. However, arylation with mixed iodonium salts is possible, as illustrated by the selected examples 25–2d and 25–2e. The authors assumed that the C–H-bond-activation reaction involved the Pd(II)↔Pd(IV) catalytic cycle.
Organophosphates are also used as electrophiles in C–H-bond activation. In a series of studies, Ackermann and co-workers investigated vinyl acetates, phosphates, and carbonates as electrophiles for the cobalt-catalyzed C–H bond activation of indoles (Scheme 26a) [110,111]. The optimized reaction conditions included the use of an N-heterocyclic carbene (NHC) ligand, enabling the introduction of an alkenyl group from vinyl acetates at room temperature. Diethyl vinyl phosphates are effective electrophiles, as demonstrated by examples 26–2 and 26–3. Unfortunately, the scope of the reaction is limited to simple vinyl phosphates despite the availability of a wide range of functionalized vinyl phosphates. It is worth noting that cyclohexyl acetates exhibit higher reactivity compared to their cyclohexenyl phosphate counterparts. Besides cobalt-catalyzed C–H-bond activation, Cramer reported the enantioselective palladium-catalyzed cyclization of ketene aminal phosphate, leading to an indolizidine scaffold (Scheme 26b) [112].
Another cobalt-catalyzed C–H-bond activation involves aromatic imines 27–1 (Scheme 27) [113]. Similar to Ackermann’s work, the NHC ligand, whose structure is shown in Scheme 26, is also used in this case. The advantage of the imino-directing group lies in its ability for late-stage transformation, either by hydrolysis into ketone 27–2 or reduction to a secondary amine 27–3. Cyclohexenyl phosphates were used predominantly because the formation of a mixture of E and Z isomers was observed for the acyclic derivative 27–2b. Furthermore, an imine with an electron-withdrawing trifluoromethyl group was unreactive. The optimized reaction conditions were effectively applied to substrates with a 2-pyridyl directing group, as exemplified by the synthesis of compounds 27–4 and 27–5. In a subsequent publication, the same group applied cobalt-catalyzed C–H-bond activation to unprotected imines [114]. In this publication, a mechanism for the studied reaction is also presented.
In addition to previous transition-metal-catalyzed reactions of organophosphates, the concept of reactions catalyzed by transition-metal complexes was also used for the preparation of aromatic amines from triaryl phosphates (Scheme 28a). The nickel-catalyzed arylation of amines proceeds in dioxane at 110 °C [115]. The optimized reaction conditions are characterized by a wide reaction scope; however, the conditions failed to deliver a substrate with an ethoxycarbonyl group in the ortho position 28–2b. The palladium-catalyzed amination of organophosphates encompasses a broad spectrum of N-heterocyclic compounds, such as indole derivatives marked as 28–2f [116]. In addition, the same catalytic conditions include [Pd(2-butenyl)Cl]2 along with the MorDalPhos ligand to facilitate the α-arylation of aryl and heteroaryl ketones [117]. Recently, the nickel-catalyzed amination of organohalides was reported by Hong and Shi. The optimized reaction conditions are suitable for the amination of carbonate, tosylate, carbamate and organophosphate 28–4 (Scheme 28b) [118]. In addition, the synthesis of carbapenems 28–7 was realized by the substitution of the diphenyl phosphate group for the corresponding thiol (Scheme 28c) [119].
3. Organophosphate Rearrangements
Organophosphates are involved in two types of rearrangements. The first rearrangement is the [1,2]-phospha-Brook rearrangement, which follows the traditional Brook rearrangement [120,121,122]. This process starts with the reaction of a carbonyl compound with dialkylphosphite in the presence of a base (Scheme 29). The initially formed phosphonate 29–1 undergoes rearrangement to produce an alkyl phosphate 29–2, which is then amenable to a further reaction with an electrophile. Terada’s group has extensively used the [1,2]-phospha-Brook rearrangement for the synthesis of substituted furans [123,124], pyrroles [125], phenantherenes [126], indolizine [127], piperidines [128], oxindoles [129], chromenes [130], and many others [131,132,133,134,135,136,137]. The [1,2]-Phospha-Brook rearrangement has also attracted significant attention from other research groups, leading to numerous studies [138,139,140,141,142] and comprehensive reviews [143,144]. The second rearrangement associated with organophosphates is the anionic phospha-Fries rearrangement. First reported by Melvin in 1981 [145], this rearrangement typically involves the isomerization of aryl phosphates to aryl phosphonates in the presence of a base, typically LDA. Since its discovery, the anionic phospha-Fries rearrangement has been used as a cornerstone to synthesize lithium borates as promising materials for lithium-ion batteries [146] and receptors for the complexation of lanthanides [147] and other transition metals [148,149]. Recently, Snieckus’s group reported an anionic phospha Fries rearrangement of aryl tetraethylphosphorodiamidates [150,151]. A review of the phospha-Fries rearrangement was published in 2005 [152]; therefore, this chapter summarizes recent findings in this area.
The phospha-Fries rearrangement has been used for the preparation of monoarylphosphines (Scheme 30) [153]. Initially, the starting ortho-bromophenol 30–1 is converted to phosphate 30–2. Subsequently, the phospha-Fries rearrangement is initiated by a bromine–lithium exchange reaction. The formed phosphonate 30–3 is reduced, using lithium aluminum hydride to phosphine 30–4a in a total isolated yield of 39% (three steps). In addition to the model example 30–4a, three additional monoaryl phosphines 30–4b, 30–4c, and 30–4d were prepared. However, the prepared phosphines have limited stability and decompose to phenol and phosphine. The authors proposed that the decomposition of phosphines 30–4 is catalyzed by traces of acid originating from the workup of the reduction of phosphonates 30–3.
In 2014, Han and Yin published a study on a copper-catalyzed phosphorylation of 2-halophenols en route to substituted phenols [154]. As part of their research, they studied the phospha-Fries rearrangement of aryl phosphinates that contain (–)-menthol 31–1 (Scheme 31). An important aspect of their findings is that the reaction proceeded with complete retention of configuration at the phosphorus atom. Thus, the phosphate RP-31–1 phosphinate is transformed into phosphonate RP-31–3. In contrast, they also showed that the cross-coupling of 2-bromophenol with phosphinate RP-31–4 gave the phosphonate SP-31–3e in a moderately isolated yield.
The anionic phospha-Fries rearrangement was also used to prepare the ferrocene ligand rac-32–5 (Scheme 32) [155]. The starting phosphate 32–1 is isomerized to the anion 32–2. Subsequent alkylation with dimethyl sulphate yields ferrocenyl ether rac-32–3. The phosphonate group of ferrocene rac-32–3 is then reduced with lithium aluminum hydride. Subsequent arylation through Stelzer P,C cross-coupling [156] gave the desired phosphine rac-32–5. The isolated phosphine rac-32–4 has been used successfully in Suzuki coupling to synthesize the corresponding biaryls. This application is exemplified by the representative synthesis of biaryl 32–6, which involves the reaction of phenanthren-9-ylboronic acid with 2-methoxy-1-bromonaphthalene.
The same group studied the phospha-Fries rearrangement of ferrocenyl bisphosphates [157]. In this study, they found that bisphosphonates can be synthesized by sequential phospha-Fries rearrangement (Scheme 33). Initially, ferrocenyl phosphate 33–1 undergoes rearrangement to form phosphonophosphate 33–2. This intermediate is then subjected to a second phospha-Fries rearrangement to give the final product 33–3. The behavior of mixed phosphates 33–4 and 33–5 was also investigated. However, the rearrangement of phosphates 33–4 and 33–5 resulted in a product mixture demonstrating limited chemoselectivity [158].
The rearrangement of organophosphates has found applications in the total synthesis of natural products. A notable example is the Ireland–Claisen rearrangement of vinyl phosphate 34–2 used in the synthesis of clavigerins B and C (Scheme 34a) [159]. After rearrangement, the hydrolysis of the crude reaction mixture yielded carboxylic acid 34–3 with a 63% yield. Furthermore, the preparation of cyclocitrinols makes use of an optimized version of alkyl diphenyl phosphate rearrangement (Scheme 33b) [160,161]. This rearrangement begins with the oxidation of sulfide 34–4 to the corresponding sulfoxide. Then, the sulfoxide was rearranged to intermediate 34–5.
Kaabi and Besbes documented the rearrangement of aliphatic diethyl phosphates 35–1 into α-amino acids 35–2 (Scheme 35) [162]. While the scope of the reaction is confined primarily to secondary amines with simple alkyl substituents, the authors propose that the formation of an aziridinium ion 35–3 plays a crucial role as an intermediate in this transformation.
4. Transition-Metal-Free Substitution Reactions of Organophosphates
Recent studies have introduced two distinct methods for the substitution of benzyl phosphates (Scheme 36). In the first method [163], diphenylmethyllithium was used as a nucleophile for the SN2 substitution of diphenyl phosphates 36–1a. Moreover, the authors also prepared two advanced aromatic hydrocarbons, 36–2b and 36–2c. The SN2 nucleophilic substitution mechanism was confirmed using an enantiomerically pure benzyl phosphate, which underwent substitution proceeded with inversion of configuration. The same group further broadened the scope of the reaction to cycloalkenyl and propargyl phosphates [164]. In contrast, Chakravarty [165] reported the Friedel-Crafts alkylation of aromatic hydrocarbons [165]. The authors used a catalytic amount of trifluoromethanesulfonic acid (TfOH) to generate a benzyl carbocation that subsequently reacted with an aromatic hydrocarbon (ArH). The efficacy of this reaction is demonstrated through the synthesis of selected products 36–3a, 36–3b, and 36–3c. Following previous work [166], Yamamoto’s group developed a ferric triflate-catalyzed method for the formation of (difluoromethyl)(diaryl)methanes from the corresponding 2,2-difluoro-1-arylethyl phosphates [167].
In 2020, a novel cyclopropanation reaction was introduced for the preparation of cyclopropylphosphinoxides (Scheme 37) [168]. The formation of cyclopropane derivatives is achieved by the interaction of allyl diethyl phosphates 37–1 with lithium phosphides in tetrahydrofuran. Upon the completion of the reaction, the resulting phosphines 37–2 are oxidized to stable phosphinoxides 37–3. An interesting aspect of this reaction is its sensitivity to the choice of solvent. For example, the use of cyclopentyl methyl ether (CPME) results in the formation of an SN2 nucleophilic substitution product 37–4, whereas tetrahydrofuran preferentially yields the cyclopropanation product 37–3. Further experiments indicate that the selectivity between the cyclopropanation product 37–3 and SN2 product 37–4 is influenced by the electrophilicity at the β-position of the allylic moiety.
Scheme 38 illustrates the application of the catalytic activation of the phosphate group for β-selective glycosylation [169,170]. Central to this process is a bifunctional and chiral catalyst. This catalyst serves a dual role: it activates the phosphate group via coordination and facilitates in the proton abstraction from the alcohol. When the phosphate group is activated by Lewis acid, followed by a reaction with a corresponding alcohol, the product is predominantly in the thermodynamically more stable α-anomer form. Selected examples of tested alcohols show that phenols can also be used.
Intramolecular substitution at the phosphate group at 2-oxoindolinyl phosphates 39–1 has been used to synthesize spiroindolines 39–3 (Scheme 39) [171]. In this study, the authors proposed that the reaction starts with the activation of carbonate 39–2 through the formation of allyl complex B. Complex B then reacts with deprotonated 2-oxindoline A to form the expected intermediate C. Subsequent intramolecular substitution at the phosphate moiety results in the final product 39–3. However, it is important to note that the reaction’s scope is limited to a few functional groups, which are enumerated in Scheme 39. An exploratory experiment focusing on the chiral induction of spirooxindoline yielded moderate enantioselectivity. A comparable methodology has been used for the synthesis of pyrano [2,3-b]indol-2-ones, with the oxindoline phosphates being generated in situ [172].
The conversion of isatin into 2-oxindol-3-yl phosphates facilitates the synthesis of 3-aryl-2-oxindoles, as illustrated in Scheme 40 [173]. The starting phosphate 40–2 was synthesized via a base-catalyzed phospha-Brook rearrangement. This is followed by the preparation of 3-aryl-2-oxindole 40–3 by Friedel-Crafts alkylation, initiated via a catalytic amount of trifluoromethanesulfonic acid. An important intermediate during the conversion of 40–2 to 40–3 is cation A (due to the delocalization of the cation). The reaction can be carried out in either acetonitrile or hexafluoropropan-2-ol (HFIP), but higher yields are typically achieved in HFIP. Due to the specific requirements of the Friedel-Crafts alkylation, only electron-rich aromatic compounds are suitable for this reaction, as illustrated by selected examples. The synthesized 3-aryl-2-oxindole 40–3 offers the potential for further transformations. For example, 2-oxindole 40–3 d was converted into the corresponding carbonate 40–4 by nucleophilic substitution. This was followed by a palladium-catalyzed asymmetric intramolecular allylic alkylation in the presence of (R,R)-ANDEN-phenyl-Trost ligand to produce 2-oxindole derivative 40–5 with a chiral quaternary center.
It has been demonstrated that heteroaromatic diethyl phosphates are effective in nucleophilic substitution reactions (Scheme 41) [174]. The starting compound 41–1 was converted to phosphate 41–2 by the reaction of 2-hydroxypyrimidines with diethyl phosphonate. The resulting phosphate 41–2 subsequently undergoes a reaction with dialkylamines or 4-tolylthiol, yielding the trisubstituted pyrimidine 41–3 in high yields. However, the scope of this reaction is limited to pyrimidine derivative 41–2.
5. Transition-Metal-Catalyzed Allylic and Benzylic Substitution of Organophosphates
The method of the transition-metal-catalyzed substitution of organophosphates typically involves the reaction of allyl or benzyl phosphates with a suitable nucleophile in the presence of a catalytic amount of a transition-metal complex (Scheme 42a,b). Since transition-metal-catalyzed allyl substitution has been the subject of several reviews [175,176,177,178,179], the aim of this section is to concisely summarize the fundamental applications of allyl and benzyl phosphates in the realm of transition-metal-catalyzed allylic substitution.
A recent example of transition-metal-catalyzed allylic substitution of allyl phosphates is the asymmetric copper(I)-catalyzed substitution between allyl diethyl phosphates 43–1 and alkenyl boronates (Scheme 43) [180]. In this reaction, the starting phosphate 43–1 is efficiently transformed into skipped dienes 43–6 in high yields and excellent enantioselectivity. The authors further demonstrated the versatility of this methodology by applying it to heterocycloalkenyl boronates. The product structure 43–6 aligns with an SN2′ nucleophilic substitution that is the predominant reaction pathway. Optimized reaction conditions were used for the formal synthesis of natural substances, such as heliespirone A and heliannuol E. The same group highlighted the unique effectiveness of ligand 1 in the enantioselective SN2′ substitution of allyl diethyl phosphate 43–1 with cuprated ethylboronic acid pinacol ester. This ester is synthesized via the in situ addition of an LnCu-H complex to a vinylboronic acid pinacol ester [181]. The use of allenyl boronate 43–4 and propargyl boronate 43–5 in the same reaction was explored by Hoveyda (Scheme 43) [182]. The dienyl phosphate 43–1 is converted into the corresponding products 43–2 and 43–3, with the outcome depending on the structure of the boronate. Once again, the reaction predominantly follows an SN2′ pathway with high enantioselectivity, which is influenced by the chosen ligand. The observed reactivity of allyl phosphate 43–1 with boronates 43–4 and 43–5 contrasts with a previous study [183]. In this study, an NHC ligand combined with copper(II) chloride was used to transform the phosphate 43–1 and alkyne 43–5 into disubstituted acetylene following SN2′ regioselectivity.
Beyond boronic acids, copper-catalyzed allylic substitution has been successfully extended to include gem-diboryl alkanes (Scheme 44) [184]. This reaction catalyzed by an NHC ligand achieves an excellent SN2′/SN2 substitution ratio. In addition to gem-diborylmethane, the optimized reaction conditions allow for the use of gem-diborylethane 44–3, as evidenced by the structure of the synthesized derivative 44–2c. Similarly, Hoveyda and co-workers [185], have reported concurrent findings, employing a chiral ligand to synthesize enantiomerically pure products.
Allyl phosphates are effective in the allylation of C-nucleophiles derived from carbonyl compounds. A typical example of this reaction is the asymmetric allylation of 2-acylimidazoles 45–2, facilitated by Ni/Pd dual catalysis (Scheme 45) [186]. In this process, a nickel complex activates the imidazole derivative 45–2, while a palladium catalyst converts the allyl phosphate 45–1 into a π-allyl complex A. The formed π-allyl complex A than alkylates the activated imidazole B. A significant feature of this work is the high number of synthesized compounds, along with the high isolated yields and high enantiomeric excess. Furthermore, the imidazole group can be readily activated through alkylation with methyl triflate. Subsequently, the hydrolysis of the activated imidazole produces acid 45–5, while the reaction with ethylmagnesium bromide results in ketone 45–4. In both cases, products 45–5 and 45–4 are obtained with the retention of the absolute configuration at the stereogenic center.
The concept of transition-metal-catalyzed asymmetric allylic substitution was applied for the desymmetrization of meso-bisphosphates (Scheme 46) [187]. In this study, bisphosphates undergo monoselective alkylation leading solely to the formation of cycloalkenyl phosphates. The reaction was optimized using cyclopentenyl derivative 46–1, giving the corresponding enantiomer 46–2a. The method allows for the desymmetrization of both five- and seven-membered rings, yielding products with uniformly high enantiomeric purity, as shown in the selected examples. The authors postulated that AgNTf2 is pivotal in the generation of catalytic moiety A from copper chloride. The alkyl zirconium reagent, produced in situ from the Schwarz reagent and alkene, undergoes Zr–Cu transmetalation. Subsequent oxidative addition and reductive elimination steps form intermediate D and regenerate catalyst A, along with product 46–2.
Allyl phosphates have been used in the Cu-H catalyzed hydroallylation of vinyl arenes and propene (Scheme 47) [188,189]. Both studies elucidate a similar mechanism. Initially, the reaction starts with the hydrocupration of the terminal double bond, followed by an enantioselective allylic substitution. This process results in the formation of complex C, which is then converted to complex A by a two-step substitution process, which involves the substitution of the phosphate ligand with tBuOLi followed by the introduction of the hydride ion. Each study employed different reducing agents. Buchwald selected dimethoxymethylsilane, while Xiong favored polymethylhydrosiloxane (PMHS). Furthermore, Buchwald used the (S,S)-Ph-BPE ligand [189], whereas Xiong used the enantiomeric (R,R)-Ph-BPE ligand [188]. In both cases, products 47–2 and 47–3 were obtained with high enantiomeric purity.
Recently, Okhuma published the synthesis of isonitriles based on a palladium-catalyzed reaction of allyl diethyl phosphates 48–1 with trimethylsilyl cyanide (Scheme 48a) [190]. The authors precisely optimized the course of the reaction and found that the use of allyl phosphate 48–1 is crucial for the successful formation of isonitriles 48–2. In contrast, allyl acetates yielded only the corresponding nitriles. However, it is important to note that the reaction scope is somewhat limited and the tolerance for various functional groups is relatively low. Although a complete mechanism has not yet been fully elucidated, the authors propose that isocyanation is likely catalyzed by a Pd(II) complex rather than a Pd(0) complex. The same group extended the reaction scope of the isocyanation reaction to include benzyl diethyl phosphates (Scheme 48b,c) [191,192]. Yamaguchi’s group established conditions for the nickel-catalyzed cyanation of phenol derivatives (Scheme 48d) [193]. Although the study focused on aryl carbamates and pivalates, other substrates, such as 2-naphthyl tosylate, trifluorosulfonate, and diethyl phosphate 48–7, were shown to be effectively used.
An interesting variation of asymmetric allyl alkylation is the intramolecular approach. Hou and co-workers exploited palladium-catalyzed cyclization to transform 1,2-disubstituted benzenes 49–1 into benzocyclopentanones 49–2 (Scheme 49) [194]. The reaction typically yields products with high diastereomeric and enantiomeric purity, which makes the developed methodology attractive with respect to a wide range of natural compounds with the benzocyclopentanone motif. Furthermore, the selected benzocyclopentanone 49–2a was reduced with triethylsilane in trifluoroacetic acid to benzocyclopentane 49–3 in an almost quantitative yield. Additionally, a novel approach based on palladium-isothiourea relay catalysis facilitated the allylation of α-amino acids using diethyl allyl phosphates [195].
Trost and co-workers developed an asymmetric allylic alkylation catalyzed by copper along with a bifunctional ligand. The bifunctional ligand combines NHC and prolinol moieties connected via a phenolic spacer. At the beginning of the reaction, the active ligand is synthesized in situ from the ligand (Ag/Zn) by its conversion to a Cu/Zn heterobimetallic complex. The developed method was successfully applied to the asymmetric synthesis of (+)-sporochnol A (50–4) (Scheme 50) [196].
Transition-metal-catalyzed reactions of benzyl phosphates with organometallic reagents were also studied. In the first report, simple arylsilanes were used to synthesize diarylmethanes (Scheme 51a) [197]. This reaction is characterized by high yields of diarylmethanes represented by a general structure 51–3, although the reaction scope is somewhat limited to a few robust functional groups. In contrast, Koert and co-workers explored the reaction of benzyl-type organophosphate 51–4 with organocuprate (Scheme 51b) [198]. This reaction predominantly gives the γ-isomer 51–5 via SN2′ pathway, while the reaction scope is limited to organocuprates derived from Grignard reagents. Chirality transfer experiments were only partially successful, as evident from the synthesis of compound 51–5a.
Dearomative cross-coupling reactions have emerged as a crucial methodology for the preparation of partially aromatic or fully dearomatized arenes [199] and heteroaromatic compounds [200,201,202]. An illustrative case of this approach for benzyl phosphates is the transformation of phosphate 52–1 into dihydronaphthalenes 52–2 (Scheme 52) [203]. Products 52–2 are prone to isomerization when exposed to silica gel. In a selected example, it was demonstrated that further modification, such as the Simmons–Smith reaction or oxidation, can be successfully carried out. Additionally, the incorporation of a nitrile group at the benzyl position of the starting substance 52–1 improves the stability of the dihydronaphthalene products. However, this modification simultaneously reduces the reactivity of phosphate [204].
Benzyl phosphates and carbonates have been used for the benzylation of azlactones (Scheme 53) [205]. Through comprehensive studies of reaction conditions, it was discovered that the (R,R)- or (S,S)-dppba ligands are highly effective for the asymmetric benzylation of starting phosphates 53–1a and 53–1b. The reaction outcome of this reaction depends on the structure of the electrophilic reagents. The electron-neutral aryl and heteroaryl carbonates of the benzyl type undergo alkylation without a base. However, phosphates 53–1a and 53–1b were alkylated in the presence of cesium carbonate, indicating that the carbonate group acts as a base during alkylation. The enantiomeric purity of the alkylation products was evaluated by the hydrolysis of azlactones to amino acid 53–3. The authors also suggested a mechanism for the benzylation process that involves the corresponding η3-complex or a η1-complex, which then isomerizes to a η3-complex.
The enantioselective palladium-catalyzed benzylation of carboxylic acid esters was achieved using the (S)-BTM ligand. The optimized reaction conditions were used for the formal synthesis of the thrombin inhibitor DX-9065A (Scheme 54) [206]. The starting pentafluorophenyl ester 54–1 was benzylated under optimized reaction conditions to give the enantiomerically pure product 54–2. The isolated pentafluorophenyl ester 54–2 was then converted to ethyl ester 54–3 in a quantitative yield. Finally, the synthesis of the inhibitor DX-9065A can be accomplished by following a previously reported procedure [207].
In 2022, the thioetherification of allyl, benzyl, and propargyl phosphates was published (Scheme 55) [208]. The reaction’s scope was exclusively evaluated using silylated thiophenol. It is crucial, however, to highlight that the reaction conditions were restricted to unfunctionalized phosphates. The complete inversion of the configuration in the chiral benzyl phosphate 55–1 suggests that the reaction mechanism involves the activation of both reactants by a heterogeneous catalyst, followed by an SN2 substitution process.
Recently, a novel synthesis of di- and trisubstituted alkenes from benzyl phosphates 56–1 and N-tosylhydrazones 56–2 was reported (Scheme 56) [209]. From a mechanistic perspective, two aspects are pivotal. Initially, the mechanism involves the oxidative addition of a palladium catalyst to create complex A, while concurrently, the hydrazone is converted in situ into diazo compound 56–4. This compound subsequently interacts with complex A to form complex B. Finally, migratory insertion, followed by β-H elimination, yields the reaction product 56–3. However, the method’s limitation lies in its applicability primarily to symmetric trisubstituted alkenes 56–3a and 56–3b. Stereoselective synthesis in this context is challenging because it results in a mixture of stereoisomers 56–3c. In contrast, the formation of disubstituted alkene 56–3d occurs exclusively as the (E) stereoisomer.
6. Oxidation and Reduction Reactions of Organophosphates
Vinyl phosphates are amenable to modification via oxidation processes. Under osmium-catalyzed Sharpless epoxidation conditions [210], the oxidation of both acyclic and cyclic vinyl phosphates 57–1 leads to the formation of enantiomerically pure hydroxyketones 57–2 or 57–3 (Scheme 57) [211,212,213]. The efficacy of commercially available (DHQ)2PHAL and (DHQD)2PHAL ligands has been compared with ligands 57–4 and 57–5. Epoxides A and B are common intermediates during the oxidation of vinyl phosphates, which are then hydrolyzed to yield hydroxyketones 57–2 and 57–3. The observed enantiomeric excess for both (S)- and (R)-alcohols (57–2 and 57–3) ranges from 13% to 100%, across both commercially available and experimentally tested ligands. In a related study, Jones and co-workers explored the epoxidation and hydroxylation of cyclic vinyl phosphate acetate and trialkylsilyl ethers. They observed that vinyl phosphate and trialkylsilyl ethers performed significantly worse than the corresponding vinyl acetate [214].
As a continuation of their previous work [215], Lei and co-workers developed a method for the oxidative addition reaction of vinyl phosphates 58–1 to β-keto sulfides 58–2 (Scheme 58) [216]. This reaction is conducted in an oxygen-rich environment. The proposed mechanism begins with the generation of an S-radical, which interacts with the activated double bond in the vinyl phosphate to form intermediate A. Oxygen then reacts with thiophenol to produce hydroperoxide B, which is reduced to intermediate C. The final step is the elimination of (EtO)2(O)POH, resulting in the formation of sulfide 58–2. It is crucial to recognize that the applicability of this reaction is limited to substrates featuring simple functional groups (Scheme 58).
Phosphates derived from 2-oxindoles can undergo reduction by means of hydroiodic acid (Scheme 59) [217]. This reduction process is initiated by the protonation of the phosphate group, leading to intermediate A. Subsequently, intermediate A is attacked by the iodide anion, resulting in the formation of 3-iodo indole derivative B. The reduction is completed by a subsequent substitution at the iodine atom. However, due to harsh reaction conditions, tolerance for functional groups is restricted to simple and rather robust groups, such as the nitro group in 59–2a and halogen in 59–2b.
Primary and secondary alkyl phosphates can be chemoselectively reduced with various hydrides (Scheme 60) [218]. The authors employed a chemoselective reduction strategy targeting a phosphate 602 derived from lithhocholanyl alcohol 60–1 in a one-pot setup. This phosphate 60–2 was then reduced to monophosphate 60–3 by treatment with lithium triethylborohydride. Finally, the diphenyl phosphate group was removed by treatment with sodium bis(2-methoxyethoxy)aluminum hydride.
Electrochemical synthesis offers another approach for organophosphate reduction to secondary alcohols from diphenyl benzyl phosphates 61–1 (Scheme 61) [219]. This reduction is carried out in an undivided cell equipped with a stainless steel (SST) anode and a graphite cathode, all at room temperature. A critical step in this synthesis is the reduction of phosphate 61–1 to anion A, which then reacts with a carbonyl group. The reduction potential for the phosphate group is established at 2.8 V via cyclic voltammetry. The optimized conditions are versatile, supporting a wide array of functional groups. Beyond aldehydes, this method has been successfully applied to ketones 61–3d, alkyl 61–3g, and propargyl 61–3f phosphates. In addition, Morzycki reported the electrochemical cholesterylation of cholesteryl diphenyl phosphate [220].
7. Conclusions
In this review, we have highlighted the significant aspects of organophosphates and their applications in organic synthesis. Organophosphates are primarily utilized in cross-coupling reactions for C–C-bond formation, particularly in transition-metal-catalyzed allyl substitutions. Additionally, the oxidation and reduction of vinyl and alkyl phosphates have become increasingly relevant. Although not explicitly discussed in this review, organophosphates’ role in organic synthesis, particularly in competition with alternative electrophiles featuring activated C–O bonds such as tosylates, acetates, and carbonates, merits attention. In specific instances, organophosphates may outperform these alternatives in stereoselective synthesis, notably in the formation of tetrasubstituted alkenes. However, this advantage is often challenged by the fact that acetates and tosylates frequently yield comparable results. Tosylates or acetates hold an edge due to their simpler 1H and 13C NMR spectra, in addition to the cost effectiveness and availability of their precursors. In addition, it is important to note the regulatory considerations surrounding the use of many organophosphorus compounds, given their documentation in the context of chemical-weapons conventions.
However, there are notable limitations in the area of multicomponent reactions involving organophosphates. This issue stems from the fact that organophosphates possess three distinct carboneous substituents. Regrettably, in most reactions, only one of these groups is typically utilized, which considerably restricts the atom economy of organophosphates.
The distinctive structure and properties of the phosphate group pave the way for innovative approaches in sustainable chemistry, particularly considering the natural abundance of phosphates. Currently, the potential of organophosphate applications in organic synthesis, such as their electrochemical transformations, remains largely untapped. Prospective advancements in organophosphate chemistry involve catalytic reactions in which phosphate electrophiles are formed in a catalytic manner.
Author Contributions
Conceptualization, T.T., writing original draft and editing—P.O. and T.T. Both authors have read and agreed to the published version of the manuscript.
Funding
The work is supported by Operational Programme Johannes Amos Comenius financed by European Structural and Investment Funds and the Czech Ministry of Education, Youth and Sports (Project No. SENDISO - CZ.02.01.01/00/22_008/0004596).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lorke, D.E.; Petroianu, G.A. Reversible cholinesterase inhibitors as pretreatment for exposure to organophosphates. A review. J. Appl. Toxicol. 2019, 39, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Beynon, K.I.; Hutson, D.H.; Wright, A.N. The Metabolism and Degradation of Vinyl Phosphate Insecticides; Springer: New York, NY, USA, 1973; pp. 55–142. [Google Scholar]
- Lorke, D.E.; Petroianu, G.A. Minireview: Does in-vitro testing of oximes help predict their in-vivo action after paraoxon exposure? J. Appl. Toxicol. 2009, 29, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Kaonga, C.C.; Chidya, R.C.G.; Kosamu, I.B.M.; Abdel-dayem, S.M.; Mapoma, H.W.T.; Thole, B.; Mbewe, R.; Sakugawa, H. Trends in usage of selected fungicides in Japan between 1962 and 2014: A review. Int. J. Environ. Sci. Technol. 2018, 15, 1801–1814. [Google Scholar] [CrossRef]
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Atobe, M. Organic electrosynthesis in flow microreactor. Curr. Opin. Electrochem. 2017, 2, 1–6. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Šljukić, B.; Santos, D.M.F.; Sequeira, C.A.C. Organic Electrosynthesis: From Laboratorial Practice to Industrial Applications. Org. Proc. Res. Dev. 2017, 21, 1213–1226. [Google Scholar] [CrossRef]
- Marken, F.; Cresswell, A.J.; Bull, S.D. Recent Advances in Paired Electrosynthesis. Chem. Rec. 2021, 21, 2585–2600. [Google Scholar] [CrossRef]
- Siu, J.C.; Fu, N.; Lin, S. Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery. Acc. Chem. Res. 2020, 53, 547–560. [Google Scholar] [CrossRef]
- Yuan, Y.; Lei, A. Is electrosynthesis always green and advantageous compared to traditional methods? Nat. Commun. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zheng, Y.; Fang, T.; Chen, Y.; Zhu, Y.; Liang, Q.; Sheng, H.; Li, Z.; Chen, C.; Wang, X. Photocatalysis: An overview of recent developments and technological advancements. Sci. China Chem. 2020, 63, 149–181. [Google Scholar] [CrossRef]
- Gisbertz, S.; Pieber, B. Heterogeneous Photocatalysis in Organic Synthesis. ChemPhotoChem 2020, 4, 456–475. [Google Scholar] [CrossRef]
- Melchionna, M.; Fornasiero, P. Updates on the Roadmap for Photocatalysis. ACS Catal. 2020, 10, 5493–5501. [Google Scholar] [CrossRef]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- Knappke, C.E.I.; Grupe, S.; Gärtner, D.; Corpet, M.; Gosmini, C.; von Wangelin, A.J. Reductive Cross-Coupling Reactions between Two Electrophiles. Chem. Eur. J. 2014, 20, 6828–6842. [Google Scholar] [CrossRef] [PubMed]
- Noël, T.; Buchwald, S.L. Cross-coupling in flow. Chem. Soc. Rev. 2011, 40, 5010–5029. [Google Scholar] [CrossRef]
- So, C.M.; Kwong, F.Y. Palladium-catalyzed cross-coupling reactions of aryl mesylates. Chem. Soc. Rev. 2011, 40, 4963–4972. [Google Scholar] [CrossRef]
- Thapa, S.; Shrestha, B.; Gurung, S.K.; Giri, R. Copper-catalysed cross-coupling: An untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. [Google Scholar] [CrossRef]
- Tobrman, T. Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions. Chemistry 2023, 5, 2288–2321. [Google Scholar] [CrossRef]
- Čubiňák, M.; Edlová, T.; Polák, P.; Tobrman, T. Indolylboronic Acids: Preparation and Applications. Molecules 2019, 24, 3523. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Ghanbarian, M.; Ghalavand, N.; Nazari, N. Current Applications of the Sonogashira Reaction in the Synthesis of Heterocyclic Compounds: An Update. Curr. Org. Chem. 2018, 22, 1420–1457. [Google Scholar] [CrossRef]
- Malapit, C.A.; Howell, A.R. Recent Applications of Oxetanes in the Synthesis of Heterocyclic Compounds. J. Org. Chem. 2015, 80, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Oeser, P.; Koudelka, J.; Petrenko, A.; Tobrman, T. Recent Progress Concerning the N-Arylation of Indoles. Molecules 2021, 26, 5079. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Jain, S.C. “In Water” Syntheses of Heterocyclic Compounds. Mini-Rev. Org. Chem. 2011, 8, 455–464. [Google Scholar] [CrossRef]
- Veisi, H.; Ghorbani-Vaghei, R. Recent progress in the application of N-halo reagents in the synthesis of heterocyclic compounds. Tetrahedron 2010, 66, 7445–7463. [Google Scholar] [CrossRef]
- Volkova, Y.; Baranin, S.; Zavarzin, I. A3 Coupling Reaction in the Synthesis of Heterocyclic Compounds. Adv. Synth. Catal. 2021, 363, 40–61. [Google Scholar] [CrossRef]
- Buttard, F.; Sharma, J.; Champagne, P.A. Recent advances in the stereoselective synthesis of acyclic all-carbon tetrasubstituted alkenes. Chem. Commun. 2021, 57, 4071–4088. [Google Scholar] [CrossRef]
- Flynn, A.B.; Ogilvie, W.W. Stereocontrolled Synthesis of Tetrasubstituted Olefins. Chem. Rev. 2007, 107, 4698–4745. [Google Scholar] [CrossRef]
- Mukherjee, N.; Planer, S.; Grela, K. Formation of tetrasubstituted C–C double bonds via olefin metathesis: Challenges, catalysts, and applications in natural product synthesis. Org. Chem. Front. 2018, 5, 494–516. [Google Scholar] [CrossRef]
- Paek, S.M. Synthesis of tetrasubstituted alkenes via metathesis. Molecules 2012, 17, 3348–3358. [Google Scholar] [CrossRef] [PubMed]
- Edlová, T.; Čubiňák, M.; Tobrman, T. Cross-Coupling Reactions of Double or Triple Electrophilic Templates for Alkene Synthesis. Synthesis 2021, 53, 255–266. [Google Scholar] [CrossRef]
- Negishi, E.-I.; Huang, Z.; Wang, G.; Mohan, S.; Wang, C.; Hattori, H. Recent Advances in Efficient and Selective Synthesis of Di-, Tri-, and Tetrasubstituted Alkenes via Pd-Catalyzed Alkenylation−Carbonyl Olefination Synergy. Acc. Chem. Res. 2008, 41, 1474–1485. [Google Scholar] [CrossRef]
- Polák, P.; Váňová, H.; Dvořák, D.; Tobrman, T. Recent progress in transition metal-catalyzed stereoselective synthesis of acyclic all-carbon tetrasubstituted alkenes. Tetrahedron Lett. 2016, 57, 3684–3693. [Google Scholar] [CrossRef]
- Reiser, O. Palladium-Catalyzed Coupling Reactions for the Stereoselective Synthesis of Tri- and Tetrasubstituted Alkenes. Angew. Chem. Int. Ed. 2006, 45, 2838–2840. [Google Scholar] [CrossRef]
- Krishnakumar, V.K.; Sharma, M.M. Synthesis of Triaryl Phosphates via Phase-Transfer Catalysis. Synthesis 1983, 1983, 558–559. [Google Scholar] [CrossRef]
- Zhong, C.; Huang, Y.; Zhang, H.; Zhou, Q.; Liu, Y.; Lu, P. Enantioselective Synthesis of 3-Substituted Cyclobutenes by Catalytic Conjugate Addition/Trapping Strategies. Angew. Chem. Int. Ed. 2020, 59, 2750–2754. [Google Scholar] [CrossRef]
- Kotek, V.; Polák, P.; Tobrman, T. Efficient and simple preparation of functionalized 1,1-dibromoenol phosphates. Monat. Chem. 2016, 147, 405–412. [Google Scholar] [CrossRef]
- Kawada, H.; Ikoma, A.; Ogawa, N.; Kobayashi, Y. Activation of Marginally Reactive Boron Enolates by MeLi for the Formation of Enol Phosphates and Synthesis of the Δ9-THC Intermediate. J. Org. Chem. 2015, 80, 9192–9199. [Google Scholar] [CrossRef]
- Perkow, W. Umsetzungen mit Alkylphosphiten. I. Mitteil.: Umlagerungen bei der Reaktion mit Chloral und Bromal. Chem. Ber. 1954, 87, 755–758. [Google Scholar] [CrossRef]
- Adamek, J. Special Issue “Organophosphorus Chemistry: A New Perspective”. Molecules 2023, 28, 4752. [Google Scholar] [CrossRef]
- Keglevich, G. Organophosphorus Chemistry 2021. Molecules 2023, 28, 394. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.R. Organophosphorus chemistry. Beilstein J. Org. Chem. 2014, 10, 2087–2088. [Google Scholar] [CrossRef]
- Han, L.-B.; Yang, S.-D.; Waterman, R.; Weigand, J.J. Love in the Time of COVID. J. Org. Chem. 2020, 85, 14273–14275. [Google Scholar] [CrossRef]
- Fiorito, D.; Folliet, S.; Liu, Y.; Mazet, C. A General Nickel-Catalyzed Kumada Vinylation for the Preparation of 2-Substituted 1,3-Dienes. ACS Catal. 2018, 8, 1392–1398. [Google Scholar] [CrossRef]
- Braconi, E.; Cramer, N. Crossed Regio- and Enantioselective Iron-Catalyzed [4+2]-Cycloadditions of Unactivated Dienes. Angew. Chem. Int. Ed. 2022, 61, e202112148. [Google Scholar] [CrossRef] [PubMed]
- Braconi, E.; Götzinger, A.C.; Cramer, N. Enantioselective Iron-Catalyzed Cross-[4+4]-Cycloaddition of 1,3-Dienes Provides Chiral Cyclooctadienes. J. Am. Chem. Soc. 2020, 142, 19819–19824. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Zhong, H.; Macaulay, R.L.; Chirik, P.J. Regio- and Diastereoselective Iron-Catalyzed [4+4]-Cycloaddition of 1,3-Dienes. J. Am. Chem. Soc. 2019, 141, 8557–8573. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Ng, J.J.W.; Chiba, S. Generation of Allylmagnesium Reagents by Hydromagnesiation of 2-Aryl-1,3-dienes. Angew. Chem. Int. Ed. 2023, 62, e202217735. [Google Scholar] [CrossRef]
- Ohta, R.; Shio, Y.; Akiyama, T.; Yamada, M.; Harada, K.; Arisawa, M. Ligand-free reductive coupling of aldehydes with 1,3-dienes using a sulfur-modified Au-supported nickel nanoparticle catalyst. New J. Chem. 2023, 47, 7694–7700. [Google Scholar] [CrossRef]
- Zhao, H.; Caldora, H.P.; Turner, O.; Douglas, J.J.; Leonori, D. A Desaturative Approach for Aromatic Aldehyde Synthesis via Synergistic Enamine, Photoredox and Cobalt Triple Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202201870. [Google Scholar] [CrossRef]
- Li, C.; Shin, K.; Liu, R.Y.; Buchwald, S.L. Engaging Aldehydes in CuH-Catalyzed Reductive Coupling Reactions: Stereoselective Allylation with Unactivated 1,3-Diene Pronucleophiles. Angew. Chem. Int. Ed. 2019, 58, 17074–17080. [Google Scholar] [CrossRef] [PubMed]
- Poisson, P.-A.; Tran, G.; Besnard, C.; Mazet, C. Nickel-Catalyzed Kumada Vinylation of Enol Phosphates: A Comparative Mechanistic Study. ACS Catal. 2021, 11, 15041–15050. [Google Scholar] [CrossRef]
- Fiorito, D.; Simon, M.; Thomas, C.M.; Mazet, C. Access to Highly Stereodefined 1,4-cis-Polydienes by a [Ni/Mg] Orthogonal Tandem Catalytic Polymerization. J. Am. Chem. Soc. 2021, 143, 13401–13407. [Google Scholar] [CrossRef] [PubMed]
- Desfeux, C.; Besnard, C.; Mazet, C. [n]Dendralenes as a Platform for Selective Catalysis: Ligand-Controlled Cu-Catalyzed Chemo-, Regio-, and Enantioselective Borylations. Org. Lett. 2020, 22, 8181–8187. [Google Scholar] [CrossRef] [PubMed]
- Saglam, M.F.; Fallon, T.; Paddon-Row, M.N.; Sherburn, M.S. Discovery and Computational Rationalization of Diminishing Alternation in [n]Dendralenes. J. Am. Chem. Soc. 2016, 138, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Zhang, Z.; Da, Y.-X.; Quan, Z.-J.; Wang, X.-C. Iron-Catalyzed Kumada Cross-Coupling Reactions of Pyrimidin-2-yl Phosphates: An Efficient Approach to C2-Functionalized Pyrimidines. Asian J. Org. Chem. 2015, 4, 538–544. [Google Scholar] [CrossRef]
- Li, Z.; Liu, L.; Sun, H.-m.; Shen, Q.; Zhang, Y. Alkyl Grignard cross-coupling of aryl phosphates catalyzed by new, highly active ionic iron(ii) complexes containing a phosphine ligand and an imidazolium cation. Dalton Trans. 2016, 45, 17739–17747. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, B.; Sun, H.; Shen, Q.; Zhang, Y. Ionic iron(III) complexes bearing a dialkylbenzimidazolium cation: Efficient catalysts for magnesium-mediated cross-couplings of aryl phosphates with alkyl bromides. Appl. Organometal. Chem. 2017, 31, e3671. [Google Scholar] [CrossRef]
- Ren, J.-A.; Chen, X.; Gui, C.; Miao, C.; Chu, X.-Q.; Xu, H.; Zhou, X.; Ma, M.; Shen, Z.-L. Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Phosphates with Aryl Bromides. Adv. Synth. Catal. 2023, 365, 2511–2515. [Google Scholar] [CrossRef]
- Cui, M.; Oestreich, M. Synthesis of Silylated Cyclobutanone and Cyclobutene Derivatives Involving 1,4-Addition of Zinc-Based Silicon Nucleophiles. Chem. Eur. J. 2021, 27, 16103–16106. [Google Scholar] [CrossRef]
- Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Convenient Syntheses of Enantiopure 1,2-Oxazin-4-yl Nonaflates and Phosphates and Their Palladium-Catalyzed Cross-Couplings. Synthesis 2013, 45, 2752–2762. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, J.; Liu, Y.; Lu, P. Synthesis of Dibenzo[a,e]cyclooctene-5,11(6H,12H)-diones via the Elusive Benzocyclobutenone Anion. Synthesis 2021, 53, 4477–4483. [Google Scholar] [CrossRef]
- Kotek, V.; Dvořáková, H.; Tobrman, T. Modular and Highly Stereoselective Approach to All-Carbon Tetrasubstituted Alkenes. Org. Lett. 2015, 17, 608–611. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Li, Y.; Brown, M.K. Stereoselective Synthesis of All-Carbon Tetrasubstituted Alkenes from In Situ Generated Ketenes and Organometallic Reagents. Org. Lett. 2013, 15, 1610–1613. [Google Scholar] [CrossRef]
- Wang, C.-S.; Tan, P.S.L.; Ding, W.; Ito, S.; Yoshikai, N. Regio- and Stereoselective Synthesis of Enol Carboxylate, Phosphate, and Sulfonate Esters via Iodo(III)functionalization of Alkynes. Org. Lett. 2022, 24, 430–434. [Google Scholar] [CrossRef]
- Bauer, A.; Maulide, N. A Stereoselective Reductive Hosomi–Sakurai Reaction. Org. Lett. 2018, 20, 1461–1464. [Google Scholar] [CrossRef]
- Meyer, D.; Renaud, P. Enantioselective Hydroazidation of Trisubstituted Non-Activated Alkenes. Angew. Chem. Int. Ed. 2017, 56, 10858–10861. [Google Scholar] [CrossRef]
- Simlandy, A.K.; Lyu, M.-Y.; Brown, M.K. Catalytic Arylboration of Spirocyclic Cyclobutenes: Rapid Access to Highly Substituted Spiro[3.n]alkanes. ACS Catal. 2021, 11, 12815–12820. [Google Scholar] [CrossRef]
- Mizuta, S.; Galicia-López, O.; Engle, K.M.; Verhoog, S.; Wheelhouse, K.; Rassias, G.; Gouverneur, V. Trifluoromethylation of Allylsilanes under Copper Catalysis. Chem. Eur. J. 2012, 18, 8583–8587. [Google Scholar] [CrossRef]
- Narita, K.; Fujisaki, N.; Sakuma, Y.; Katoh, T. A novel approach to oxazole-containing diterpenoid synthesis from plant roots: Salviamines E and F. Org. Biomol. Chem. 2019, 17, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Cahiez, G.; Guerret, O.; Moyeux, A.; Dufour, S.; Lefevre, N. Eco-Friendly and Industrially Scalable Synthesis of the Sex Pheromone of Lobesia botrana. Important Progress for the Eco-Protection of Vineyard. Org. Process Res. Dev. 2017, 21, 1542–1546. [Google Scholar] [CrossRef]
- Ikoma, A.; Ogawa, N.; Kondo, D.; Kawada, H.; Kobayashi, Y. Synthesis of (−)-Piperitylmagnolol Featuring ortho-Selective Deiodination and Pd-Catalyzed Allylation. Org. Lett. 2016, 18, 2074–2077. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, H.; Oikawa, H.; Oguri, H. Biogenetically inspired synthesis and skeletal diversification of indole alkaloids. Nat. Chem. 2014, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Yang, Z.-K.; Minami, H.; Kojima, K.; Saito, T.; Wang, C.; Uchiyama, M. Revisitation of Organoaluminum Reagents Affords a Versatile Protocol for C–X (X = N, O, F) Bond-Cleavage Cross-Coupling: A Systematic Study. ACS Catal. 2017, 7, 3988–3994. [Google Scholar] [CrossRef]
- Nakatsuji, H.; Ashida, Y.; Hori, H.; Sato, Y.; Honda, A.; Taira, M.; Tanabe, Y. (E)- and (Z)-stereodefined enol phosphonates derived from β-ketoesters: Stereocomplementary synthesis of fully-substituted α,β-unsaturated esters. Org. Biomol. Chem. 2015, 13, 8205–8210. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, H.; Wu, Q.; Bi, X.; Shi, E.; Xiao, J. Stereoselective synthesis of (E)-α,β-unsaturated esters: Triethylamine-catalyzed allylic rearrangement of enol phosphates. RSC Adv. 2023, 13, 13511–13515. [Google Scholar] [CrossRef]
- Kotek, V.; Polák, P.; Dvořáková, H.; Tobrman, T. Aluminum Chloride Promoted Cross-Coupling of Trisubstituted Enol Phosphates with Organozinc Reagents En Route to the Stereoselective Synthesis of Tamoxifen and Its Analogues. Eur. J. Org. Chem. 2016, 2016, 5037–5044. [Google Scholar] [CrossRef]
- Polák, P.; Tobrman, T. The synthesis of polysubstituted indoles from 3-bromo-2-indolyl phosphates. Org. Biomol. Chem. 2017, 15, 6233–6241. [Google Scholar] [CrossRef]
- Polák, P.; Tobrman, T. Novel Selective Approach to Terminally Substituted [n]Dendralenes. Eur. J. Org. Chem. 2019, 2019, 957–968. [Google Scholar] [CrossRef]
- Koudelka, J.; Tobrman, T. Synthesis of 2-Substituted Cyclobutanones by a Suzuki Reaction and Dephosphorylation Sequence. Eur. J. Org. Chem. 2021, 2021, 3260–3269. [Google Scholar] [CrossRef]
- Edlová, T.; Dvořáková, H.; Eigner, V.; Tobrman, T. Substrate-Controlled Regioselective Bromination of 1,2-Disubstituted Cyclobutenes: An Application in the Synthesis of 2,3-Disubstituted Cyclobutenones. J. Org. Chem. 2021, 86, 5820–5831. [Google Scholar] [CrossRef]
- Čubiňák, M.; Bigeon, J.; Galář, P.; Ondič, L.; Tobrman, T. The Synthesis of Tetrasubstituted Cycloalkenes Bearing π-Conjugated Substituents and Their Optical Properties. ChemistrySelect 2021, 6, 9904–9910. [Google Scholar] [CrossRef]
- Čubiňák, M.; Tobrman, T. Room-Temperature Negishi Reaction of Trisubstituted Vinyl Phosphates for the Synthesis of Tetrasubstituted Alkenes. J. Org. Chem. 2020, 85, 10728–10739. [Google Scholar] [CrossRef] [PubMed]
- Fihri, A.; Bouhrara, M.; Nekoueishahraki, B.; Basset, J.-M.; Polshettiwar, V. Nanocatalysts for Suzuki cross-coupling reactions. Chem. Soc. Rev. 2011, 40, 5181–5203. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Hashemi, E. Recent applications of the Suzuki reaction in total synthesis. Tetrahedron 2012, 68, 9145–9178. [Google Scholar] [CrossRef]
- Maluenda, I.; Navarro, O. Recent Developments in the Suzuki-Miyaura Reaction: 2010–2014. Molecules 2015, 20, 7528–7557. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Islam, M.M.; Islam, S.M. Suzuki–Miyaura reaction by heterogeneously supported Pd in water: Recent studies. RSC Adv. 2015, 5, 42193–42221. [Google Scholar] [CrossRef]
- Rossi, R.; Bellina, F.; Lessi, M. Selective Palladium-Catalyzed Suzuki–Miyaura Reactions of Polyhalogenated Heteroarenes. Adv. Synth. Catal. 2012, 354, 1181–1255. [Google Scholar] [CrossRef]
- Chen, H.; Huang, Z.; Hu, X.; Tang, G.; Xu, P.; Zhao, Y.; Cheng, C.-H. Nickel-Catalyzed Cross-Coupling of Aryl Phosphates with Arylboronic Acids. J. Org. Chem. 2011, 76, 2338–2344. [Google Scholar] [CrossRef]
- Gigant, N.; Honraedt, A.; Gras, E.; Gillaizeau, I. Efficient Cross-Coupling of Dioxazaborocanes with α-Phosphate Enamides. Eur. J. Org. Chem. 2014, 2014, 7889–7894. [Google Scholar] [CrossRef]
- Senra, J.D.; Silva, A.C.; Santos, R.V.; Malta, L.F.B.; Simas, A.B.C. Palladium on Layered Double Hydroxide: A Heterogeneous System for the Enol Phosphate Carbon-Oxygen Bond Activation in Aqueous Media. J. Chem. 2017, 2017, 8418939. [Google Scholar] [CrossRef]
- Leidy, M.R.; Mason Hoffman, J.; Pongdee, R. Preparation of C-arylglycals via Suzuki–Miyaura cross-coupling of dihydropyranylphosphates. Tetrahedron Lett. 2013, 54, 6889–6891. [Google Scholar] [CrossRef] [PubMed]
- Mole, J.; Philip, R.M.; Anilkumar, G. Nickel-catalyzed (hetero)aryl borylations: An update. ARKIVOC 2022, 2022, 165–199. [Google Scholar] [CrossRef]
- Steven, A. Micelle-Mediated Chemistry in Water for the Synthesis of Drug Candidates. Synthesis 2019, 51, 2632–2647. [Google Scholar] [CrossRef]
- Jin, S.; Dang, H.T.; Haug, G.C.; He, R.; Nguyen, V.D.; Nguyen, V.T.; Arman, H.D.; Schanze, K.S.; Larionov, O.V. Visible Light-Induced Borylation of C–O, C–N, and C–X Bonds. J. Am. Chem. Soc. 2020, 142, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Cheung, M.S.; Lin, Z.; Li, P. Metal-free borylation of electron-rich aryl (pseudo)halides under continuous-flow photolytic conditions. Org. Chem. Front. 2016, 3, 875–879. [Google Scholar] [CrossRef]
- Begliomini, S.; Sernissi, L.; Scarpi, D.; Occhiato, E.G. A Short, Chemo-Enzymatic Synthesis of Both Enantiomers of trans-3-Hydroxy pipecolic Acid. Eur. J. Org. Chem. 2014, 2014, 5448–5455. [Google Scholar] [CrossRef]
- Rey-Rodriguez, R.; Jestin, G.; Gandon, V.; Grelier, G.; Retailleau, P.; Darses, B.; Dauban, P.; Gillaizeau, I. Intermolecular Rhodium(II)-Catalyzed Allylic C(sp3)–H Amination of Cyclic Enamides. Adv. Synth. Catal. 2018, 360, 513–518. [Google Scholar] [CrossRef]
- Adamson, N.J.; Park, S.; Zhou, P.; Nguyen, A.L.; Malcolmson, S.J. Enantioselective Construction of Quaternary Stereogenic Centers by the Addition of an Acyl Anion Equivalent to 1,3-Dienes. Org. Lett. 2020, 22, 2032–2037. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, P.; Dong, L.; Wang, W.; Duan, S.; Wang, B.; Gong, X.; Ye, L.; Wang, H.; Tian, J. Discovery of the Next-Generation Pan-TRK Kinase Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 10286–10296. [Google Scholar] [CrossRef]
- Kurimoto, Y.; Nasu, T.; Fujii, Y.; Asano, K.; Matsubara, S. Asymmetric Cycloetherification of in Situ Generated Cyanohydrins through the Concomitant Construction of Three Chiral Carbon Centers. Org. Lett. 2019, 21, 2156–2160. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Muto, T.; Sekine, K.; Sasaki, M. Total Synthesis and Structure Revision of Didemnaketal B. Chem. Eur. J. 2014, 20, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Sakamoto, K.; Muto, T.; Sasaki, M. Concise synthesis of the C15–C38 fragment of okadaic acid, a specific inhibitor of protein phosphatases 1 and 2A. Tetrahedron 2015, 71, 6369–6383. [Google Scholar] [CrossRef]
- Sallio, R.; Lebrun, S.; Gigant, N.; Gillaizeau, I.; Deniau, E. Asymmetric Synthesis of 2-Heteroaryl Cyclic Amines: Total Synthesis of (–)-Anabasine. Eur. J. Org. Chem. 2014, 2014, 4381–4388. [Google Scholar] [CrossRef]
- Hu, X.-H.; Yang, X.-F.; Loh, T.-P. Selective Alkenylation and Hydroalkenylation of Enol Phosphates through Direct C–H Functionalization. Angew. Chem. Int. Ed. 2015, 54, 15535–15539. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.H.; Lee, T.S.; Kim, E.J.; Moon, B.; Kang, J. Palladium(II)-catalyzed ortho-arylation via phosphate-group-directed C–H activation. Tetrahedron 2013, 69, 5152–5159. [Google Scholar] [CrossRef]
- Chan, L.Y.; Cheong, L.; Kim, S. Pd(II)-Catalyzed ortho-Arylation of Aryl Phosphates and Aryl Hydrogen Phosphates with Diaryliodonium Triflates. Org. Lett. 2013, 15, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- Moselage, M.; Sauermann, N.; Richter, S.C.; Ackermann, L. C–H Alkenylations with Alkenyl Acetates, Phosphates, Carbonates, and Carbamates by Cobalt Catalysis at 23 °C. Angew. Chem. Int. Ed. 2015, 54, 6352–6355. [Google Scholar] [CrossRef]
- Sauermann, N.; Loup, J.; Kootz, D.; Yatham, V.R.; Berkessel, A.; Ackermann, L. Triazolylidene Ligands Allow Cobalt-Catalyzed C–H/C–O Alkenyl ations at Ambient Temperature. Synthesis 2017, 49, 3476–3484. [Google Scholar] [CrossRef]
- Grosheva, D.; Cramer, N. Ketene Aminal Phosphates: Competent Substrates for Enantioselective Pd(0)-Catalyzed C–H Functionalizations. ACS Catal. 2017, 7, 7417–7420. [Google Scholar] [CrossRef]
- Lee, P.-S.; Xu, W.; Yoshikai, N. Directed C–H Alkenylation of Aryl Imines with Alkenyl Phosphates Promoted by a Cobalt–N-Heterocyclic Carbene Catalyst. Adv. Synth. Catal. 2017, 359, 4340–4347. [Google Scholar] [CrossRef]
- Xu, W.; Yoshikai, N. Cobalt-catalyzed directed C–H alkenylation of pivalophenone N–H imine with alkenyl phosphates. Beilstein J. Org. Chem. 2018, 14, 709–715. [Google Scholar] [CrossRef]
- Huang, J.-H.; Yang, L.-M. Nickel-Catalyzed Amination of Aryl Phosphates through Cleaving Aryl C–O Bonds. Org. Lett. 2011, 13, 3750–3753. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, X.; So, C.M. Palladium-Catalyzed C(sp2)–N Bond Cross-Coupling with Triaryl Phosphates. J. Org. Chem. 2019, 84, 6366–6376. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Z.; So, C.M. Exploration of Aryl Phosphates in Palladium-Catalyzed Mono-α-arylation of Aryl and Heteroaryl Ketones. J. Org. Chem. 2019, 84, 6337–6346. [Google Scholar] [CrossRef]
- Wang, Z.-C.; Li, Y.-Y.; Zhang, S.-Q.; Hong, X.; Shi, S.-L. Unsymmetric N-heterocyclic carbene ligand enabled nickel-catalysed arylation of bulky primary and secondary amines. Chem. Sci. 2023, 14, 4390–4396. [Google Scholar] [CrossRef]
- Valiullina, Z.R.; Galeeva, A.M.; Gimalova, F.A.; Selezneva, N.K.; Khasanova, L.S.; Mavzyutov, A.R.; Miftakhov, M.S. Synthesis and In Vitro Antibacterial Activity of New C-3-Modified Carbapenems. Russ. J. Bioorg. Chem. 2019, 45, 398–404. [Google Scholar] [CrossRef]
- Lee, N.; Tan, C.-H.; Leow, D. Asymmetric Brook Rearrangement. Asian J. Org. Chem. 2019, 8, 25–31. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, C. Radical-mediated 1,2-Brook rearrangements. Chem Catal. 2021, 1, 250–252. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.-J.; Huang, H.-M. Radical Brook Rearrangements: Concept and Recent Developments. Angew. Chem. Int. Ed. 2022, 61, e202205671. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Aita, K.; Ishikawa, S.; Terada, M. Synthesis of Tetrasubstituted Furans through One-Pot Formal [3 + 2] Cycloaddition Utilizing [1,2]-Phospha-Brook Rearrangement. Org. Lett. 2020, 22, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Ishikawa, S.; Aoki, T.; Terada, M. Synthesis of 2,3-allenylamides utilizing [1,2]-phospha-Brook rearrangement and their application to gold-catalyzed cycloisomerization providing 2-aminofuran derivatives. Chem. Commun. 2016, 52, 12513–12516. [Google Scholar] [CrossRef]
- Kondoh, A.; Iino, A.; Ishikawa, S.; Aoki, T.; Terada, M. Efficient Synthesis of Polysubstituted Pyrroles Based on [3+2] Cycloaddition Strategy Utilizing [1,2]-Phospha-Brook Rearrangement under Brønsted Base Catalysis. Chem. Eur. J. 2018, 24, 15246–15253. [Google Scholar] [CrossRef]
- Kondoh, A.; Aoki, T.; Terada, M. Synthesis of Phenanthrene Derivatives by Intramolecular Cyclization Utilizing the [1,2]-Phospha-Brook Rearrangement Catalyzed by a Brønsted Base. Chem. Eur. J. 2015, 21, 12577–12580. [Google Scholar] [CrossRef]
- Kondoh, A.; Koda, K.; Kamata, Y.; Terada, M. Synthesis of Indolizine Derivatives Utilizing [1,2]-Phospha-Brook Rearrangement/Cycloisomerization Sequence. Chem. Lett. 2017, 46, 1020–1023. [Google Scholar] [CrossRef]
- Kondoh, A.; Ojima, R.; Terada, M. Formal Fluorinative Ring Opening of 2-Benzoylpyrrolidines Utilizing [1,2]-Phospha-Brook Rearrangement for Synthesis of 2-Aryl-3-fluoropiperidines. Org. Lett. 2021, 23, 7894–7899. [Google Scholar] [CrossRef]
- Kondoh, A.; Takei, A.; Terada, M. Novel Methodology for the Efficient Synthesis of 3-Aryloxindoles: [1,2]-Phospha-Brook Rearrangement–Palladium-Catalyzed Cross-Coupling Sequence. Synlett 2016, 27, 1848–1853. [Google Scholar] [CrossRef]
- Kondoh, A.; Terada, M. Synthesis of 2,2-Disubstituted 2H-Chromenes through Carbon-Carbon Bond Formation Utilizing a [1,2]-Phospha-Brook Rearrangement under Brønsted Base Catalysis. Chem. Eur. J. 2022, 28, e202201198. [Google Scholar] [CrossRef]
- Kondoh, A.; Aoki, T.; Terada, M. Intramolecular Cyclization of Alkynyl α-Ketoanilide Utilizing [1,2]-Phospha-Brook Rearrangement Catalyzed by Phosphazene Base. Org. Lett. 2014, 16, 3528–3531. [Google Scholar] [CrossRef]
- Kondoh, A.; Aoki, T.; Terada, M. Generation and Application of Homoenolate Equivalents Utilizing [1,2]-Phospha-Brook Rearrangement under Brønsted Base Catalysis. Chem. Eur. J. 2017, 23, 2769–2773. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Aoki, T.; Terada, M. Organocatalytic Arylation of α-Ketoesters Based on Umpolung Strategy: Phosphazene-Catalyzed SNAr Reaction Utilizing [1,2]-Phospha-Brook Rearrangement. Chem. Eur. J. 2018, 24, 13110–13113. [Google Scholar] [CrossRef]
- Kondoh, A.; Hirozane, T.; Terada, M. Formal Umpolung Addition of Phosphites to 2-Azaaryl Ketones under Chiral Brønsted Base Catalysis: Enantioselective Protonation Utilizing [1,2]-Phospha-Brook Rearrangement. Chem. Eur. J. 2022, 28, e202201240. [Google Scholar] [CrossRef]
- Kondoh, A.; Ozawa, R.; Aoki, T.; Terada, M. Intramolecular addition of benzyl anion to alkyne utilizing [1,2]-phospha-Brook rearrangement under Brønsted base catalysis. Org. Biomol. Chem. 2017, 15, 7277–7281. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Tasato, N.; Aoki, T.; Terada, M. Brønsted Base-Catalyzed Transformation of α,β-Epoxyketones Utilizing [1,2]-Phospha-Brook Rearrangement for the Synthesis of Allylic Alcohols Having a Tetrasubstituted Alkene Moiety. Org. Lett. 2020, 22, 5170–5175. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Terada, M. Brønsted base-catalyzed α-oxygenation of carbonyl compounds utilizing the [1,2]-phospha-Brook rearrangement. Org. Chem. Front. 2015, 2, 801–805. [Google Scholar] [CrossRef]
- Ranga, S.; Chakravarty, M.; Chatterjee, T.; Ghosal, S. Mechanistic insights into n-BuLi mediated phospha-Brook rearrangement. New J. Chem. 2019, 43, 9886–9890. [Google Scholar] [CrossRef]
- Tan, Q.; Guo, N.; Yang, L.; Wang, F.; Feng, X.; Liu, X. Asymmetric Organocatalytic 1,6-Conjugate Addition of para-Quinone Methides Using [1,2]-Phospha-Brook Rearrangement. J. Org. Chem. 2023, 88, 9332–9342. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.S.; Pandey, C.B.; Kumar, S.; Tiwari, B. Carbene-Catalyzed Tandem [1,2]-Phospha-Brook/[1,4]-Phosphate Rearrangement: Access to α-Ketophosphates via Controlled Cross-Acyloin Condensation. J. Org. Chem. 2018, 83, 9478–9483. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ishida, Y.; Takamizu, Y.; Yasui, T. Synthesis of (Difluoromethyl)cycloalkenes from 2-Cycloalkenones by Utilizing Phospha-Brook Rearrangement. Adv. Synth. Catal. 2019, 361, 3739–3743. [Google Scholar] [CrossRef]
- Cheibas, C.; Fincias, N.; Casaretto, N.; Garrec, J.; El Kaïm, L. Passerini–Smiles Reaction of α-Ketophosphonates: Platform for Phospha-Brook/Smiles Embedded Cascades. Angew. Chem. Int. Ed. 2022, 61, e202116249. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Singh, R.P. Stereoselective Reductive Coupling Reactions Utilizing [1,2]-Phospha-Brook Rearrangement: A Powerful Umpolung Approach. J. Org. Chem. 2023, 88, 10325–10338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Miao, Z. Research Progress in [1,2]-Phospha-Brook Rearrangement Reaction. Univ. Chem. 2021, 36, 2008082. [Google Scholar] [CrossRef]
- Melvin, L.S. An efficient synthesis of 2-hydroxyphenylphosphonates. Tetrahedron Lett. 1981, 22, 3375–3376. [Google Scholar] [CrossRef]
- Delgado Rosario, E.; Rectenwald, M.F.; Gaffen, J.R.; Rheingold, A.L.; Protasiewicz, J.D. Organophosphorus decorated lithium borate and phosphate salts with extended π-conjugated backbone. Dalton Trans. 2021, 50, 6667–6672. [Google Scholar] [CrossRef] [PubMed]
- Placidi, M.P.; Botta, M.; Kálmán, F.K.; Hagberg, G.E.; Baranyai, Z.; Krenzer, A.; Rogerson, A.K.; Tóth, I.; Logothetis, N.K.; Angelovski, G. Aryl-Phosphonate Lanthanide Complexes and Their Fluorinated Derivatives: Investigation of Their Unusual Relaxometric Behavior and Potential Application as Dual Frequency 1H/19F MRI Probes. Chem. Eur. J. 2013, 19, 11644–11660. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, I.Y.; Baulina, T.Y.V.; Pasechnik, M.P.; Matveev, S.V.; Matveeva, A.G. Synthesis and Coordination Properties of Tripodal Ligand on the Triphenylphosphine Oxide Platform with Carbamoyl Side Arms. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 946–962. [Google Scholar] [CrossRef]
- Kudryavtsev, I.Y.; Bykhovskaya, O.V.; Matveeva, A.G.; Baulina, T.V.; Pasechnik, M.P.; Matveev, S.V.; Vologzhanina, A.V.; Turanov, A.N.; Karandashev, V.K.; Brel, V.K. New tripodal ligand on the triphenylphosphine oxide platform with 1,2,3-triazole side arms: Synthesis, structure, coordination, and extraction properties. Monatsh. Chem. 2020, 151, 1705–1713. [Google Scholar] [CrossRef]
- Alessi, M.; Patel, J.J.; Zumbansen, K.; Snieckus, V. The Tetraethylphosphorodiamidate (OP(O)(NEt2)2) Directed Metalation Group (DMG). Directed ortho and Lateral Metalation and the Phospha Anionic Fries Rearrangement. Org. Lett. 2020, 22, 2147–2151. [Google Scholar] [CrossRef]
- Patel, J.J.; Blackburn, T.; Alessi, M.; Sawinski, H.; Snieckus, V. Tetraethylphosphorodiamidate-Directed Metalation Group: Directed Ortho and Remote Metalation, Cross Coupling, and Remote Phospha Anionic Fries Rearrangement Reactions. Org. Lett. 2020, 22, 3860–3864. [Google Scholar] [CrossRef]
- Taylor, C.; Watson, A. The Anionic Phospho-Fries Rearrangement. Curr. Org. Chem. 2004, 8, 623–636. [Google Scholar] [CrossRef]
- Wu, S.; Deligonal, N.; Protasiewicz, J.D. An unusually unstable ortho-phosphinophenol and its use to prepare benzoxaphospholes having enhanced air-stability. Dalton Trans. 2013, 42, 14866–14874. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Li, M.; Liu, Y.; Zhou, Y.; Zhao, C.; Goto, M.; Yin, S.-F.; Han, L.-B. Stereoselective Synthesis of Phosphoryl-Substituted Phenols. Adv. Synth. Catal. 2014, 356, 781–794. [Google Scholar] [CrossRef]
- Korb, M.; Schaarschmidt, D.; Lang, H. Anionic Phospho-Fries Rearrangement at Ferrocene: One-Pot Approach to P,O-Substituted Ferrocenes. Organometallics 2014, 33, 2099–2108. [Google Scholar] [CrossRef]
- Herd, O.; Heßler, A.; Hingst, M.; Tepper, M.; Stelzer, O. Water soluble phosphines VII. Palladium-catalyzed P–C cross coupling reactions between primary or secondary phosphines and functional aryliodides—A novel synthetic route to water soluble phosphines. J. Organomet. Chem. 1996, 522, 69–76. [Google Scholar] [CrossRef]
- Korb, M.; Lang, H. Planar Chirality from the Chiral Pool: Diastereoselective Anionic Phospho-Fries Rearrangements at Ferrocene. Organometallics 2014, 33, 6643–6659. [Google Scholar] [CrossRef]
- Korb, M.; Lehrich, S.W.; Lang, H. Reactivity of Ferrocenyl Phosphates Bearing (Hetero-)Aromatics and [3]Ferrocenophanes toward Anionic Phospho-Fries Rearrangements. J. Org. Chem. 2017, 82, 3102–3124. [Google Scholar] [CrossRef]
- Kakimoto, N.; Ogura, Y.; Watanabe, H.; Takikawa, H. Total synthesis of both enantiomers of clavigerins B and C. Tetrahedron 2020, 76, 131297. [Google Scholar] [CrossRef]
- Wang, Y.; Ju, W.; Tian, H.; Sun, S.; Li, X.; Tian, W.; Gui, J. Facile Access to Bridged Ring Systems via Point-to-Planar Chirality Transfer: Unified Synthesis of Ten Cyclocitrinols. J. Am. Chem. Soc. 2019, 141, 5021–5033. [Google Scholar] [CrossRef]
- Wang, Y.; Ju, W.; Tian, H.; Tian, W.; Gui, J. Scalable Synthesis of Cyclocitrinol. J. Am. Chem. Soc. 2018, 140, 9413–9416. [Google Scholar] [CrossRef]
- Kaabi, A.; Besbes, R. Amino Phosphate Monoesters: A Convenient Source of 2-Alkylamino-3-methoxy-3-phenylpropionates via Aziridinium Ions. Synth. Commun. 2013, 43, 1587–1593. [Google Scholar] [CrossRef]
- Shinohara, R.; Kawashima, H.; Ogawa, N.; Kobayashi, Y. Substitution of Secondary Benzylic Phosphates with Diarylmethyl Anions. Tetrahedron 2019, 75, 2717–2725. [Google Scholar] [CrossRef]
- Shinohara, R.; Ogawa, N.; Kawashima, H.; Wada, K.; Saito, S.; Yamazaki, T.; Kobayashi, Y. SN2 Reaction of Diarylmethyl Anions at Secondary Alkyl and Cycloalkyl Carbons. Eur. J. Org. Chem. 2019, 2019, 1461–1478. [Google Scholar] [CrossRef]
- Pallikonda, G.; Chakravarty, M. Benzylic Phosphates in Friedel–Crafts Reactions with Activated and Unactivated Arenes: Access to Polyarylated Alkanes. J. Org. Chem. 2016, 81, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Sakai, M.; Ishida, Y.; Yasui, T. Synthesis of 1-(Difluoromethyl)alk-1-enes via Palladium-Catalyzed SN2′-Type Substitution Reaction of Difluoromethylated Allylic Phosphates with 1,3-Dicarbonyl Compounds and Imides. J. Org. Chem. 2021, 86, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Takase, T.; Kuroyanagi, E.; Yasui, T. Synthesis of difluoromethylated diarylmethanes via Fe(OTf)3-catalyzed Friedel–Crafts reaction of 2,2-difluoro-1-arylethyl phosphates. Chem. Commun. 2021, 57, 3877–3880. [Google Scholar] [CrossRef] [PubMed]
- Shintani, R.; Ohzono, A.; Shirota, K. Phosphinative cyclopropanation of allyl phosphates with lithium phosphides. Chem. Commun. 2020, 56, 11851–11854. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.M.; Li, Q.; Rötheli, A.R.; Jacobsen, E.N. Catalytic activation of glycosyl phosphates for stereoselective coupling reactions. Proc. Natl. Acad. Sci. USA 2019, 116, 35–39. [Google Scholar] [CrossRef]
- Li, Q.; Levi, S.M.; Jacobsen, E.N. Highly Selective β-Mannosylations and β-Rhamnosylations Catalyzed by Bis-thiourea. J. Am. Chem. Soc. 2020, 142, 11865–11872. [Google Scholar] [CrossRef]
- Li, Y.; Jie, J.; Li, H.; Yang, H.; Fu, H. Synthesis of Spirotetrahydrofuran Oxindoles via Palladium-Catalyzed [4 + 1] Cycloaddition of Diphenyl 2-Oxoindolin-3-yl Phosphates and 2-Methylidenetrimethylene Carbonate. Org. Lett. 2021, 23, 6499–6503. [Google Scholar] [CrossRef]
- Chen, Q.; Teng, Y.; Xu, F. Lanthanide Silylamide-Catalyzed Synthesis of Pyrano[2,3-b]indol-2-ones. Org. Lett. 2021, 23, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Rokade, B.V.; Guiry, P.J. Synthesis of α-Aryl Oxindoles by Friedel–Crafts Alkylation of Arenes. J. Org. Chem. 2020, 85, 6172–6180. [Google Scholar] [CrossRef]
- Xing, T.; Wei, K.-J.; Quan, Z.-J.; Wang, X.-C. Nucleophilic Substitution Reaction of Pyrimidin-2-yl Phosphates Using Amines and Thiols as Nucleophiles Mediated by PEG-400 as an Environmentally Friendly Solvent. Synthesis 2015, 47, 3925–3935. [Google Scholar] [CrossRef]
- Butt, N.A.; Zhang, W. Transition metal-catalyzed allylic substitution reactions with unactivated allylic substrates. Chem. Soc. Rev. 2015, 44, 7929–7967. [Google Scholar] [CrossRef] [PubMed]
- Mohammadkhani, L.; Heravi, M.M. Applications of Transition-Metal-Catalyzed Asymmetric Allylic Substitution in Total Synthesis of Natural Products: An Update. Chem. Rec. 2021, 21, 29–68. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.; Evans, P.A. Transition-Metal-Catalyzed Allylic Substitution Reactions: Stereoselective Construction of α- and β-Substituted Carbonyl Compounds. Synthesis 2013, 45, 3179–3198. [Google Scholar] [CrossRef]
- Qu, J.; Helmchen, G. Applications of Iridium-Catalyzed Asymmetric Allylic Substitution Reactions in Target-Oriented Synthesis. Acc. Chem. Res. 2017, 50, 2539–2555. [Google Scholar] [CrossRef] [PubMed]
- Sundararaju, B.; Achard, M.; Bruneau, C. Transition metal catalyzed nucleophilic allylic substitution: Activation of allylic alcohols via π-allylic species. Chem. Soc. Rev. 2012, 41, 4467–4483. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Carr, J.L.; Hoveyda, A.H. A Broadly Applicable NHC–Cu-Catalyzed Approach for Efficient, Site-, and Enantioselective Coupling of Readily Accessible (Pinacolato)alkenylboron Compounds to Allylic Phosphates and Applications to Natural Product Synthesis. J. Am. Chem. Soc. 2014, 136, 2149–2161. [Google Scholar] [CrossRef]
- Lee, J.; Torker, S.; Hoveyda, A.H. Versatile Homoallylic Boronates by Chemo-, SN2′-, Diastereo- and Enantioselective Catalytic Sequence of Cu−H Addition to Vinyl-B(pin)/Allylic Substitution. Angew. Chem. Int. Ed. 2017, 56, 821–826. [Google Scholar] [CrossRef]
- Zhou, Y.; Shi, Y.; Torker, S.; Hoveyda, A.H. SN2″-Selective and Enantioselective Substitution with Unsaturated Organoboron Compounds and Catalyzed by a Sulfonate-Containing NHC-Cu Complex. J. Am. Chem. Soc. 2018, 140, 16842–16854. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jung, B.; Torker, S.; Hoveyda, A.H. N-Heterocyclic Carbene–Copper-Catalyzed Group-, Site-, and Enantioselective Allylic Substitution with a Readily Accessible Propargyl(pinacolato)boron Reagent: Utility in Stereoselective Synthesis and Mechanistic Attributes. J. Am. Chem. Soc. 2015, 137, 8948–8964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Q.; Zhang, B.; Lu, X.; Liu, J.-H.; Lu, X.-Y.; Xiao, B.; Fu, Y. Copper-Catalyzed SN2′-Selective Allylic Substitution Reaction of gem-Diborylalkanes. Org. Lett. 2016, 18, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hoveyda, A.H. Catalytic SN2′- and Enantioselective Allylic Substitution with a Diborylmethane Reagent and Application in Synthesis. Angew. Chem. Int. Ed. 2016, 55, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, Z.; Fu, C.; Wang, G.; Zheng, C.; Wu, X. Synergistic Ni/Pd Catalysis for Asymmetric Allylic Alkylation of 2-Acyl Imidazoles. Org. Lett. 2023, 25, 5448–5453. [Google Scholar] [CrossRef] [PubMed]
- Jacques, R.; Pullin, R.D.C.; Fletcher, S.P. Desymmetrization of meso-bisphosphates using copper catalysis and alkylzirconocene nucleophiles. Nat. Commun. 2019, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Q.; Niu, J.; Guo, X.; Xiong, T.; Zhang, Q. Copper-Catalyzed Asymmetric Hydroallylation of Vinylsilanes. Eur. J. Org. Chem. 2022, 2022, e202101575. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Buchwald, S.L. Enantioselective CuH-Catalyzed Hydroallylation of Vinylarenes. J. Am. Chem. Soc. 2016, 138, 5024–5027. [Google Scholar] [CrossRef] [PubMed]
- Yurino, T.; Tani, R.; Ohkuma, T. Pd-Catalyzed Allylic Isocyanation: Nucleophilic N-Terminus Substitution of Ambident Cyanide. ACS Catal. 2019, 9, 4434–4440. [Google Scholar] [CrossRef]
- Yurino, T.; Tange, Y.; Ohkuma, T. Palladium-Catalyzed Nucleophilic Isocyanation for the Synthesis of α-Aryl-α-Isocyanoacetoamide Derivatives. Bull. Chem. Soc. Jpn. 2021, 94, 2155–2161. [Google Scholar] [CrossRef]
- Yurino, T.; Tange, Y.; Tani, R.; Ohkuma, T. Ag2O-catalysed nucleophilic isocyanation: Selective formation of less-stable benzylic isonitriles. Org. Chem. Front. 2020, 7, 1308–1313. [Google Scholar] [CrossRef]
- Takise, R.; Itami, K.; Yamaguchi, J. Cyanation of Phenol Derivatives with Aminoacetonitriles by Nickel Catalysis. Org. Lett. 2016, 18, 4428–4431. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Zheng, B.-H.; Ding, C.-H.; Hou, X.-L. Enantioselective Synthesis of 2,3-Disubstituted Indanones via Pd-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Ketones. Org. Lett. 2013, 15, 6086–6089. [Google Scholar] [CrossRef]
- Spoehrle, S.S.M.; West, T.H.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of α-Amino Acid Derivatives via Allylic Amination and [2,3]-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2017, 139, 11895–11902. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Zhang, G.; Xu, M.; Qi, X. ProPhenol Derived Ligands to Simultaneously Coordinate a Main-Group Metal and a Transition Metal: Application to a Zn−Cu Catalyzed Reaction. Chem. Eur. J. 2022, 28, e202104268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xu, J.; Gao, Y.; Li, X.; Tang, G.; Zhao, Y. Synthesis of Diarylmethanes through Palladium-Catalyzed Coupling of Benzylic Phosphates with Arylsilanes. Synlett 2014, 25, 2928–2932. [Google Scholar] [CrossRef]
- Zhang, Y.; Raugh, N.; Koert, U. Fluorotrifluoromethyl Group Installation: Tetrasubstituted Tertiary Stereocenters Containing C–F and C–CF3 Bonds via Copper-Mediated Allylic Substitution. Org. Lett. 2023, 25, 5641–5645. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Sarlah, D. Arenophile-Mediated Dearomative Functionalization Strategies. Synlett 2018, 29, 845–855. [Google Scholar] [CrossRef]
- Petrenko, A.; Mrkobrada, S.; Tobrman, T. State-or-the-Art Approaches to the Synthesis of 2H-Pyrroles. Targets Heterocycl. Syst. 2021, 25, 308–325. [Google Scholar] [CrossRef]
- Polák, P.; Tobrman, T. Dearomatization Strategy for the Synthesis of Arylated 2H-Pyrroles and 2,3,5-Trisubstituted 1H-Pyrroles. Org. Lett. 2017, 19, 4608–4611. [Google Scholar] [CrossRef]
- Huang, G.; Yin, B. Recent Developments in Transition Metal-Catalyzed Dearomative Cyclizations of Indoles as Dipolarophiles for the Construction of Indolines. Adv. Synth. Catal. 2019, 361, 405–425. [Google Scholar] [CrossRef]
- Komatsuda, M.; Muto, K.; Yamaguchi, J. Pd-Catalyzed Dearomative Allylation of Benzyl Phosphates. Org. Lett. 2018, 20, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Yanagimoto, A.; Komatsuda, M.; Muto, K.; Yamaguchi, J. Dearomative Allylation of Naphthyl Cyanohydrins by Palladium Catalysis: Catalyst-Enhanced Site Selectivity. Org. Lett. 2020, 22, 3423–3427. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Czabaniuk, L.C. Palladium-Catalyzed Asymmetric Benzylation of Azlactones. Chem. Eur. J. 2013, 19, 15210–15218. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.J.; Yang, C.; Fyfe, J.W.B.; Snaddon, T.N. Enantioselective α-Benzylation of Acyclic Esters Using π-Extended Electrophiles. Angew. Chem. Int. Ed. 2018, 57, 12102–12105. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Yokoyama, Y.; Inamura, K.; Katakura, S.-i.; Komoriya, S.; Yamaguchi, H.; Hara, T.; Iwamoto, M. Dibasic (Amidinoaryl)propanoic Acid Derivatives as Novel Blood Coagulation Factor Xa Inhibitors. J. Med. Chem. 1994, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Toyomasu, T.; Nishio, H.; Shishido, T. Gold-catalyzed thioetherification of allyl, benzyl, and propargyl phosphates. Catal. Sci. Technol. 2022, 12, 1109–1116. [Google Scholar] [CrossRef]
- Zhang, K.; Provot, O.; Alami, M.; Tran, C.; Hamze, A. Pd-Catalyzed Coupling of N-Tosylhydrazones with Benzylic Phosphates: Toward the Synthesis of Di- or Tri-Substituted Alkenes. J. Org. Chem. 2022, 87, 1249–1261. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Krawczyk, E.; Mielniczak, G.; Owsianik, K.; Łuczak, J. Asymmetric oxidation of enol phosphates to α-hydroxy ketones using Sharpless reagents and a fructose derived dioxirane. Tetrahedron Asymmetry 2012, 23, 1480–1489. [Google Scholar] [CrossRef]
- Owsianik, K.; Krawczyk, E.; Mielniczak, G.; Koprowski, M.; Sieroń, L. Three-step synthesis of chiral and sterically hindered amino alcohols based on cyclic enol phosphates. Tetrahedron 2018, 74, 7343–7350. [Google Scholar] [CrossRef]
- Krawczyk, E.; Koprowski, M.; Mielniczak, G.; Owsianik, K. Asymmetric synthesis of 5,7-O-dimethyleucomols via enantioselective oxidation of enol phosphates. Tetrahedron Asymmetry 2015, 26, 876–883. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Almutairi, S.M.; Chan, Y.; Stephenson, G.R.; Gama, Y.; Goodyear, R.L.; Douteau, A.; Allin, S.M.; Jones, G.A. Asymmetric Oxidation of Enol Derivatives to α-Alkoxy Carbonyls Using Iminium Salt Catalysts: A Synthetic and Computational Study. J. Org. Chem. 2019, 84, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, Q.; Qian, C.; Liu, C.; Liu, W.; Chen, K.; Lei, A. Solvent-Enabled Radical Selectivities: Controlled Syntheses of Sulfoxides and Sulfides. Angew. Chem. Int. Ed. 2016, 55, 1094–1097. [Google Scholar] [CrossRef]
- Wang, H.; Wang, G.; Lu, Q.; Chiang, C.-W.; Peng, P.; Zhou, J.; Lei, A. Catalyst-Free Difunctionalization of Activated Alkenes in Water: Efficient Synthesis of β-Keto Sulfides and Sulfones. Chem. Eur. J. 2016, 22, 14489–14493. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Wang, F.; Ning, R.; Kong, D.; Wu, M. A new synthetic approach to oxindoles (1,3-dihydro-2H-indol-2-ones) by reductive dephosphorylation with hydroiodic acid of 3-(diethylphosphoryloxy)- oxindoles, derived from isatins (1H-Indole-2,3-diones). ARKIVOC 2022, 2022, 135–146. [Google Scholar] [CrossRef]
- Chowdhury, S.; Standaert, R.F. Deoxygenation of Unhindered Alcohols via Reductive Dealkylation of Derived Phosphate Esters. J. Org. Chem. 2016, 81, 9957–9963. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Zhao, G.; Ramadoss, V.; Tian, L.; Wang, Y. Electrochemical Deoxygenative Barbier-Type Reaction. Org. Lett. 2022, 24, 3668–3673. [Google Scholar] [CrossRef]
- Tomkiel, A.M.; Siergiejczyk, L.; Naróg, D.; Płoszyńska, J.; Sobkowiak, A.; Morzycki, J.W. Electrochemical cholesterylation of sugars with cholesteryl diphenylphosphate. Steroids 2017, 117, 44–51. [Google Scholar] [CrossRef]
Scheme 1.
Representative examples of organophosphorus compounds related to biological systems.
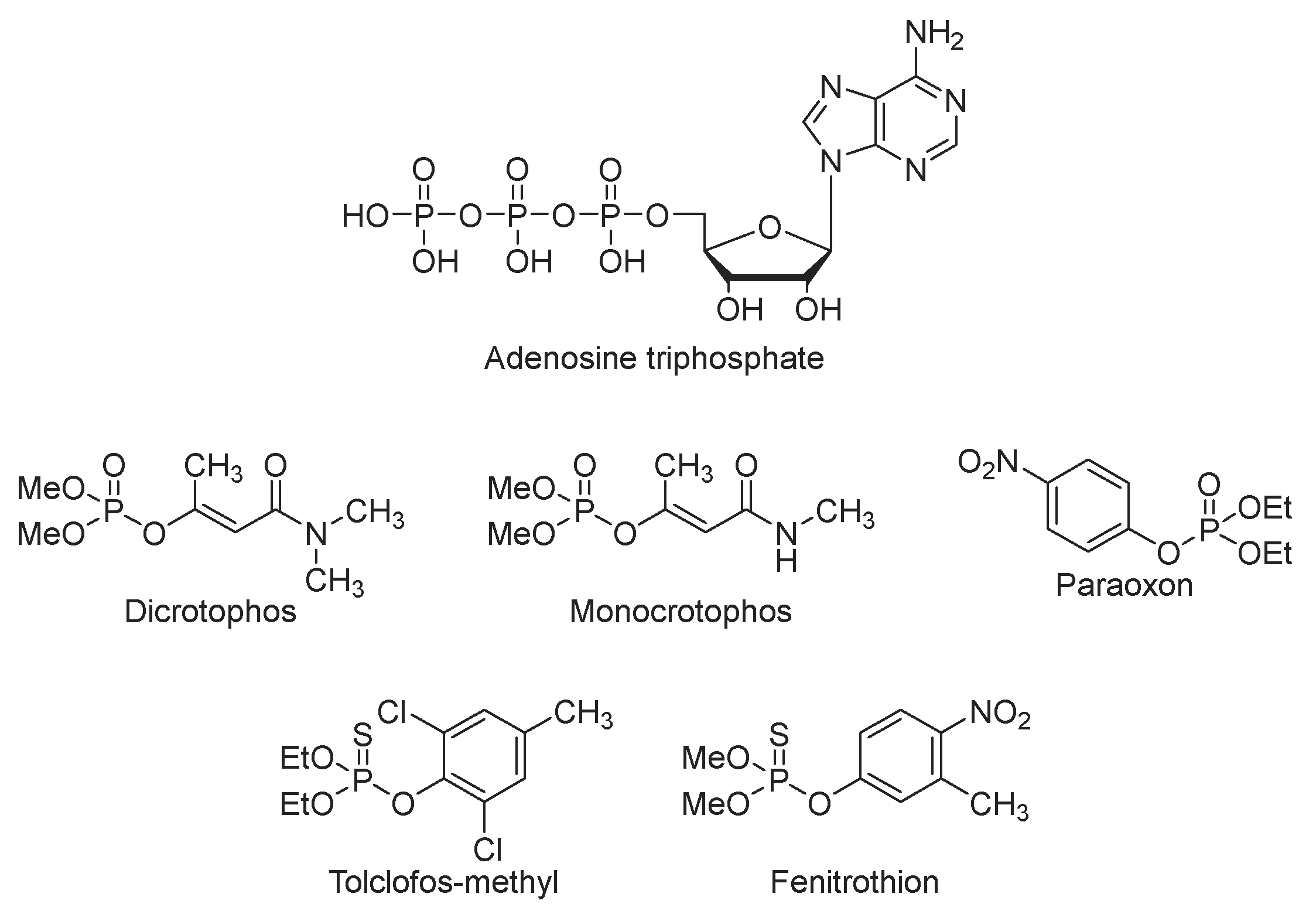
Scheme 2.
General scheme representing the synthesis and applications of organophosphates under review.
Scheme 2.
General scheme representing the synthesis and applications of organophosphates under review.

Scheme 3.
The general scheme summarizing the use of organophosphates in cross-coupling reactions.
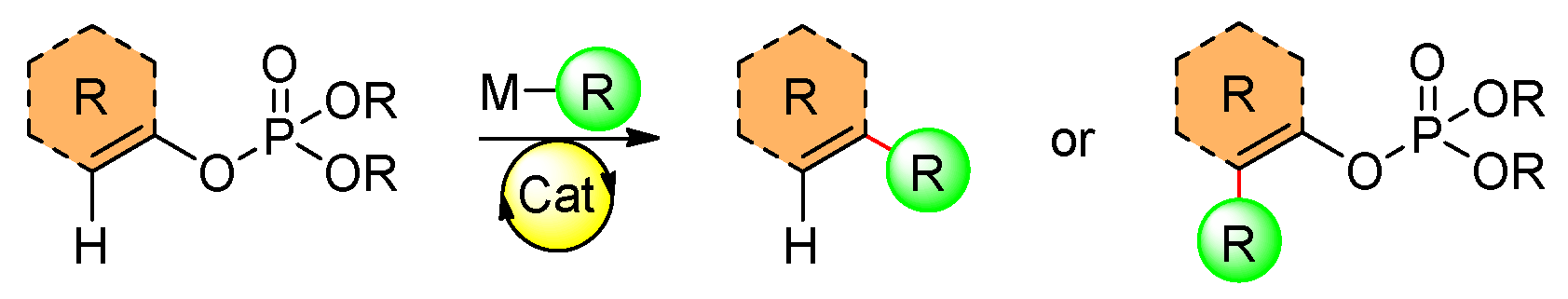
Scheme 4.
Nickel-catalyzed Kumada reaction for the synthesis of conjugated dienes.

Scheme 5.
Proposed mechanism of the nickel-catalyzed Kumada coupling of vinyl diethyl phosphates.

Scheme 6.
Magnesium-catalyzed polymerization as a side reaction during the Kumada reaction of vinyl phosphates.
Scheme 6.
Magnesium-catalyzed polymerization as a side reaction during the Kumada reaction of vinyl phosphates.
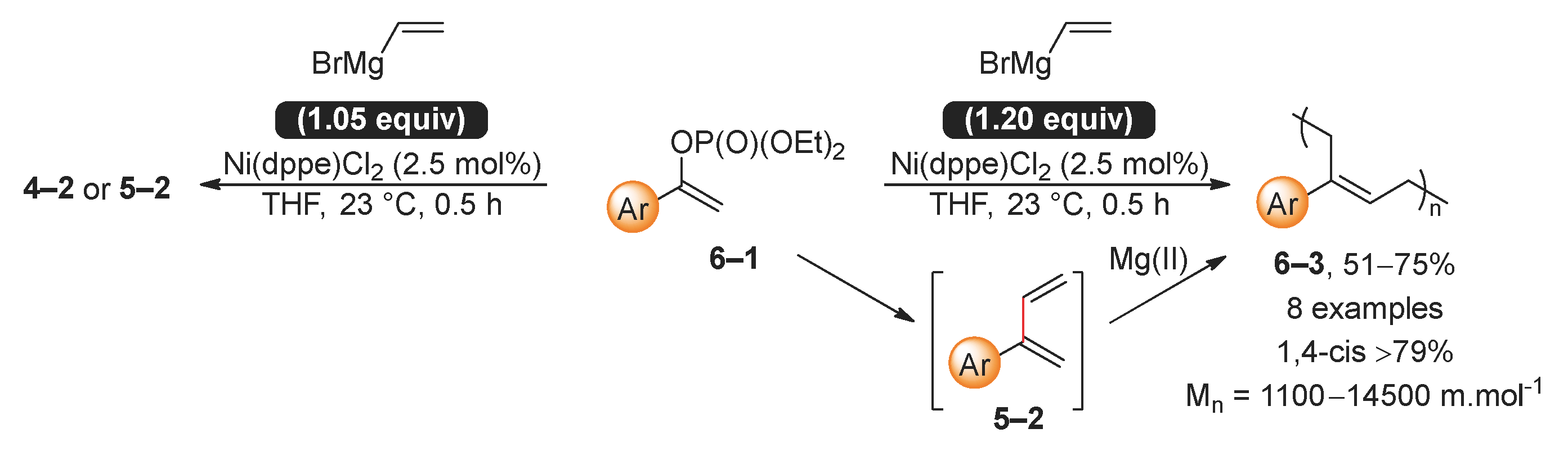
Scheme 7.
Synthesis of [n]dendralenes by the Kumada reaction of vinyl diethyl phosphates.
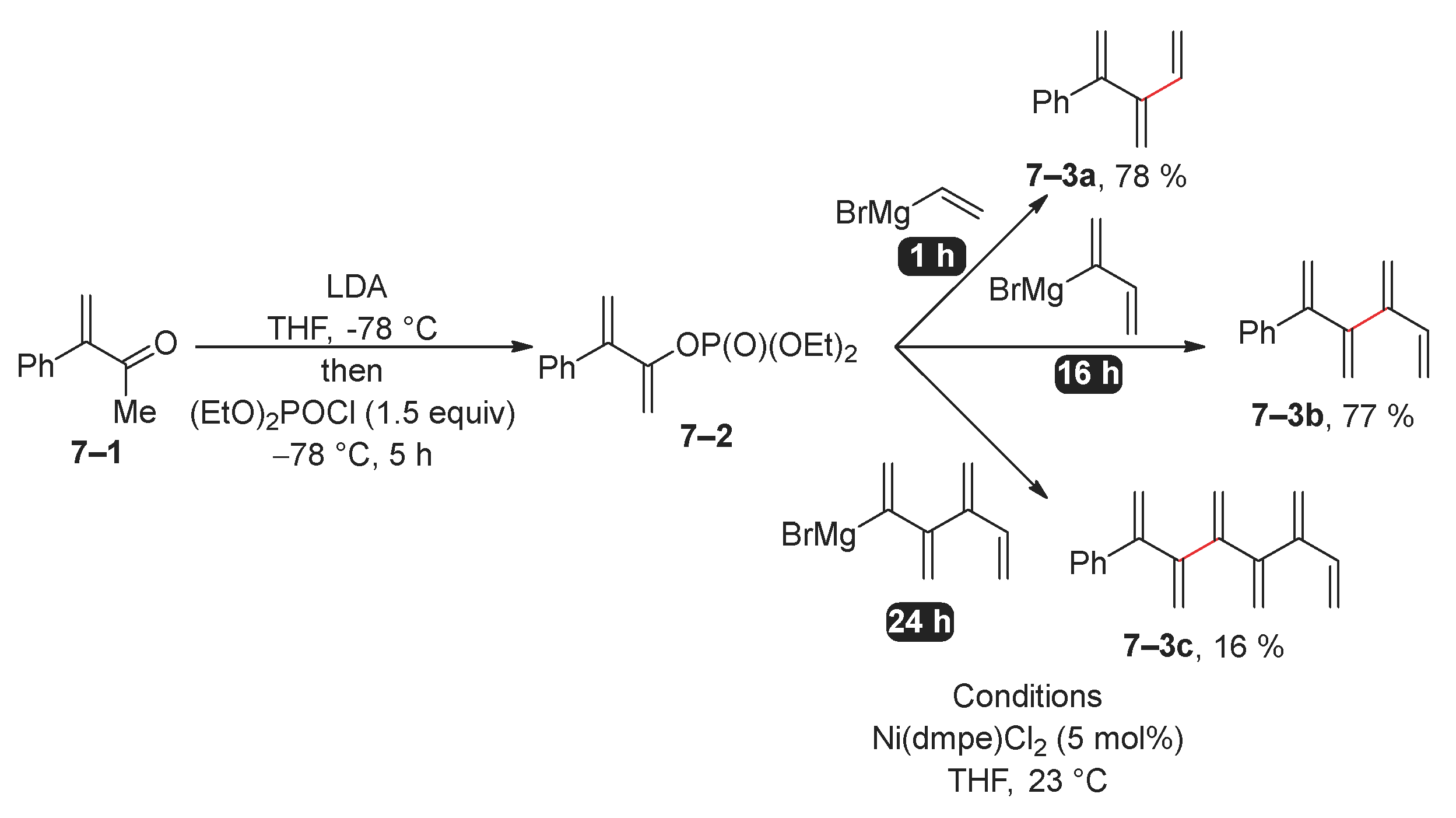
Scheme 8.
Iron-catalyzed Kumada reaction of heteroaryl diethyl phosphate.

Scheme 9.
(a) Iron-catalyzed Kumada reaction of aryl phosphates and (b) iron- [60] and nickel-catalyzed [61] reductive cross-coupling reactions of aryl phosphates.

Scheme 10.
Palladium- and nickel-catalyzed Kumada reaction for the synthesis of (a–c) cyclic alkenes and (d) acyclic tetrasubstituted alkenes.
Scheme 10.
Palladium- and nickel-catalyzed Kumada reaction for the synthesis of (a–c) cyclic alkenes and (d) acyclic tetrasubstituted alkenes.

Scheme 11.
Synthesis of key intermediate during the total synthesis of salviamines E and F.
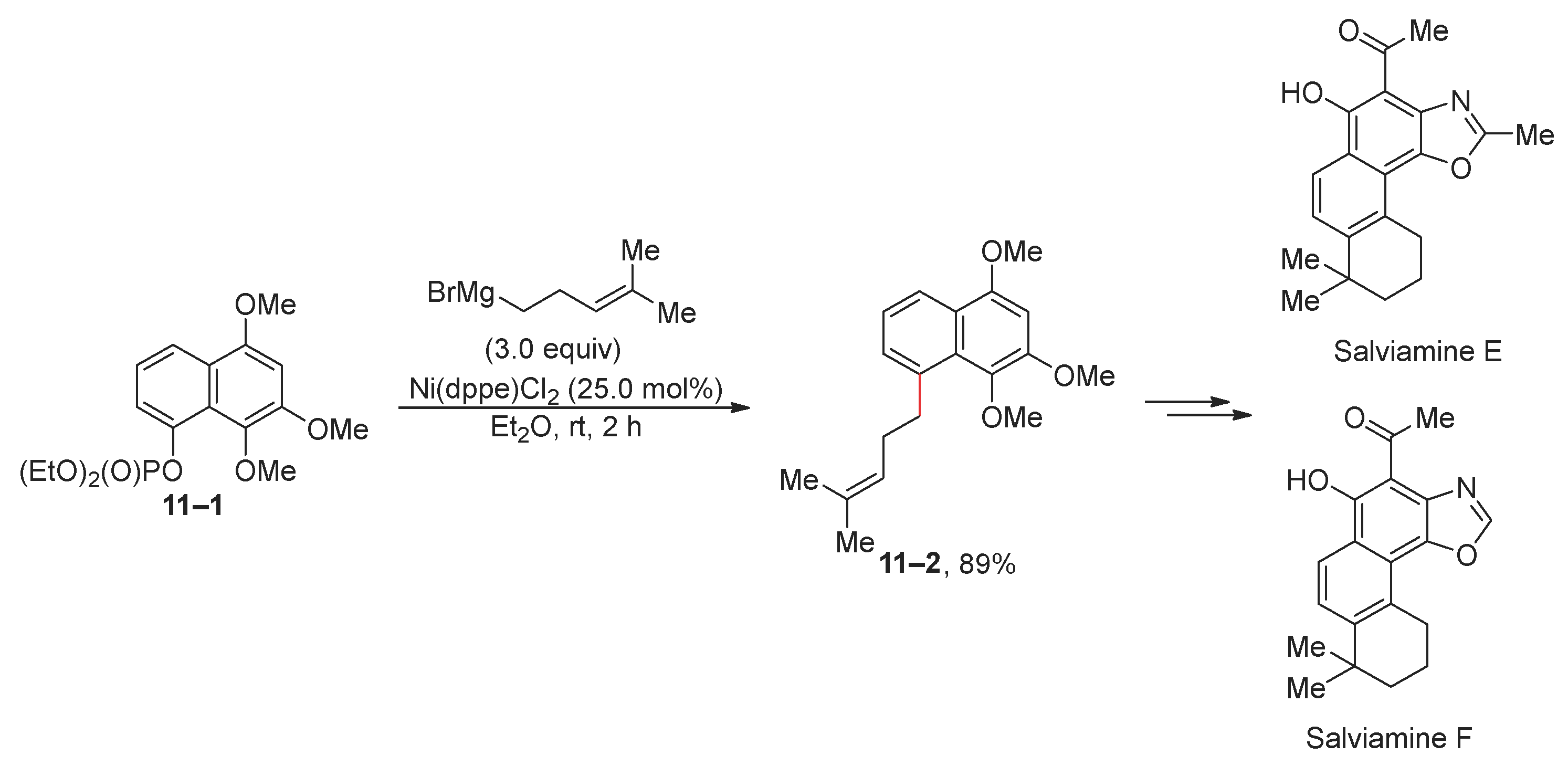
Scheme 12.
Application of the Negishi reaction for the synthesis of (a) tetrasubstituted and (b) trisubstituted alkenes.
Scheme 12.
Application of the Negishi reaction for the synthesis of (a) tetrasubstituted and (b) trisubstituted alkenes.
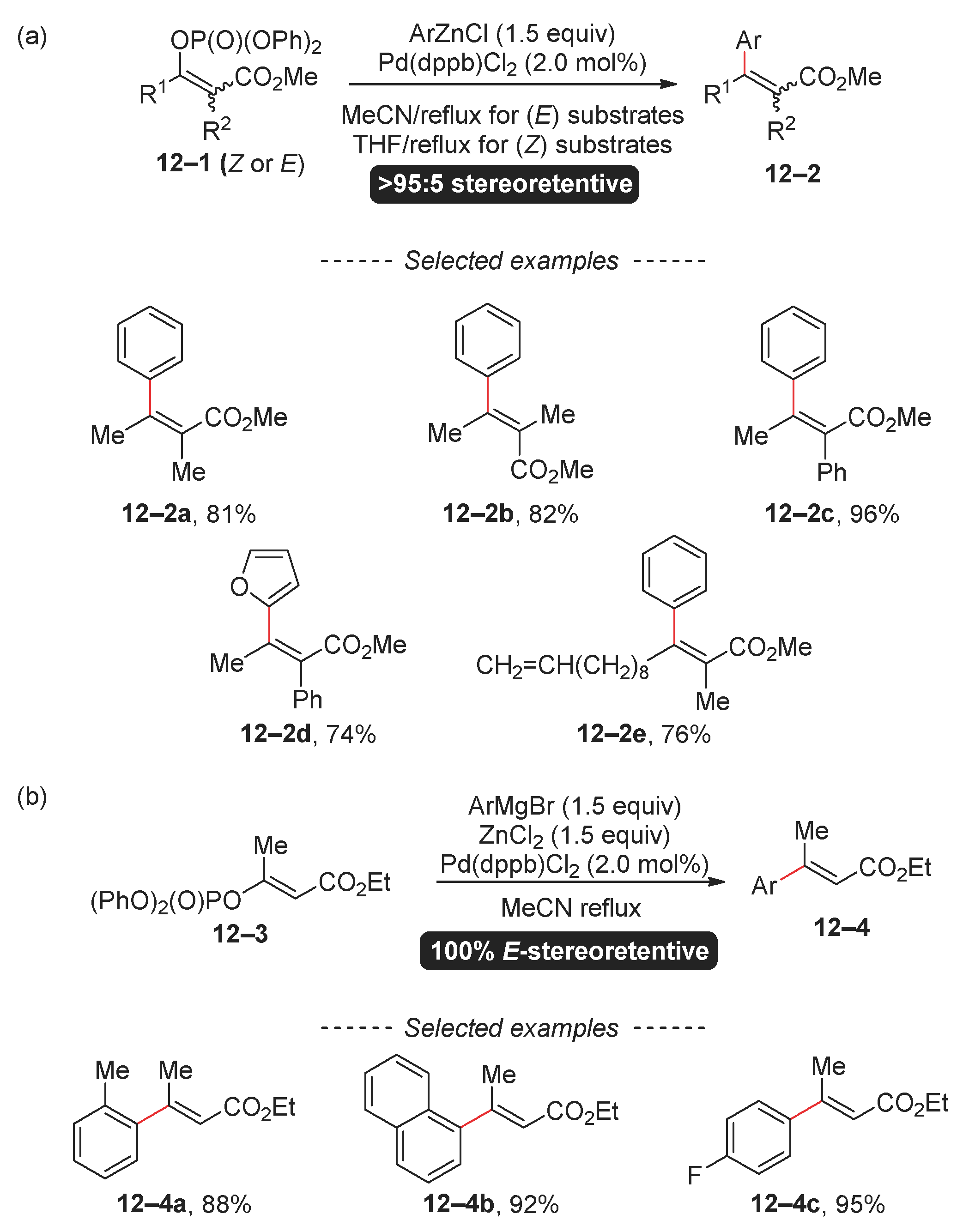
Scheme 13.
Aluminum chloride-mediated Negishi reaction of trisubstituted vinyl phosphate en route to (Z)-tamoxifen.
Scheme 13.
Aluminum chloride-mediated Negishi reaction of trisubstituted vinyl phosphate en route to (Z)-tamoxifen.
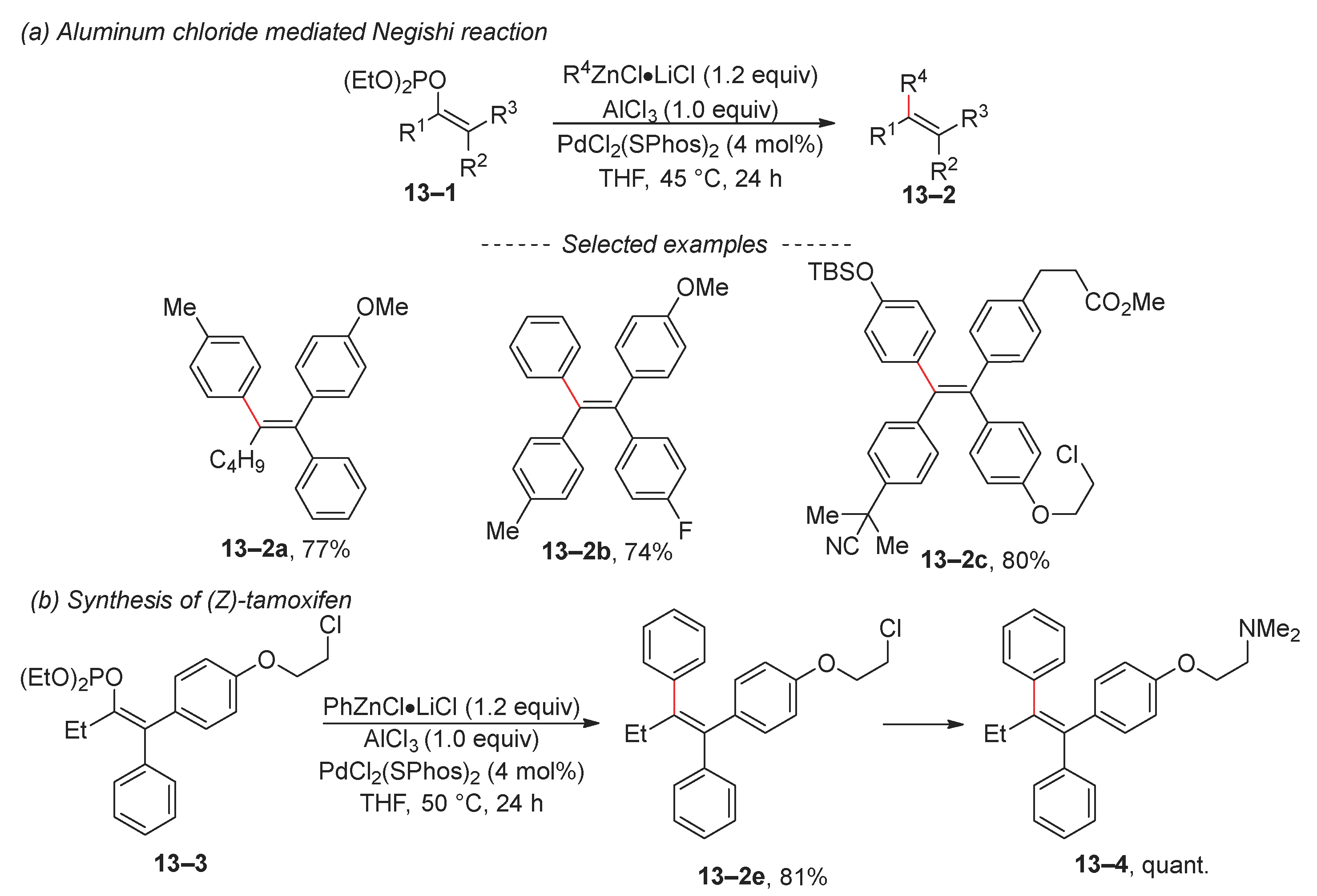
Scheme 14.
An aluminum chloride-promoted Negishi reaction for the synthesis of (a) substituted indole 14–3 and (b) [4]dendralene 14–7.
Scheme 14.
An aluminum chloride-promoted Negishi reaction for the synthesis of (a) substituted indole 14–3 and (b) [4]dendralene 14–7.
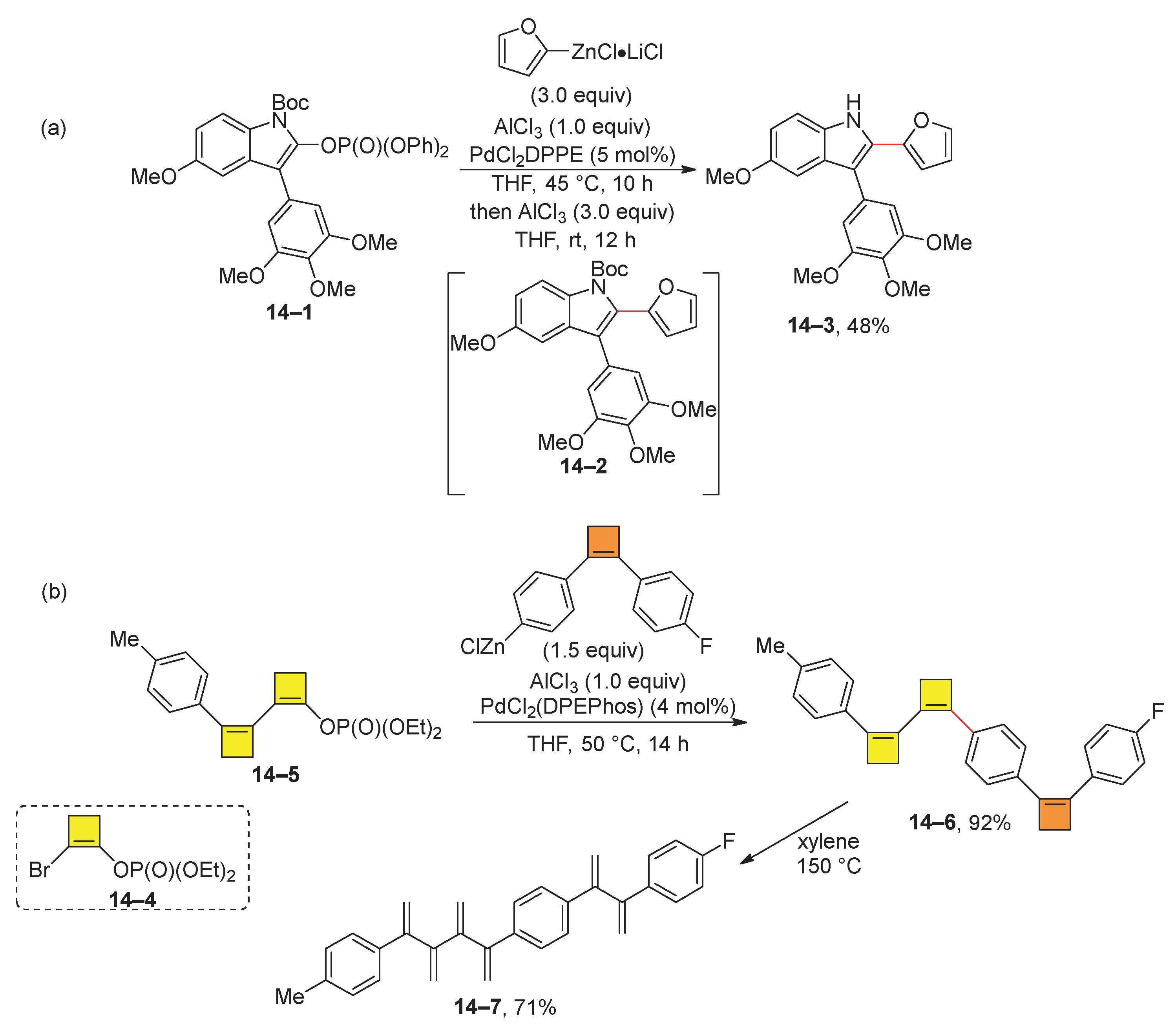
Scheme 15.
Nickel-catalyzed Suzuki reaction of aryl phosphates.

Scheme 16.
Palladium-catalyzed Suzuki reaction of phosphate enamides.

Scheme 17.
Suzuki reaction en route to C-arylglycal.
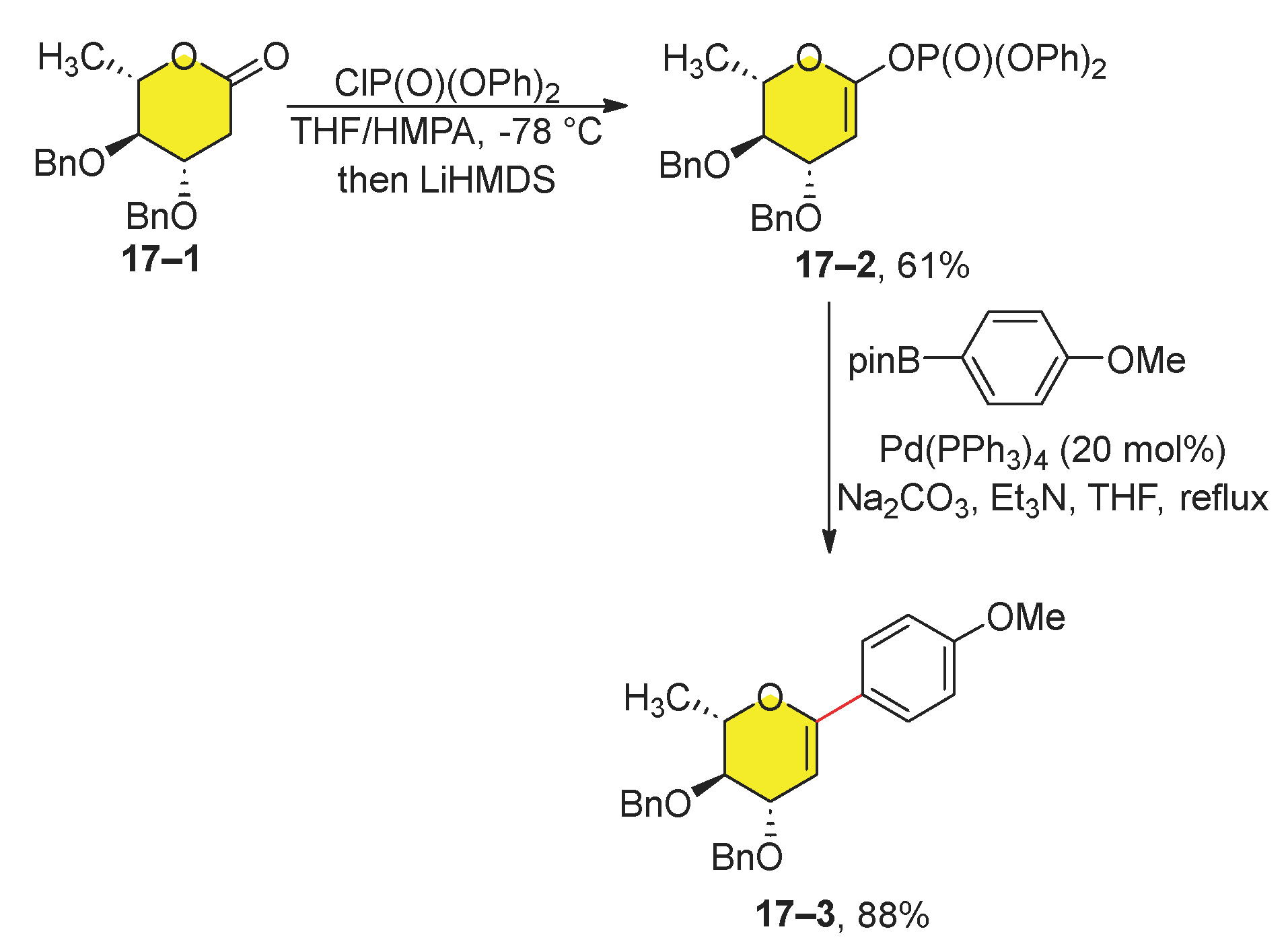
Scheme 18.
Visible light-mediated borylation of aryl and heteroaryl diethyl phosphates.
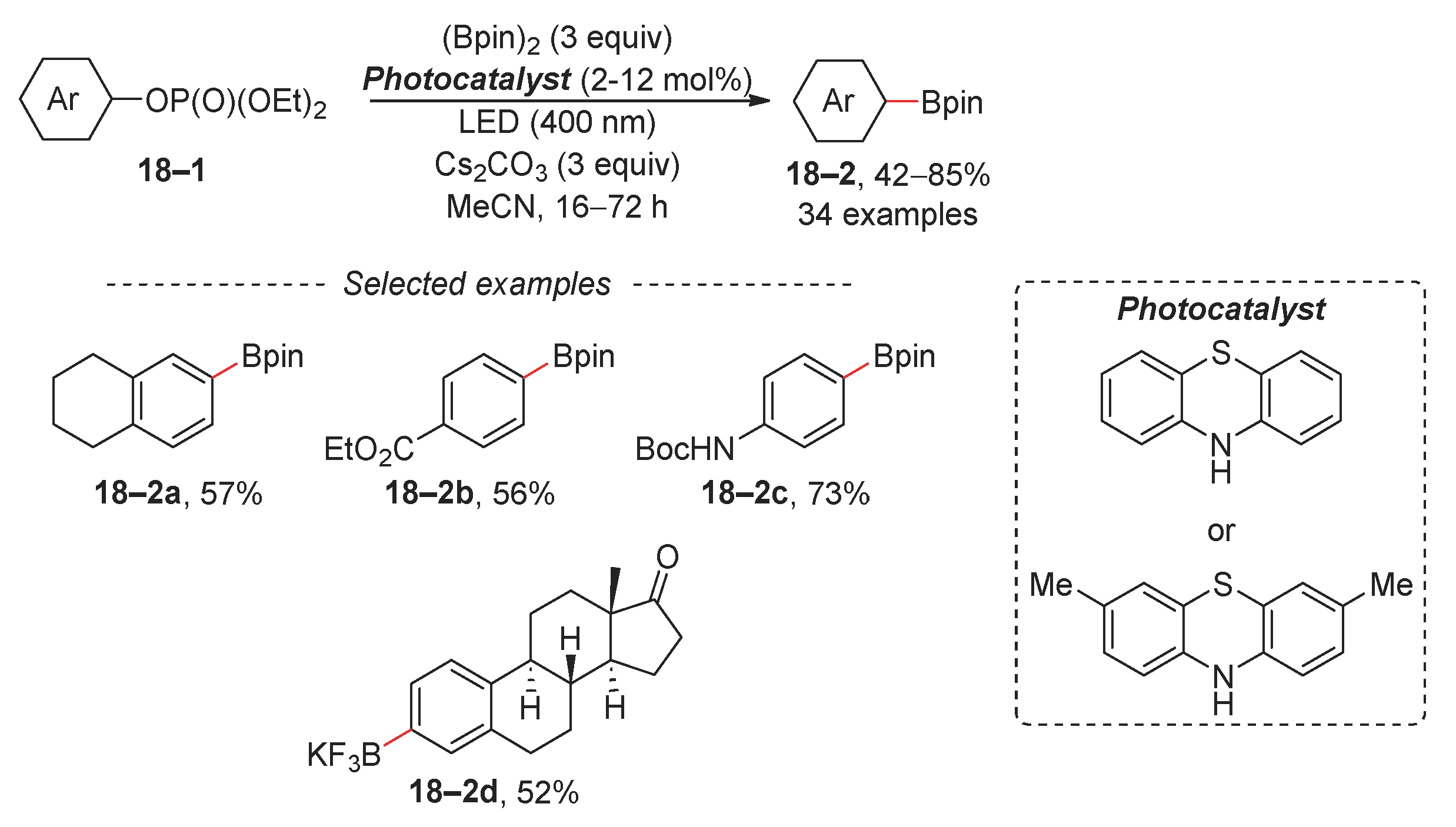
Scheme 19.
Application of the Suzuki reaction of organophosphates for the preparation of key intermediates.
Scheme 19.
Application of the Suzuki reaction of organophosphates for the preparation of key intermediates.

Scheme 20.
Suzuki coupling of cycloalkenyl phosphate en route to Didemnaketal B.
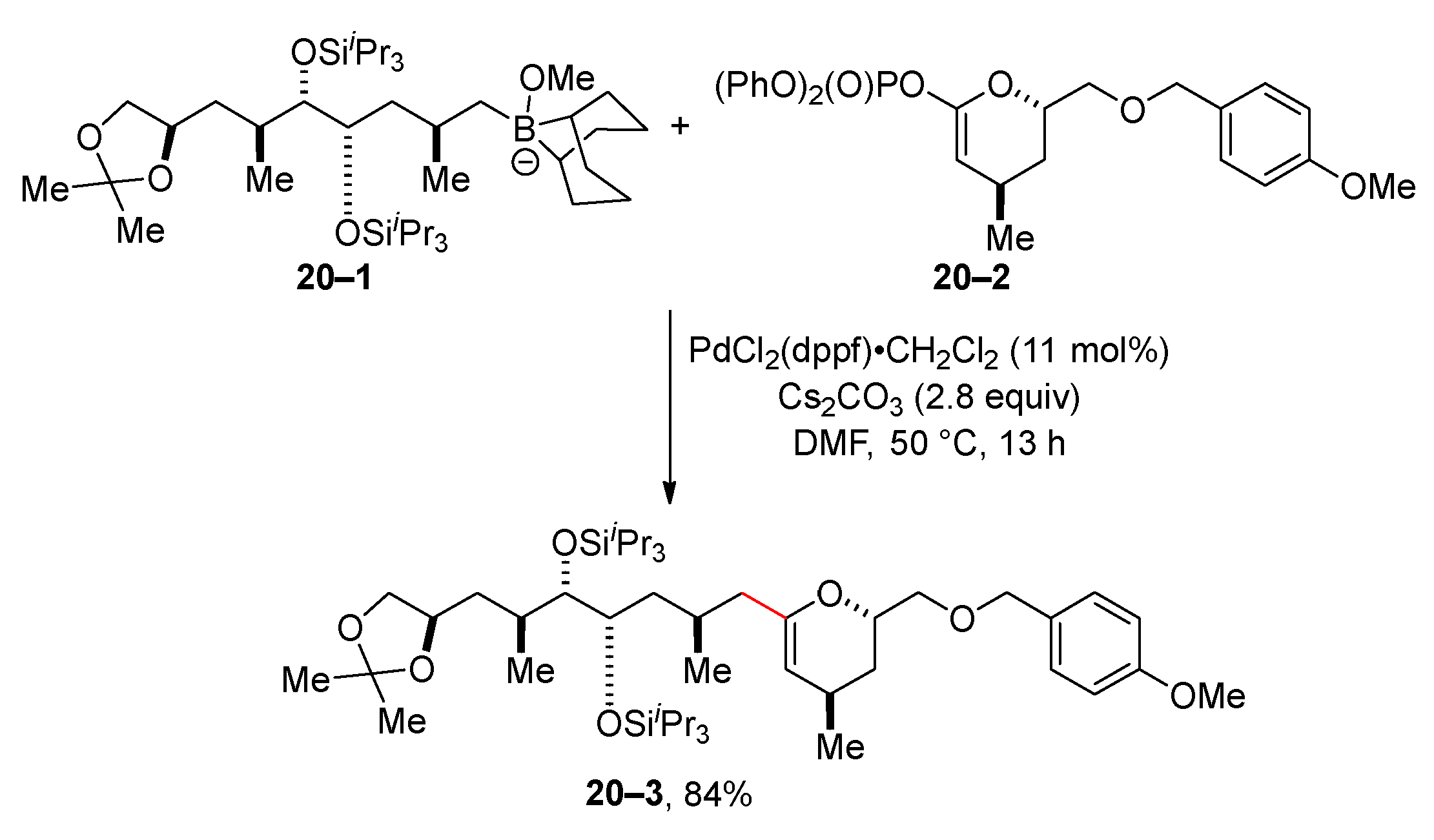
Scheme 21.
Total synthesis of (–)-anabasine.

Scheme 22.
General scheme representing the use of organophosphates for the activation of the C–H bond.
Scheme 22.
General scheme representing the use of organophosphates for the activation of the C–H bond.
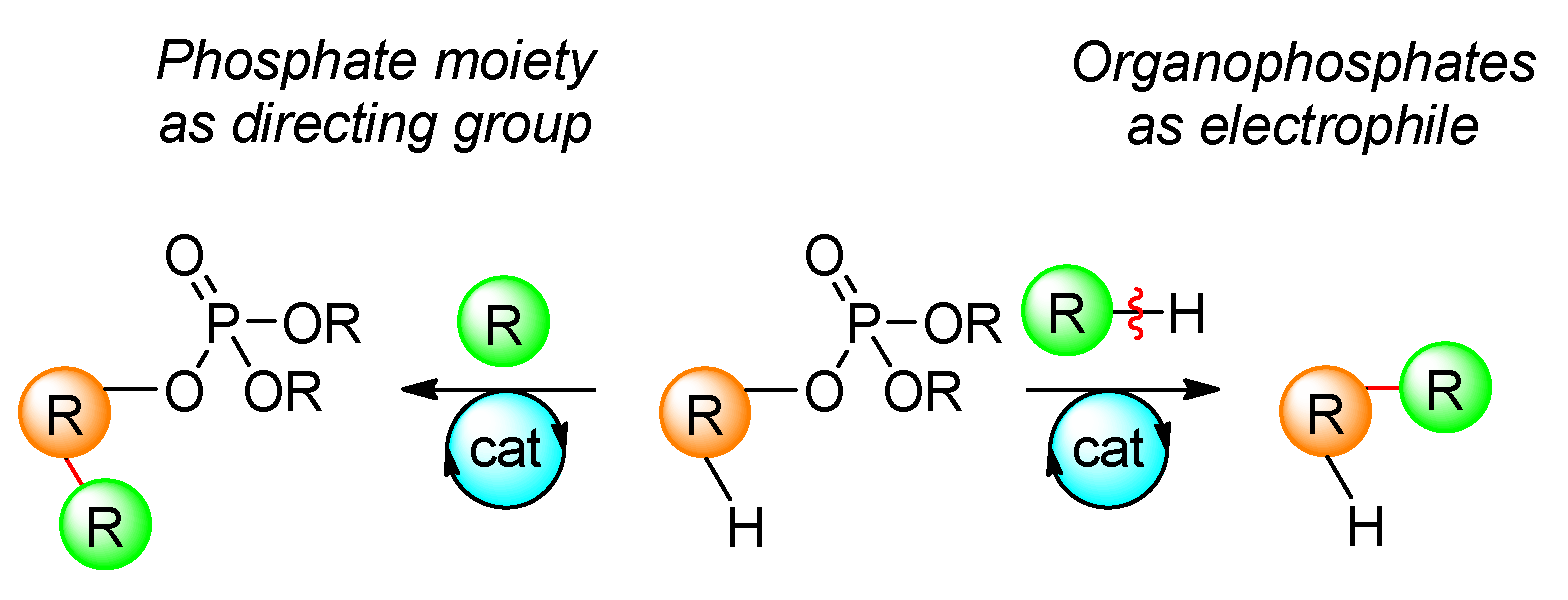
Scheme 23.
Rhodium-catalyzed vinylation of vinyl phosphates.
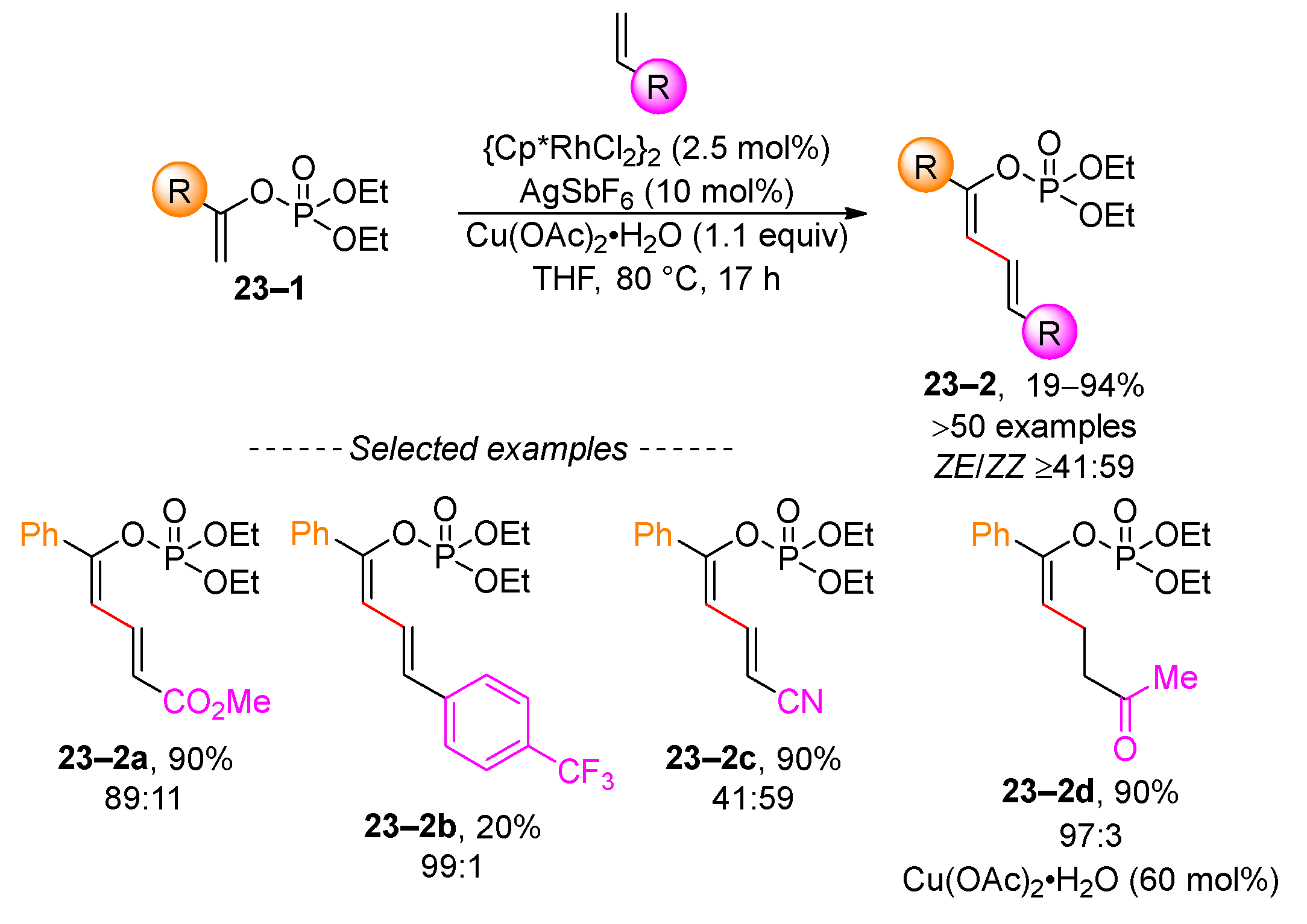
Scheme 24.
Palladium-catalyzed arylation of aryl phosphates via iodonium salts.
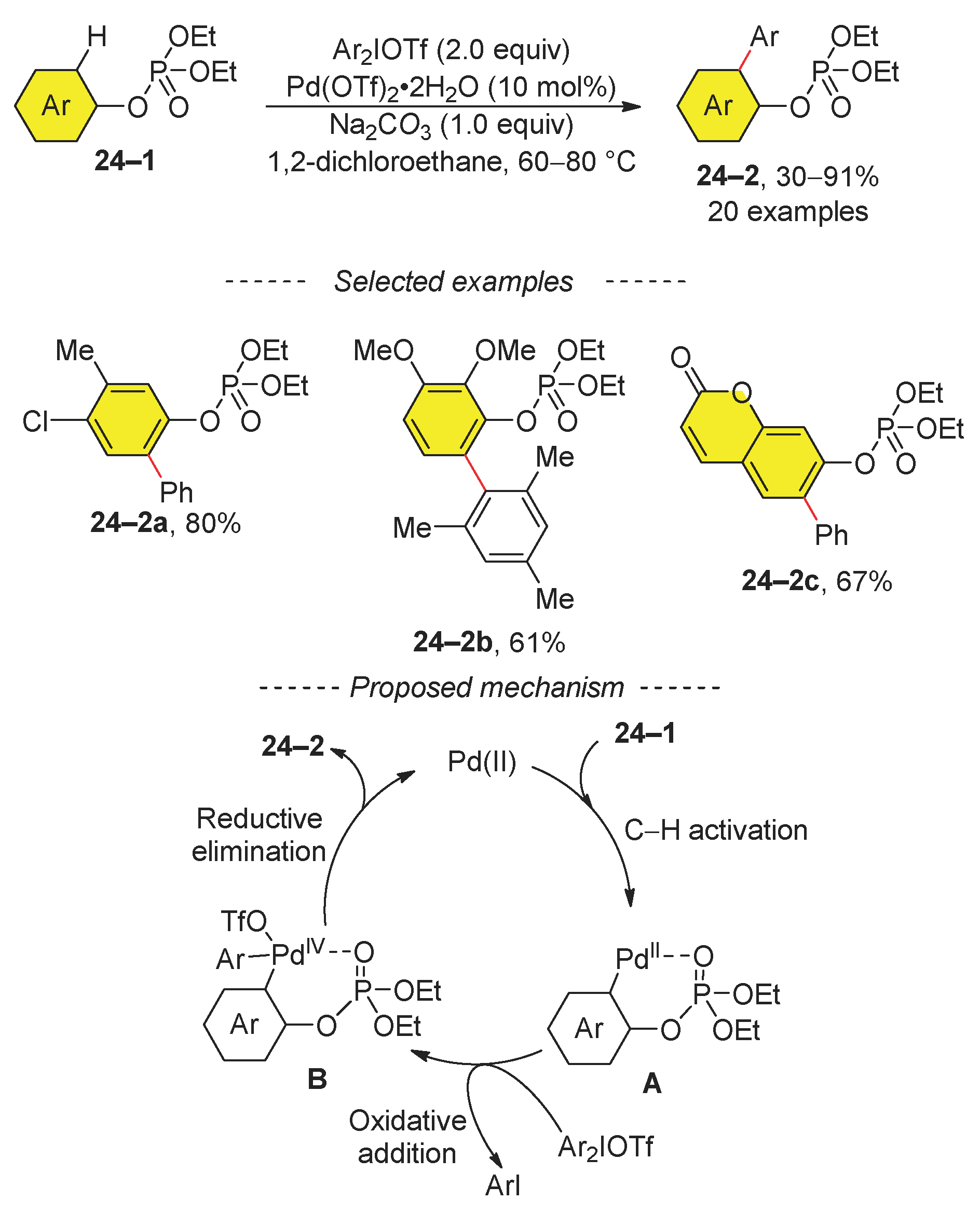
Scheme 25.
Palladium-catalyzed C–H bond activation directed via monophosphoric acid group.
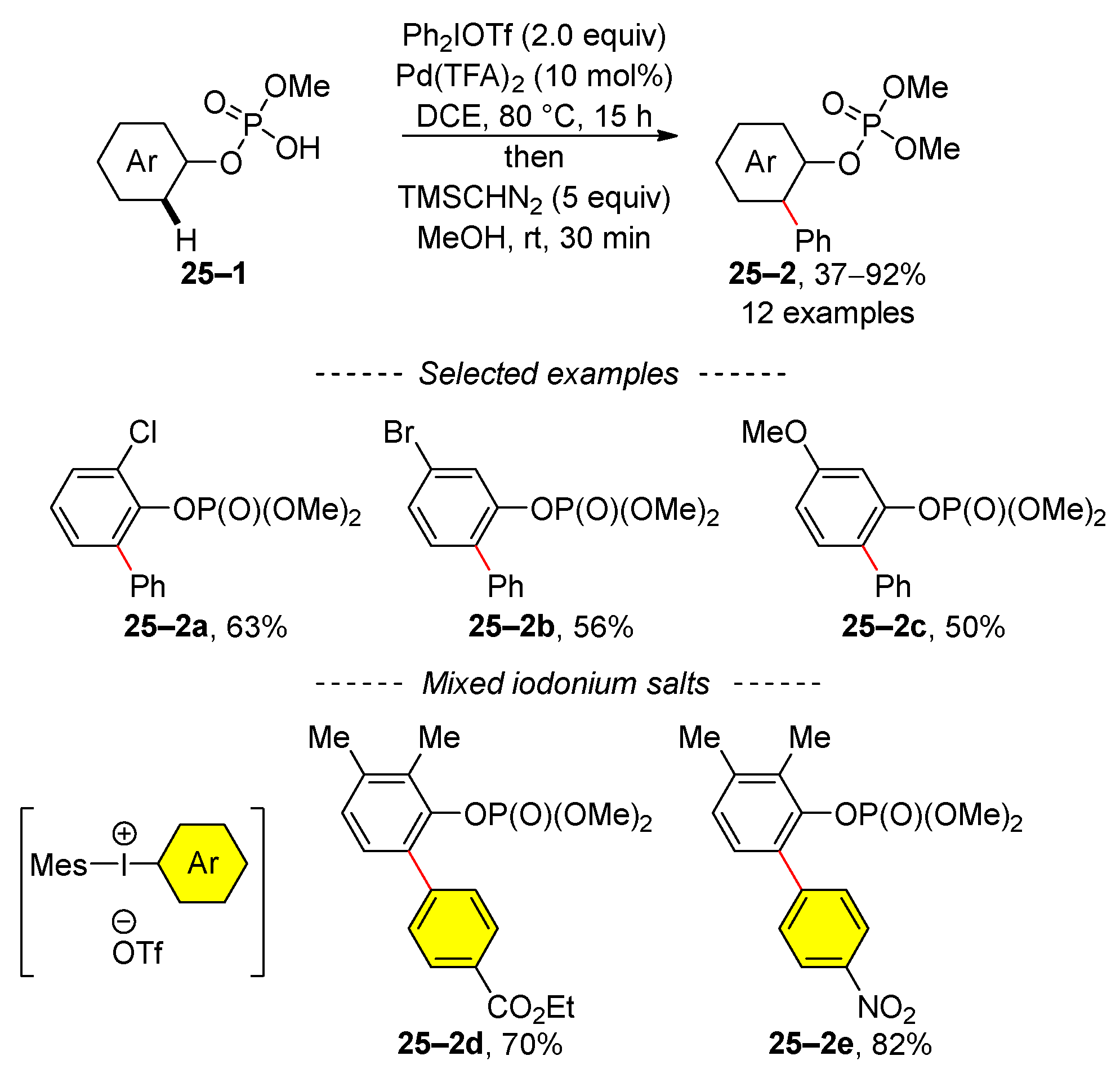
Scheme 26.
Vinyl phosphates as electrophiles in (a) cobalt-catalyzed C−H-activation of substituted indole and (b) intramolecular cyclization of vinyl phosphate.
Scheme 26.
Vinyl phosphates as electrophiles in (a) cobalt-catalyzed C−H-activation of substituted indole and (b) intramolecular cyclization of vinyl phosphate.

Scheme 27.
Cobalt-catalyzed vinylation of benzylic imines.
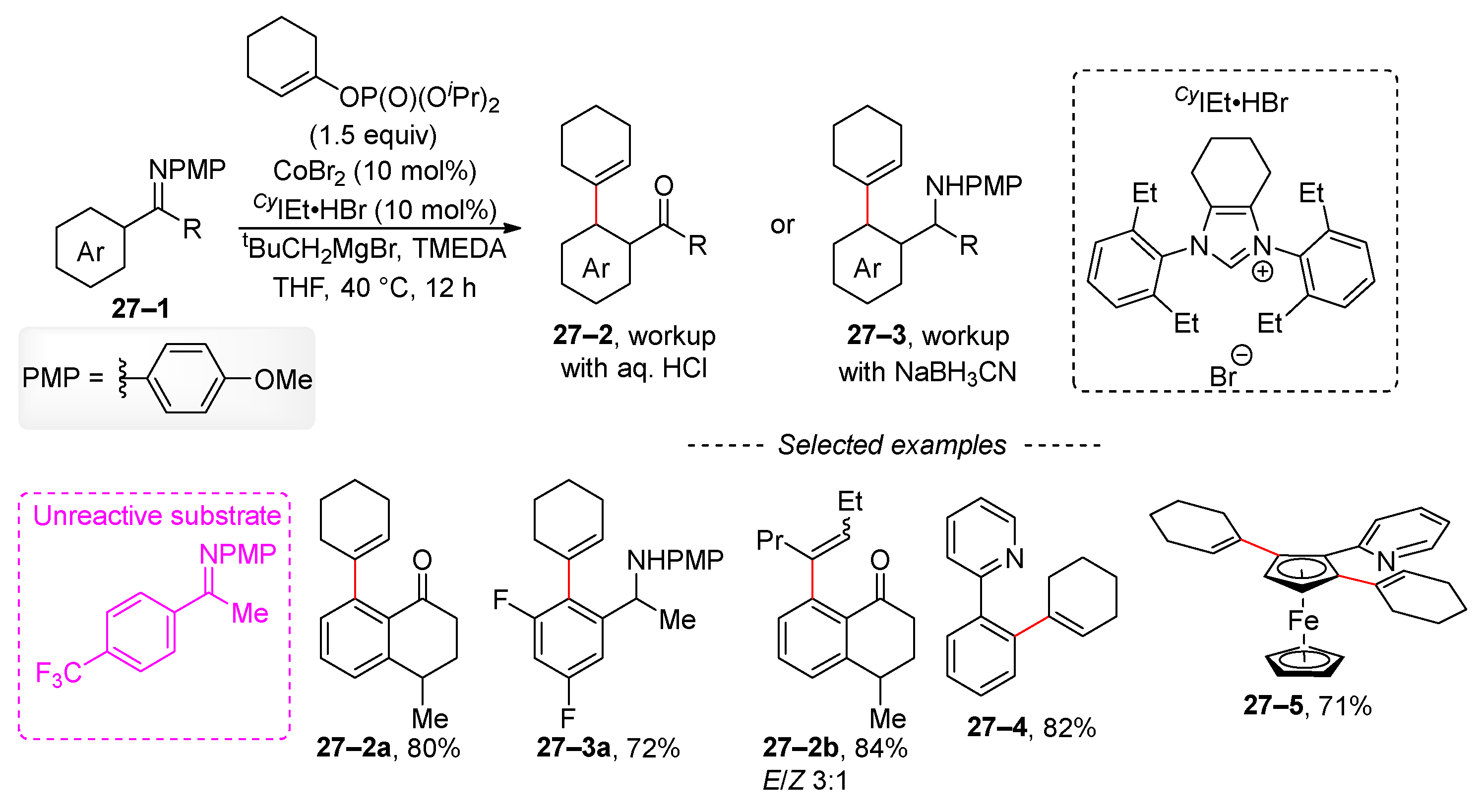
Scheme 28.
Nickel- and palladium-catalyzed synthesis of (a) of amines, (b) substituted 2-naphthylamine 28–5. Transition-metal-free synthesis of carbapenems (c).
Scheme 28.
Nickel- and palladium-catalyzed synthesis of (a) of amines, (b) substituted 2-naphthylamine 28–5. Transition-metal-free synthesis of carbapenems (c).

Scheme 29.
Schematic representation of the [1,2]-phospha-Brook and phospha-Fries rearrangements.
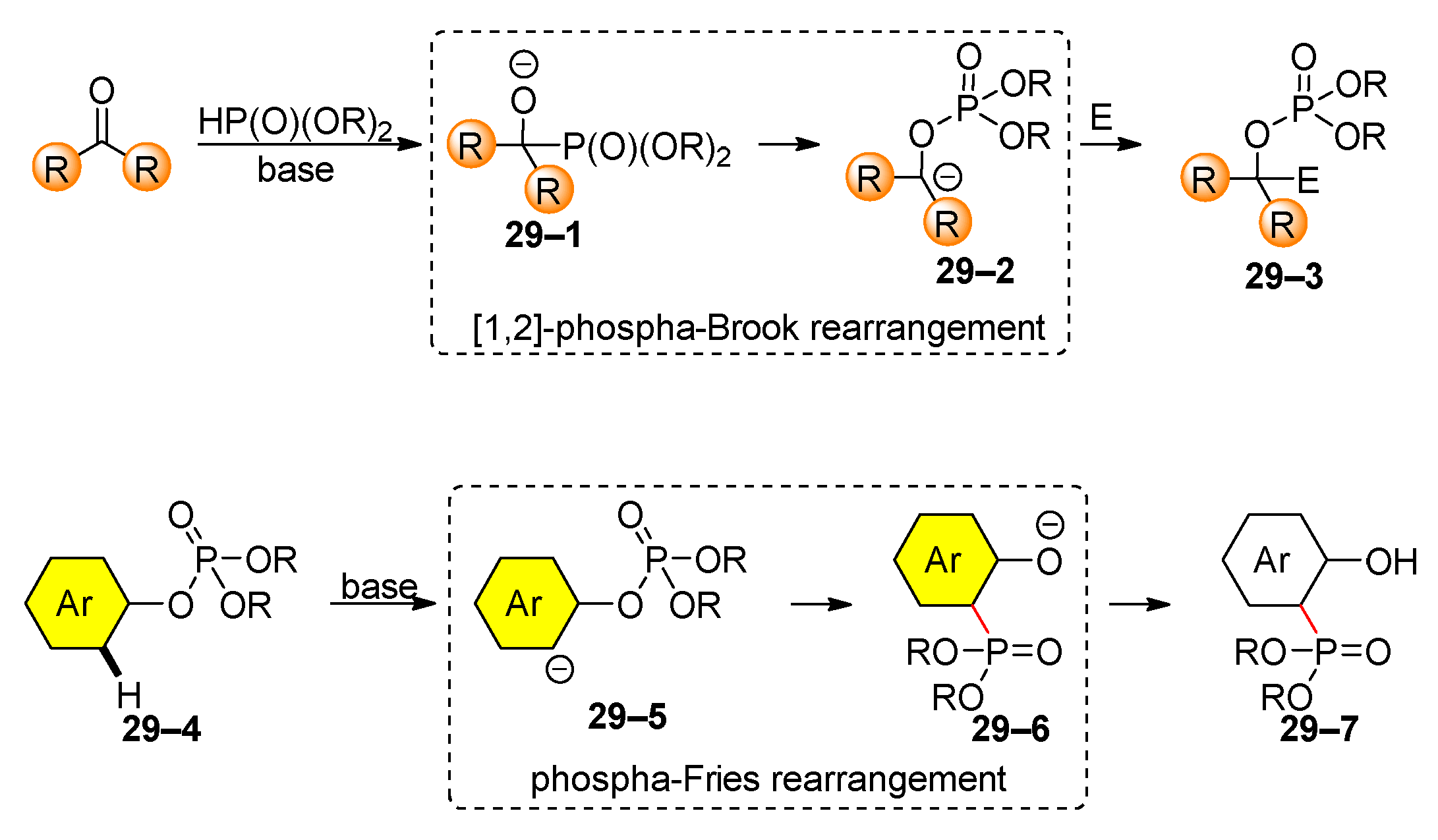
Scheme 30.
Synthesis of monoaryl phosphines by anionic phospha-Fries rearrangement.
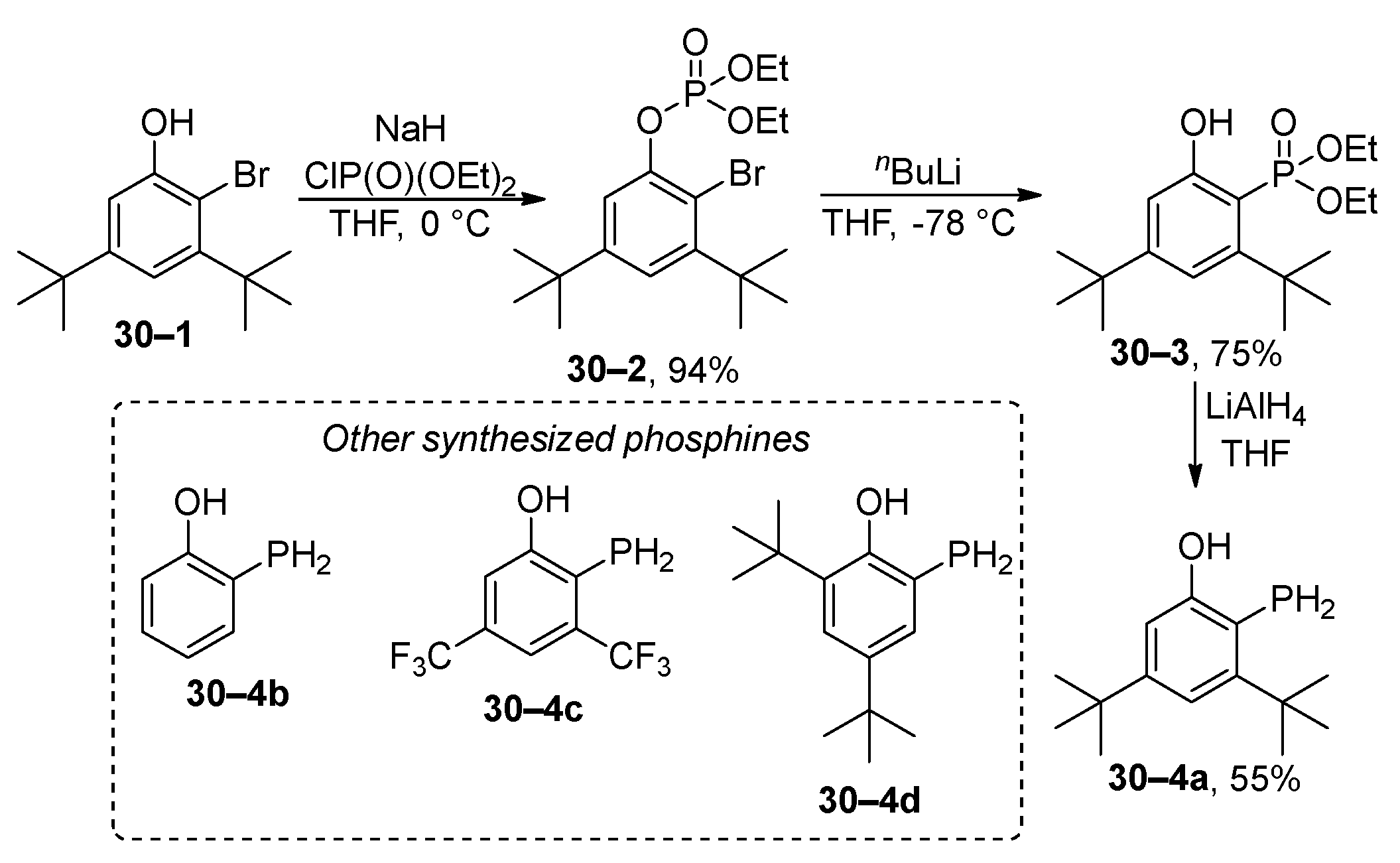
Scheme 31.
Stereoretentive phospha-Fries rearrangement.
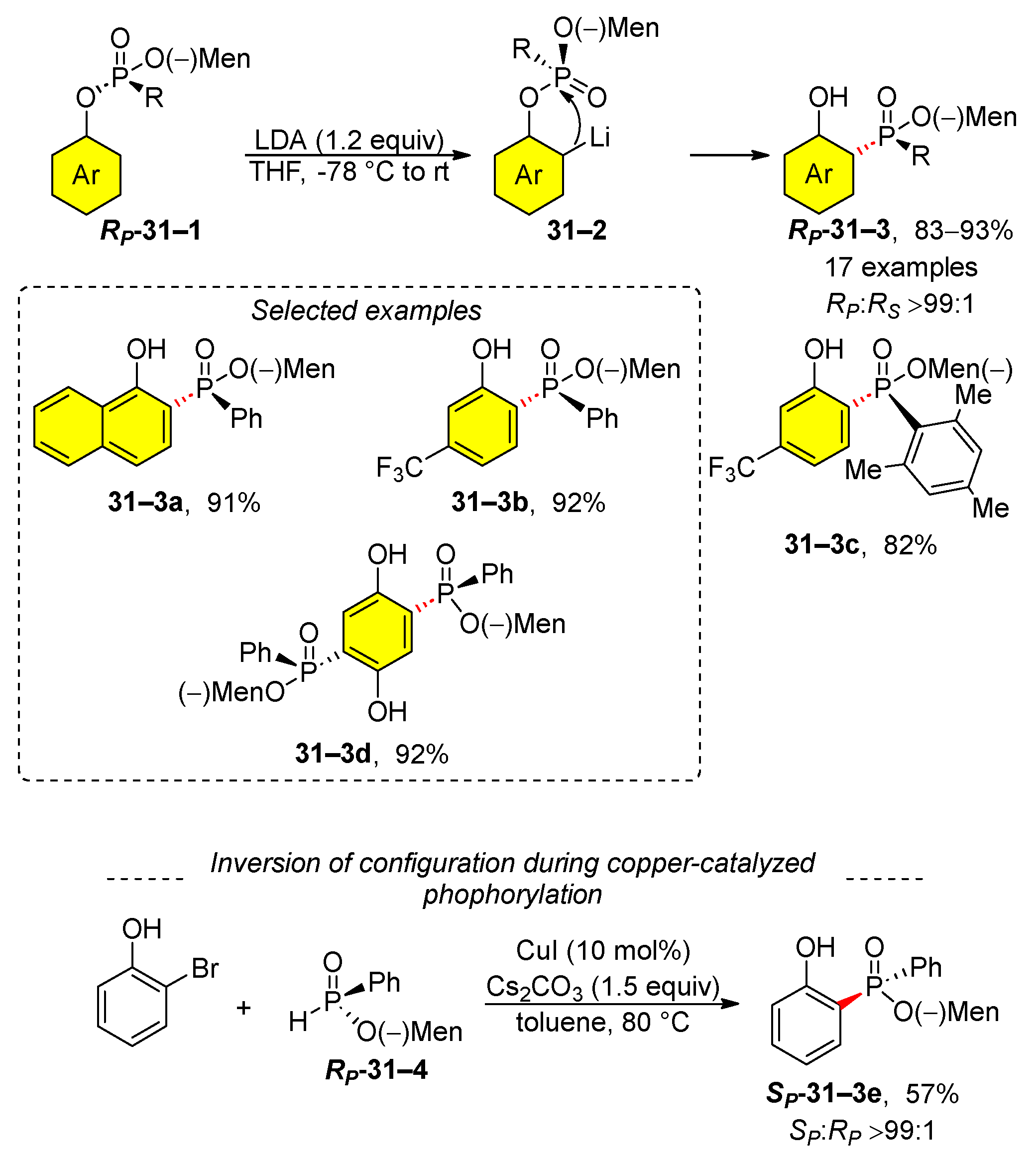
Scheme 32.
Synthesis of ferrocenyl ligand rac-32–5 and its application in the Suzuki cross-coupling reaction.
Scheme 32.
Synthesis of ferrocenyl ligand rac-32–5 and its application in the Suzuki cross-coupling reaction.
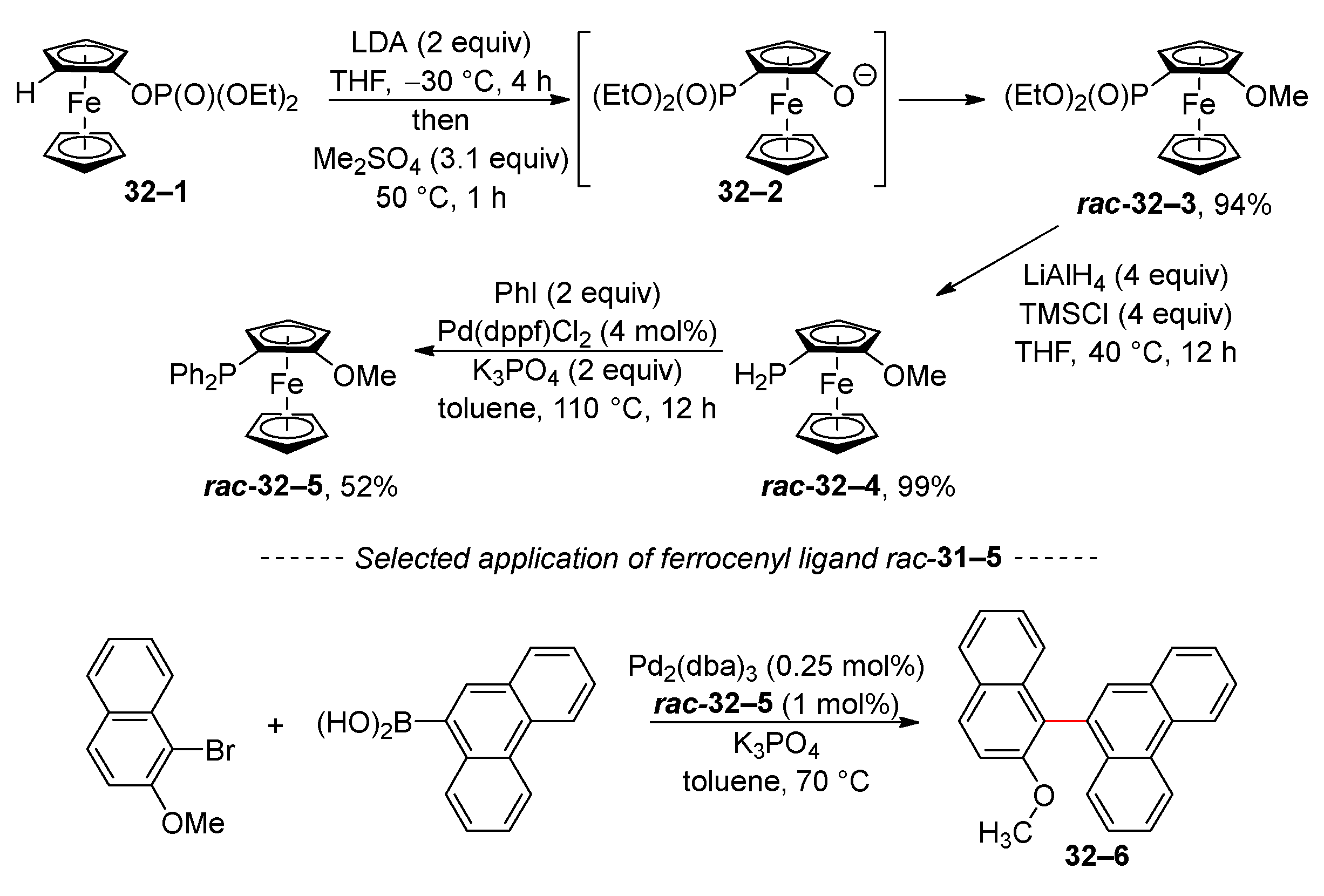
Scheme 33.
Phospha-Fries rearrangement of (a) symmetrical ferrocenyl bisphosphates and (b) unsymmetrical bisphosphates.
Scheme 33.
Phospha-Fries rearrangement of (a) symmetrical ferrocenyl bisphosphates and (b) unsymmetrical bisphosphates.

Scheme 34.
(a) Ireland-Claisen rearrangement of vinyl phosphate en route to clavigerins B and C. (b) Base-mediated phosphate elimination in the synthesis of cyclocitrinols.
Scheme 34.
(a) Ireland-Claisen rearrangement of vinyl phosphate en route to clavigerins B and C. (b) Base-mediated phosphate elimination in the synthesis of cyclocitrinols.

Scheme 35.
Rearrangement of aliphatic diethyl phosphate 34–1 to α-amino acid.

Scheme 36.
Benzyl phosphates as precursors for the preparation of aromatic hydrocarbons.
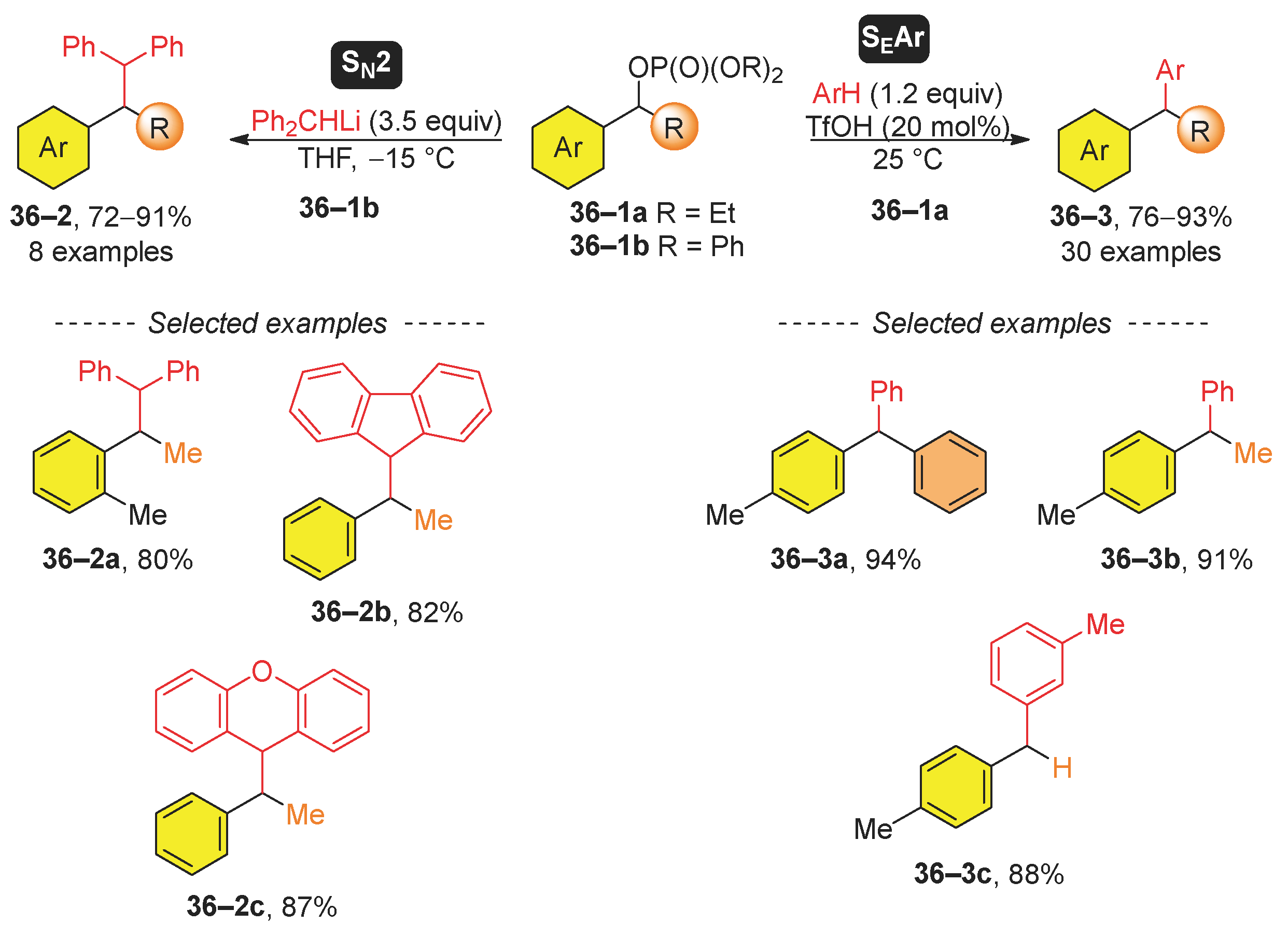
Scheme 37.
Synthesis of cyclopropylphosphines by the cyclopropanation of allyl phosphates.

Scheme 38.
β-Selective glycosylation of glycosyl phosphates through dual activation mode.

Scheme 39.
[4 + 1] cyclization of 2-oxindolyl phophates en route to spirooxindolin derivatives.
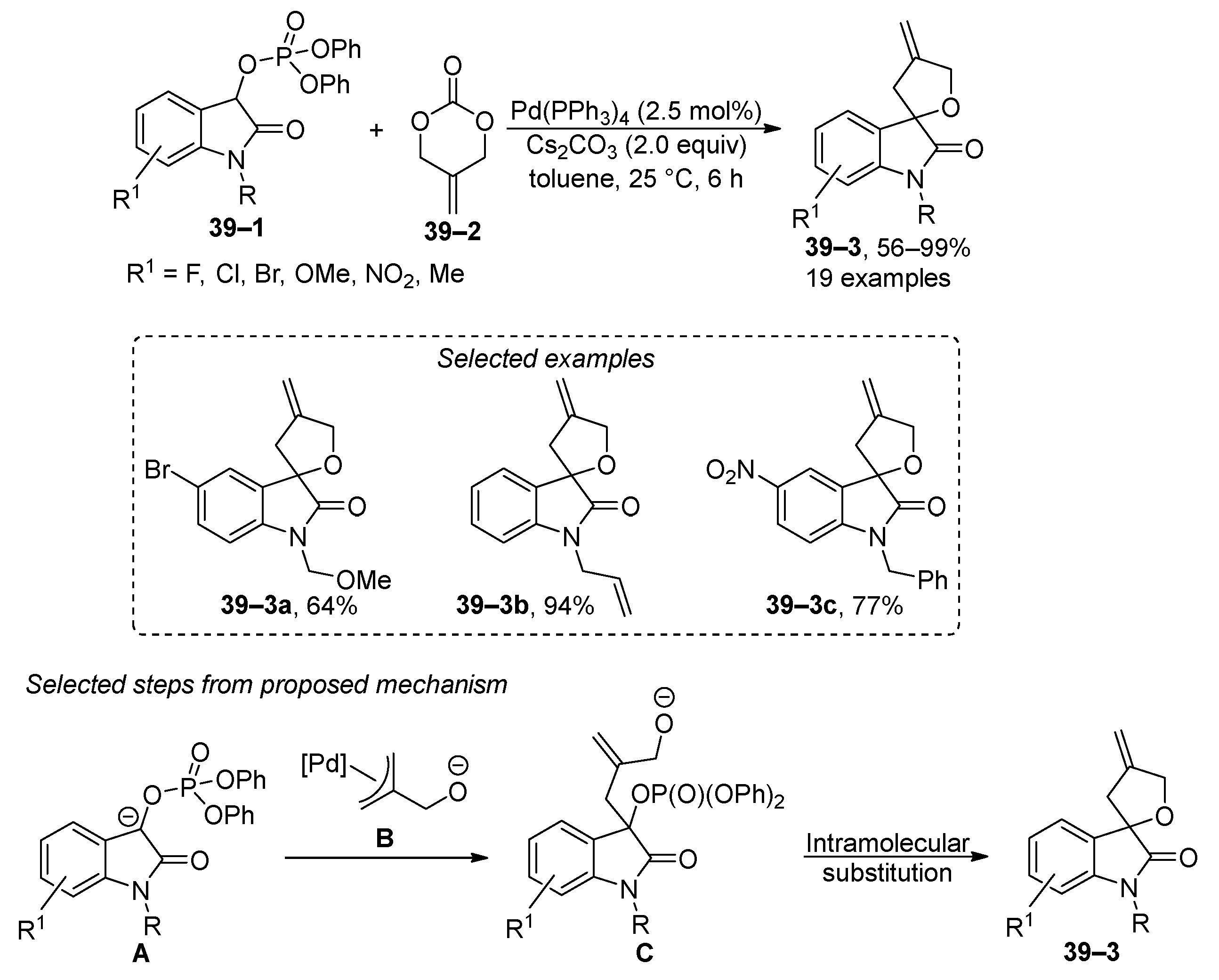
Scheme 40.
One-pot two-step synthesis of aryl 2-oxindoles from organophosphates.

Scheme 41.
Nucleophilic aromatic substitution of pyrimidyl phosphate.
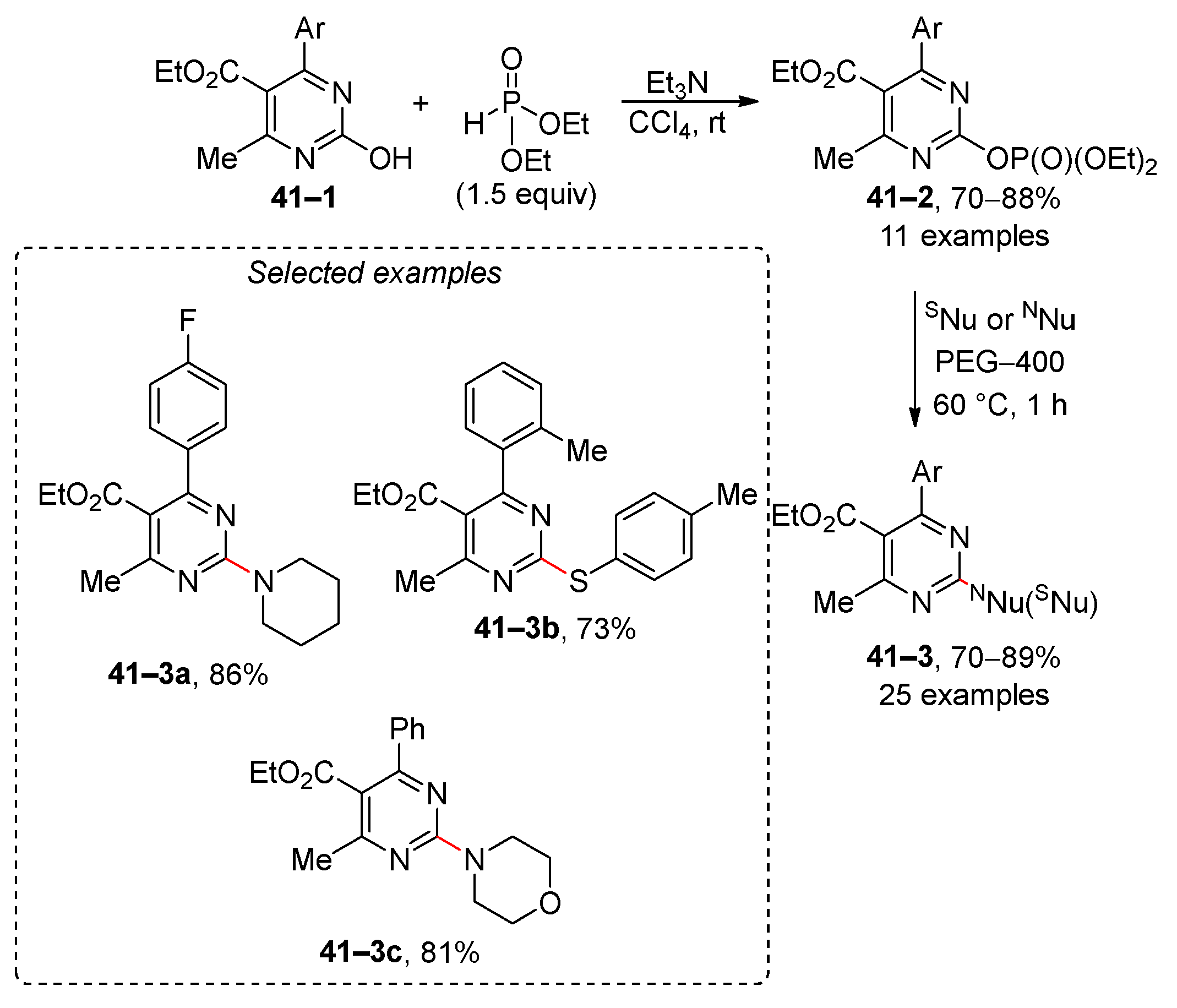
Scheme 42.
Transition-metal-catalyzed allylic substitution of (a) allyl and (b) benzyl phosphates.
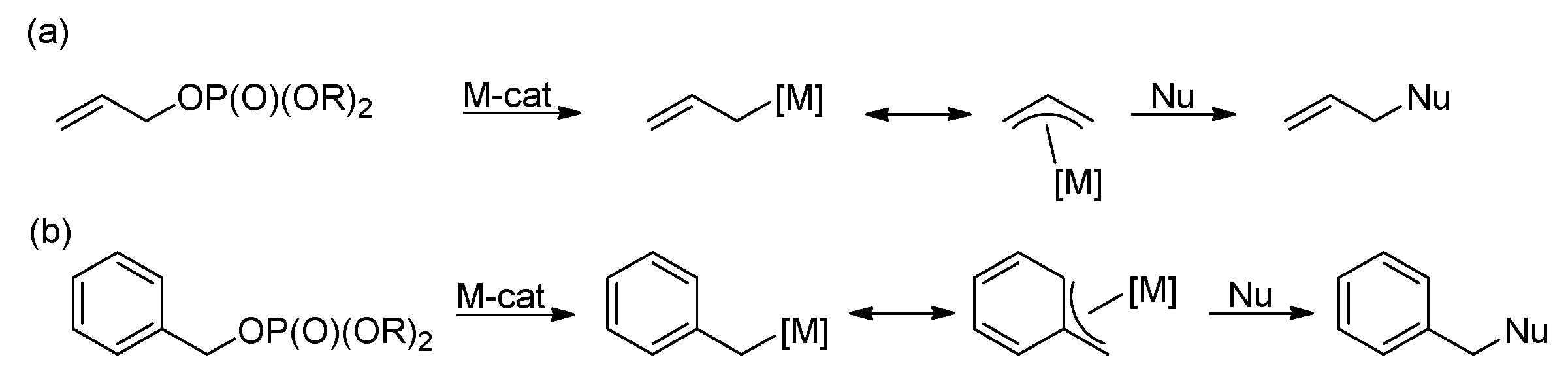
Scheme 43.
Copper-catalyzed reaction of allyl diethyl phosphates with boronic acid pinacol esters.

Scheme 44.
Gem-diborylalkanes in copper/NHC ligand-catalyzed allylic substitution.

Scheme 45.
Palladium/nickel-catalyzed allylation of 2-acylimidazole.
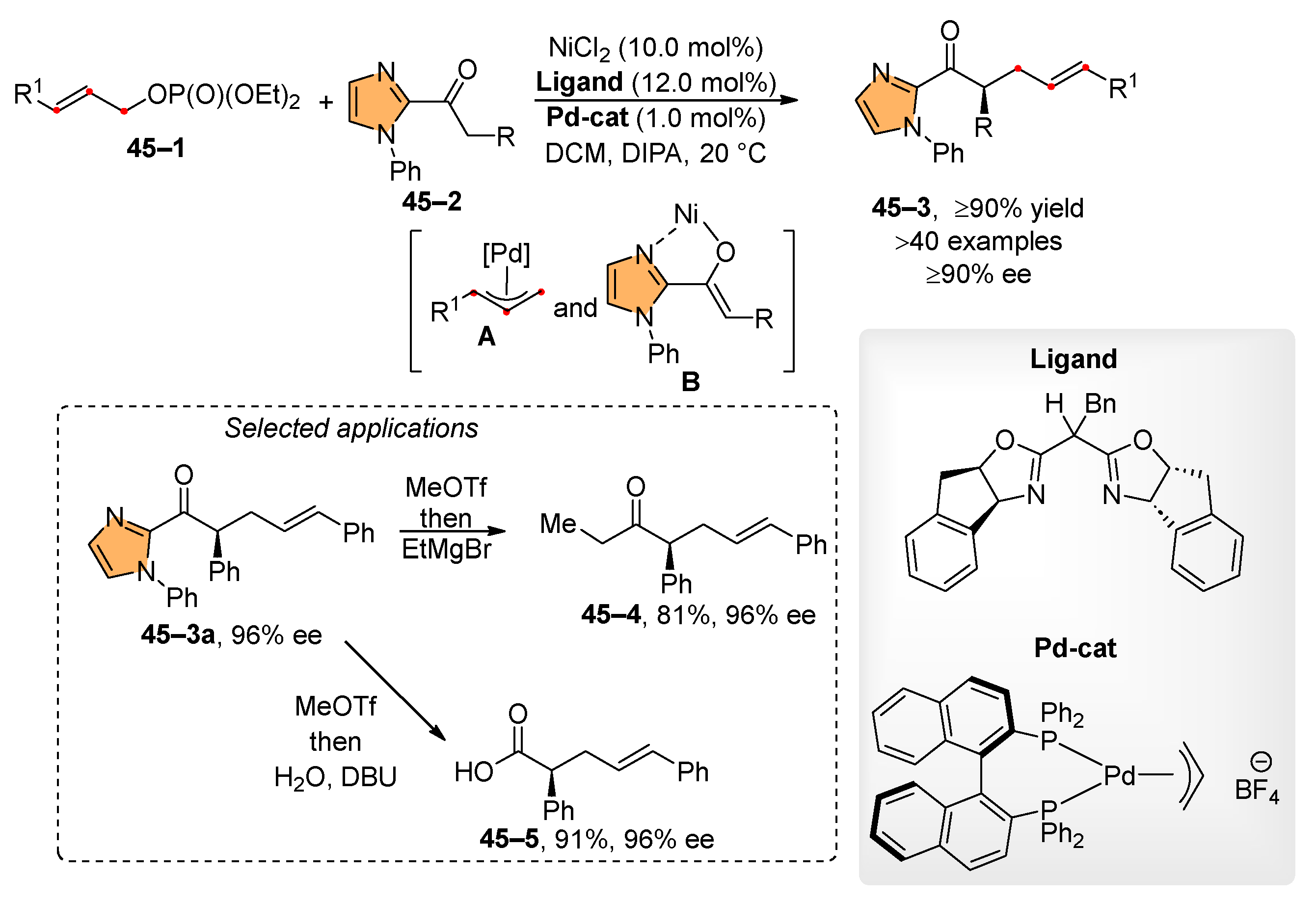
Scheme 46.
Copper-catalyzed desymmetrization of meso-bisphosphates.
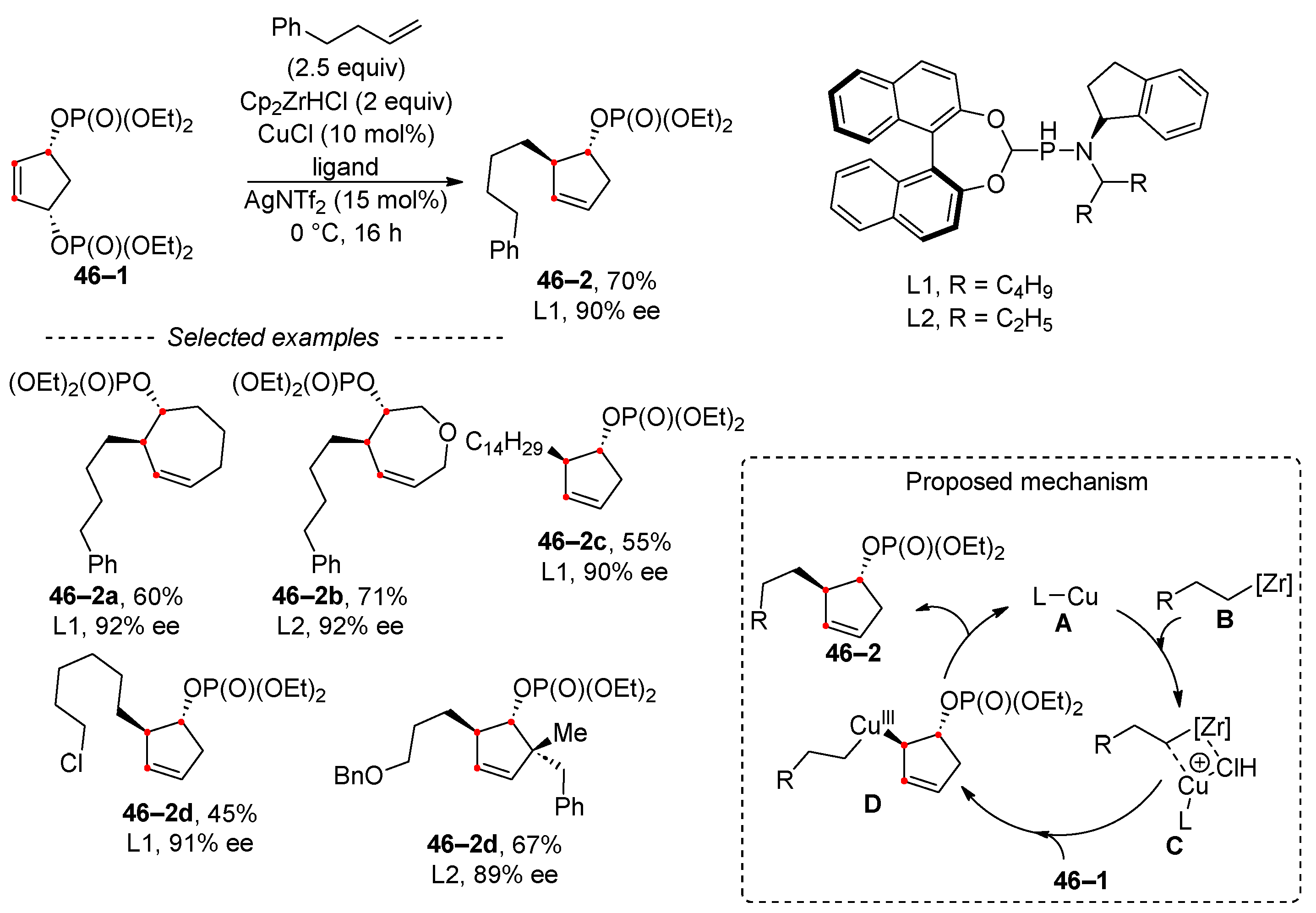
Scheme 47.
Copper–H-catalyzed hydroallylation of vinylarenes.
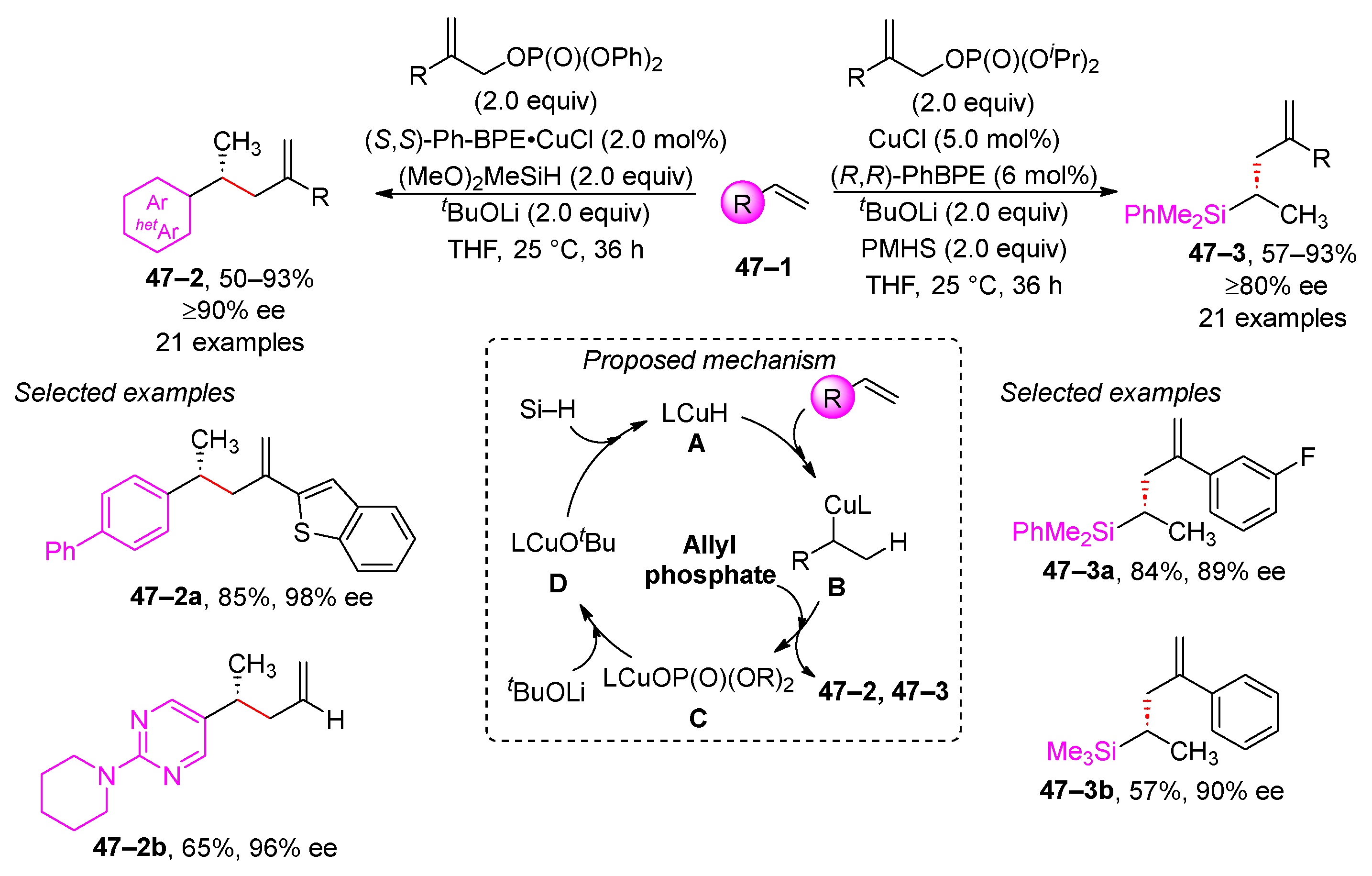
Scheme 48.
Transition-metal-catalyzed isocyanation of (a) allyl, (b,c) benzyl and (d) 2-naphthyl phosphates.
Scheme 48.
Transition-metal-catalyzed isocyanation of (a) allyl, (b,c) benzyl and (d) 2-naphthyl phosphates.

Scheme 49.
Palladium-catalyzed asymmetric cyclization of aromatic ketones.
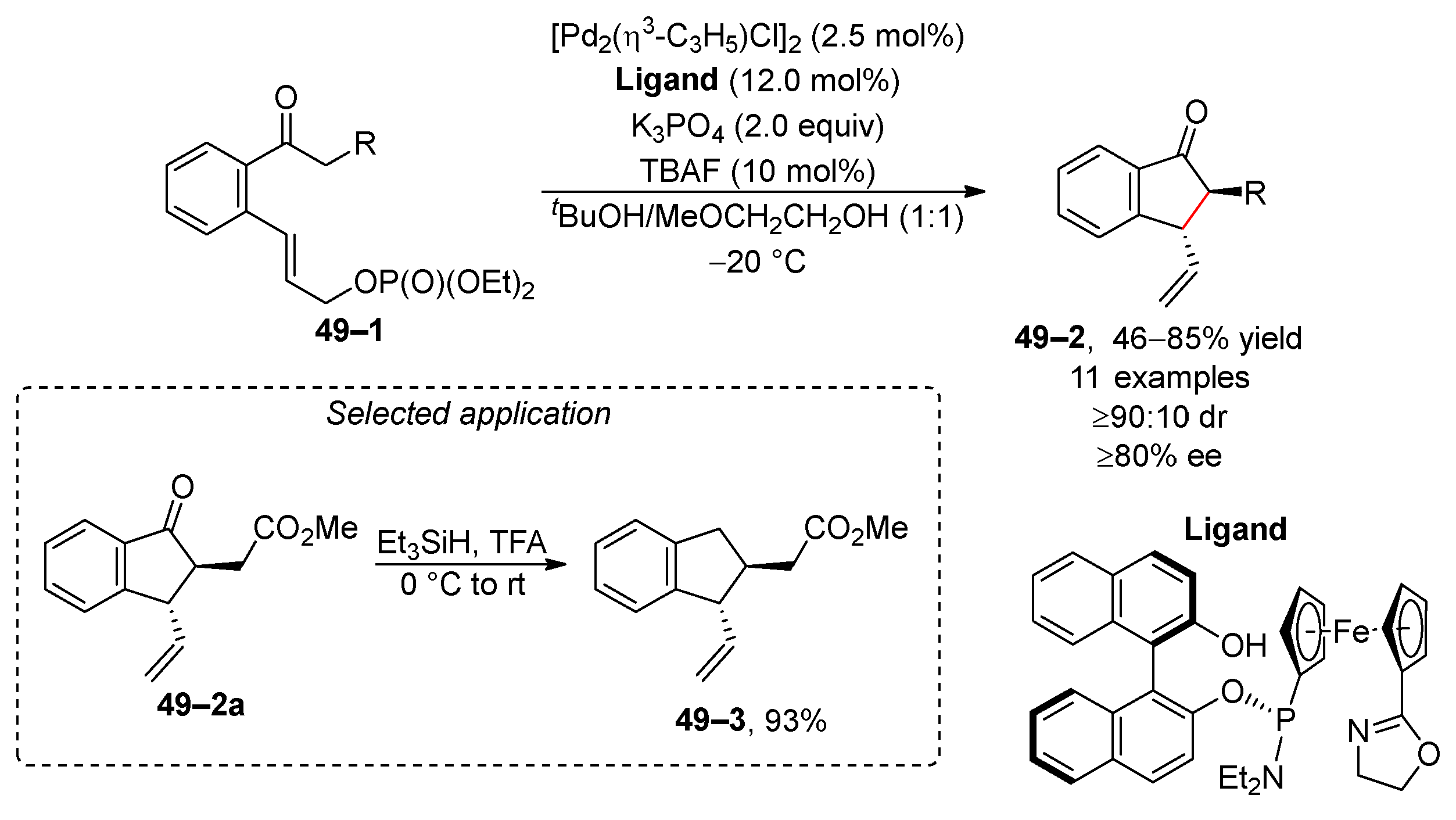
Scheme 50.
Total synthesis of (+)-sporochnol A.
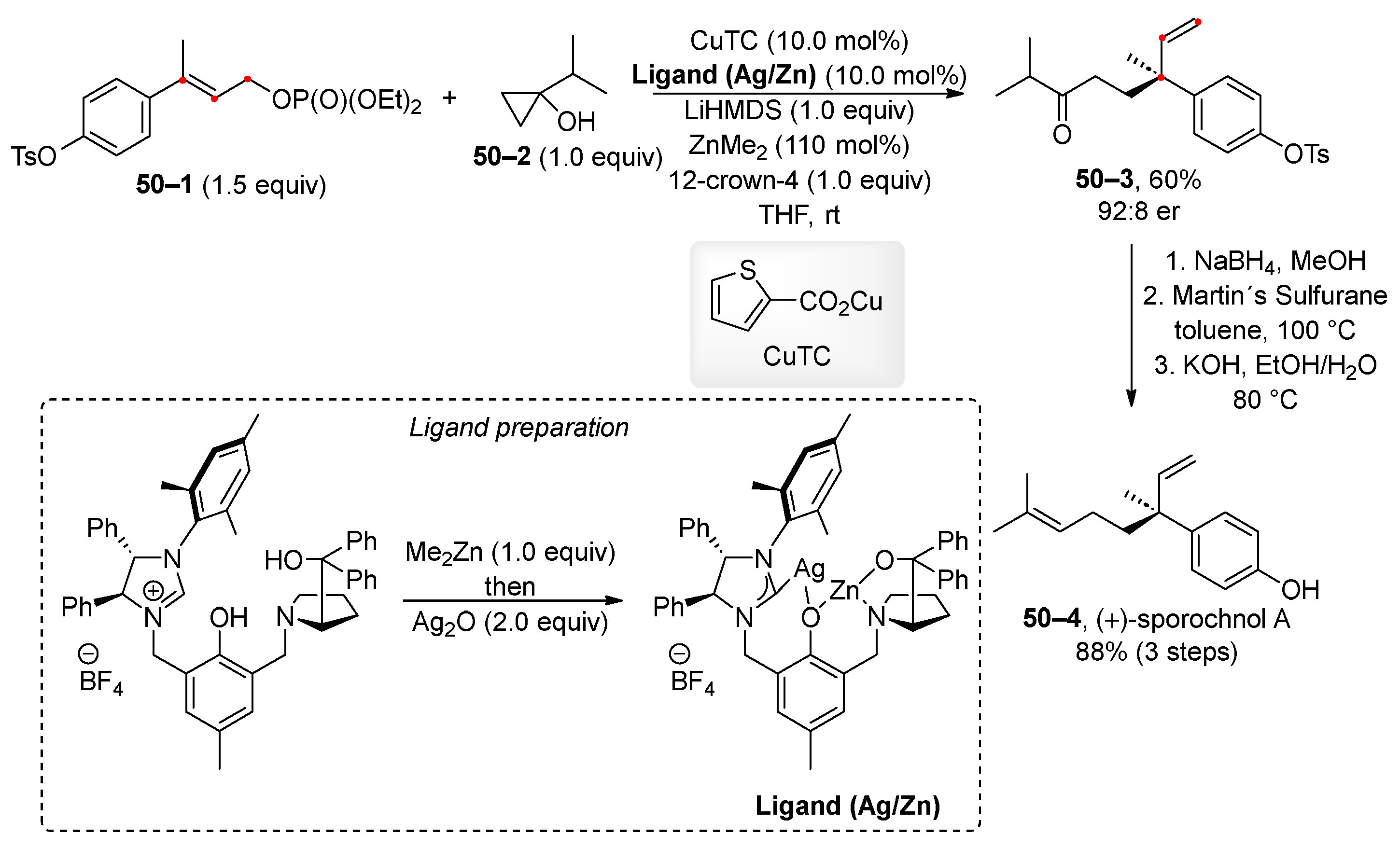
Scheme 51.
Reaction of benzyl phosphates with organometallic reagents.

Scheme 52.
Dearomative allylation of benzyl phosphates.
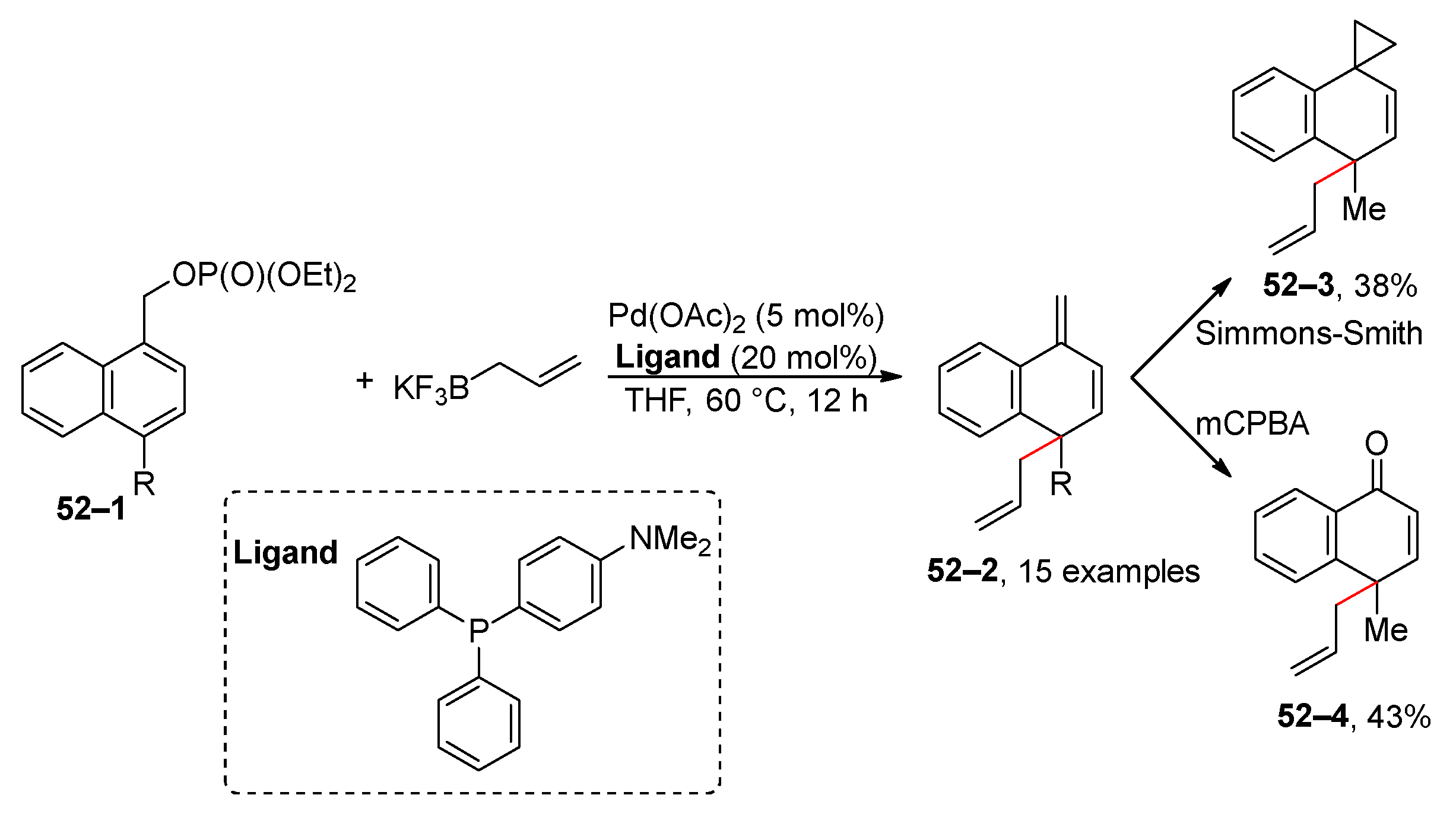
Scheme 53.
Palladium-catalyzed benzylation of carbonyl compounds en route to chiral amino acid derivatives.
Scheme 53.
Palladium-catalyzed benzylation of carbonyl compounds en route to chiral amino acid derivatives.
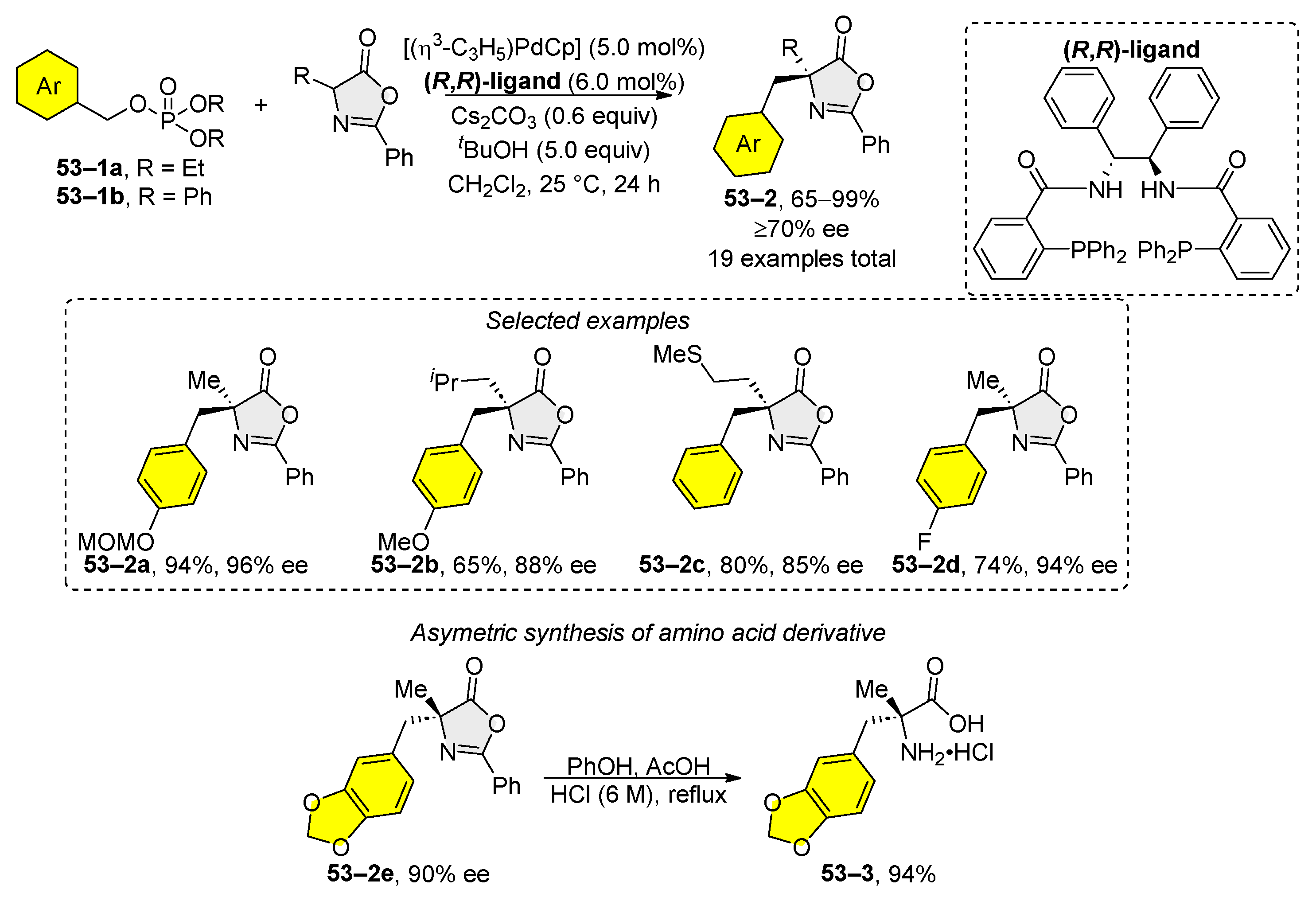
Scheme 54.
Enantioselective synthesis of the thrombin inhibitor DX-9065A by palladium-catalyzed benzylation.
Scheme 54.
Enantioselective synthesis of the thrombin inhibitor DX-9065A by palladium-catalyzed benzylation.

Scheme 55.
Gold-catalyzed enantioselective thioetherification of benzyl phosphate.
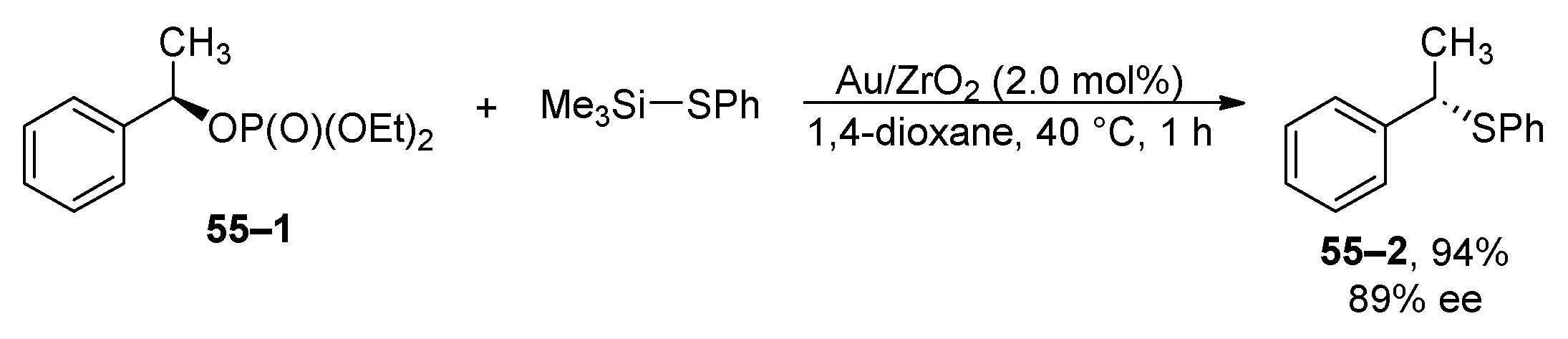
Scheme 56.
Palladium-catalyzed cross-coupling of N-tosylhydrazones with benzyl phosphates.
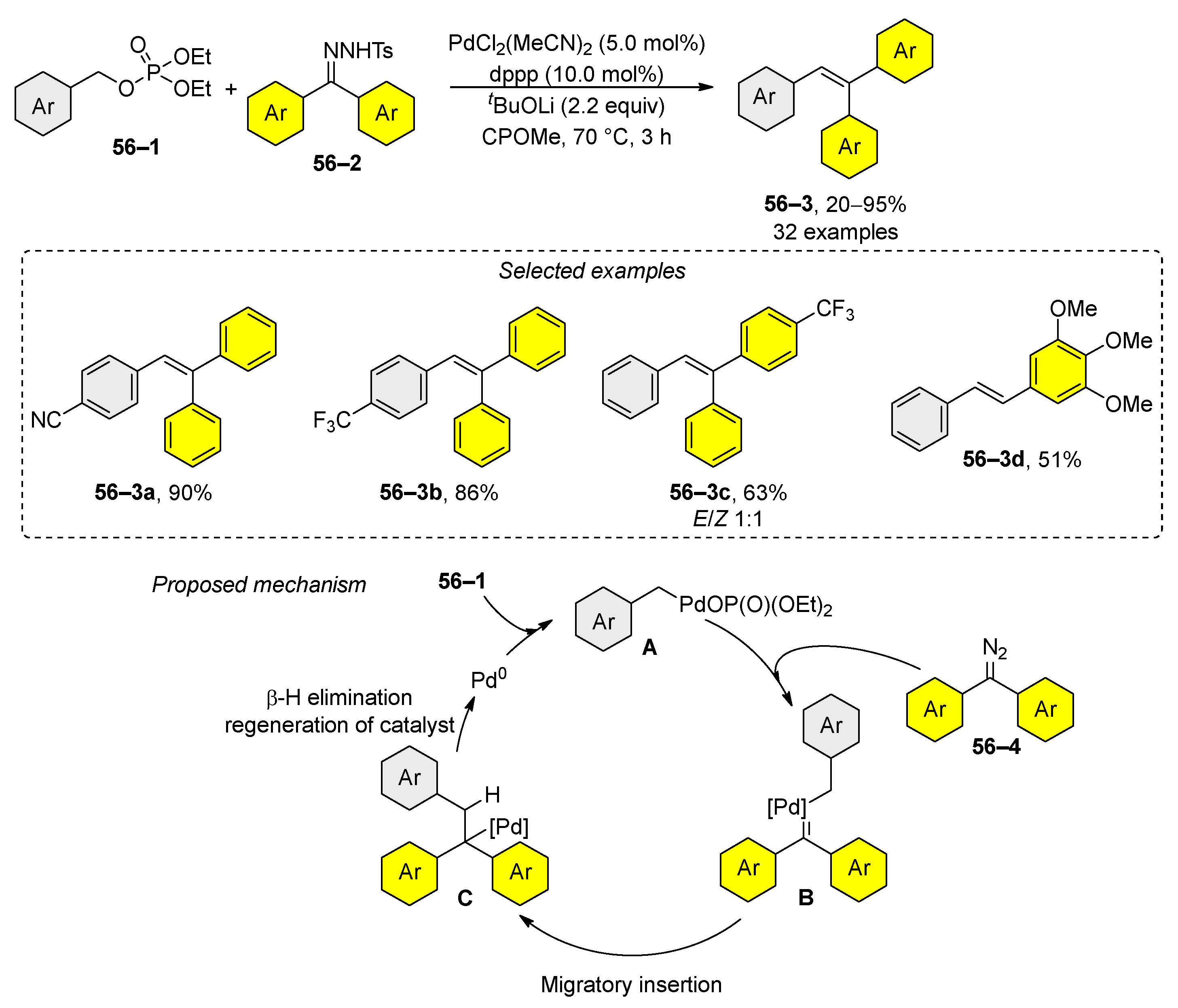
Scheme 57.
Enantioselective hydroxylation of cyclic enol phosphates.
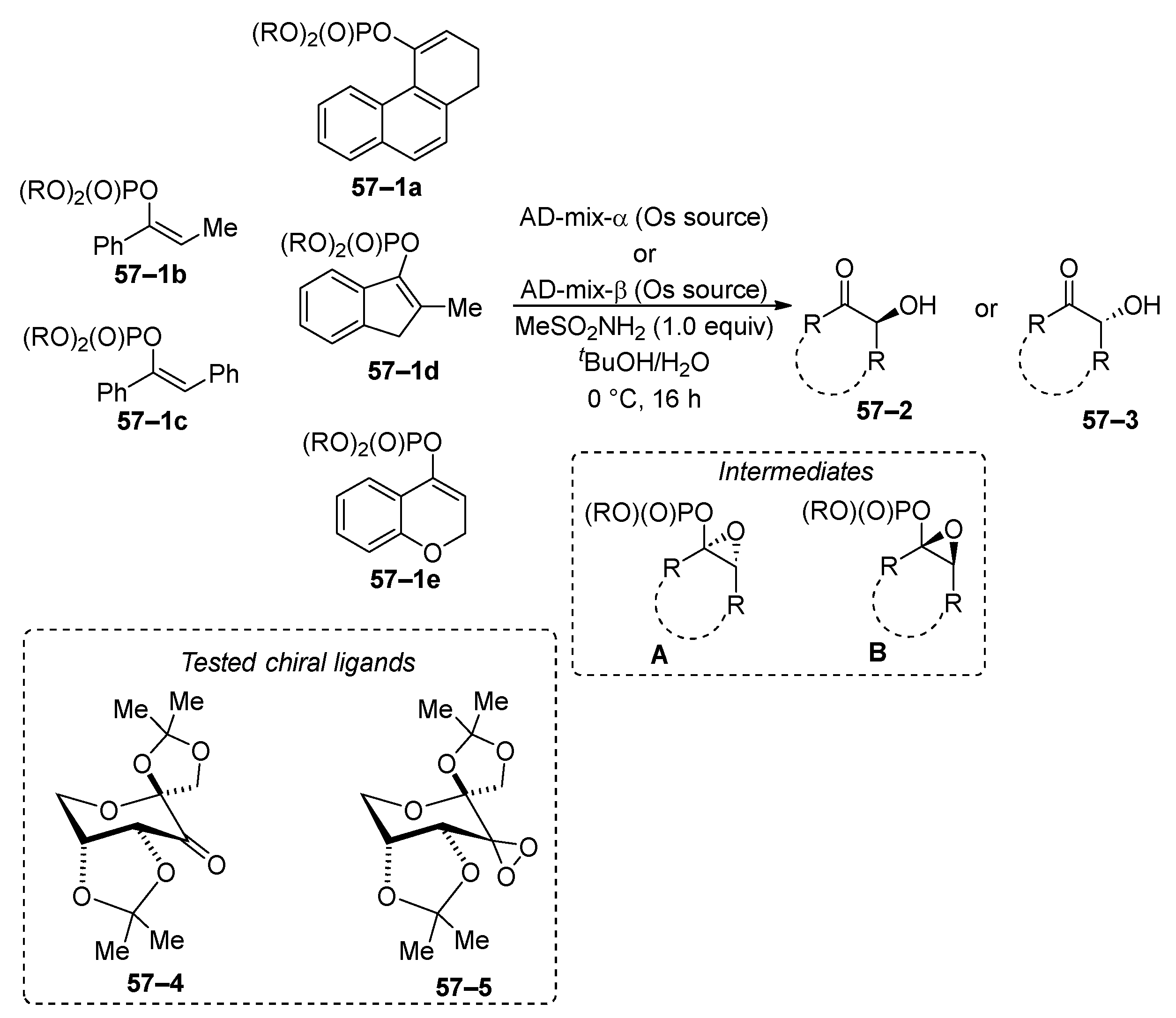
Scheme 58.
Transition-metal-free conversion of vinyl phosphates to β-keto sulfides.
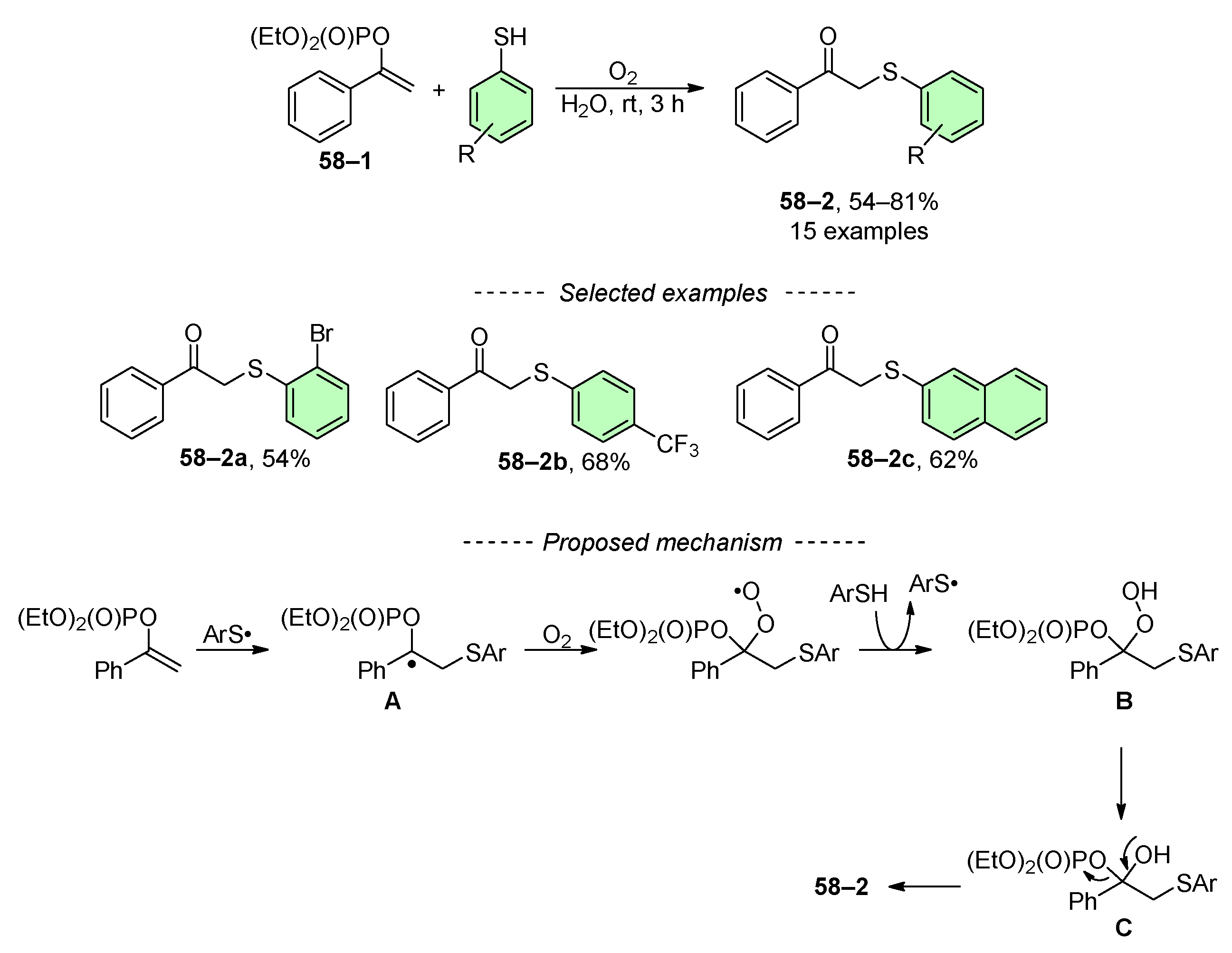
Scheme 59.
Synthesis of 2-oxindoles by reductive dephosphorylation of 2-oxindolyl phosphates.
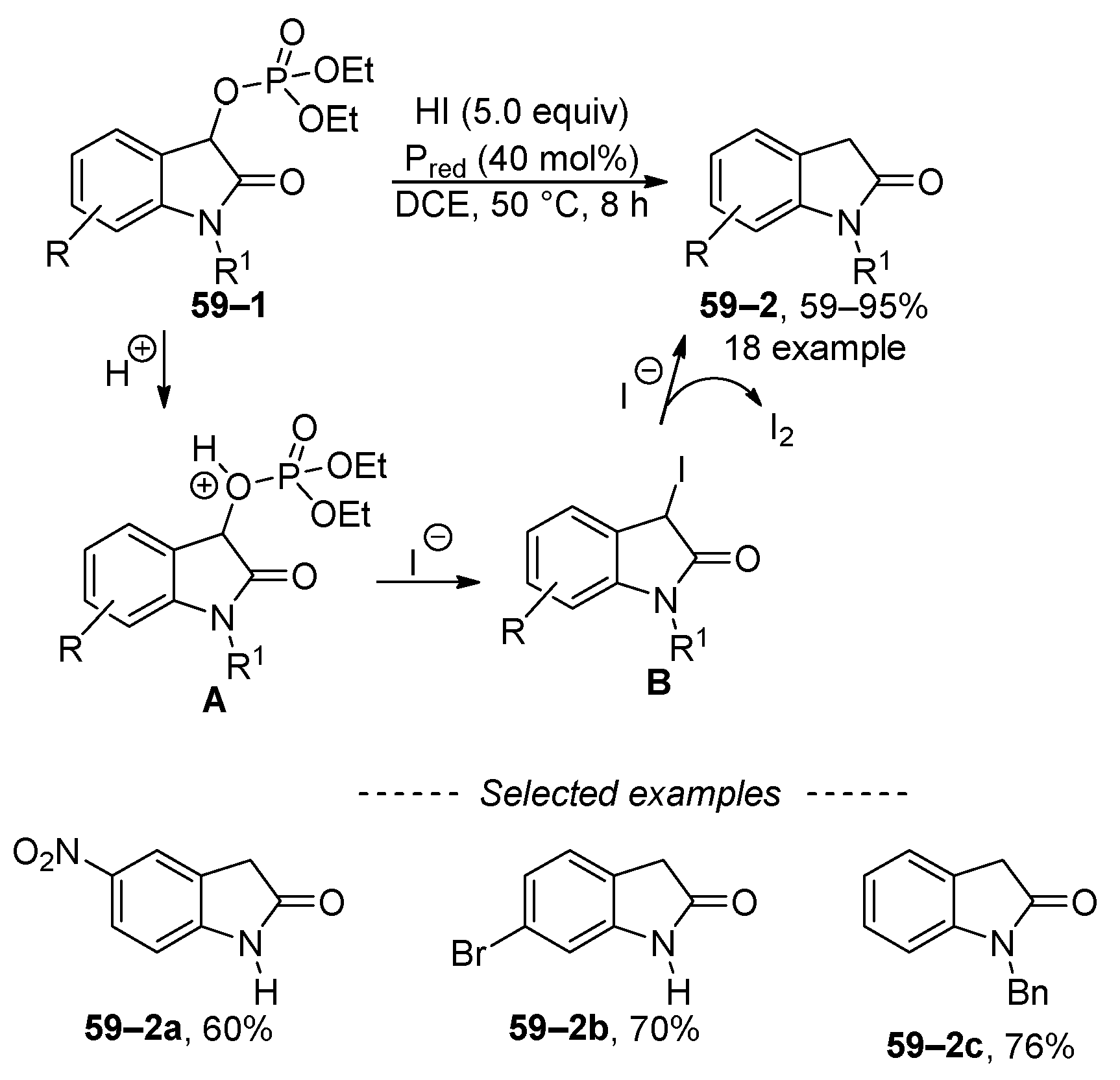
Scheme 60.
Chemoselective reduction of primary alkyl phosphate by lithium triethylborohydride.
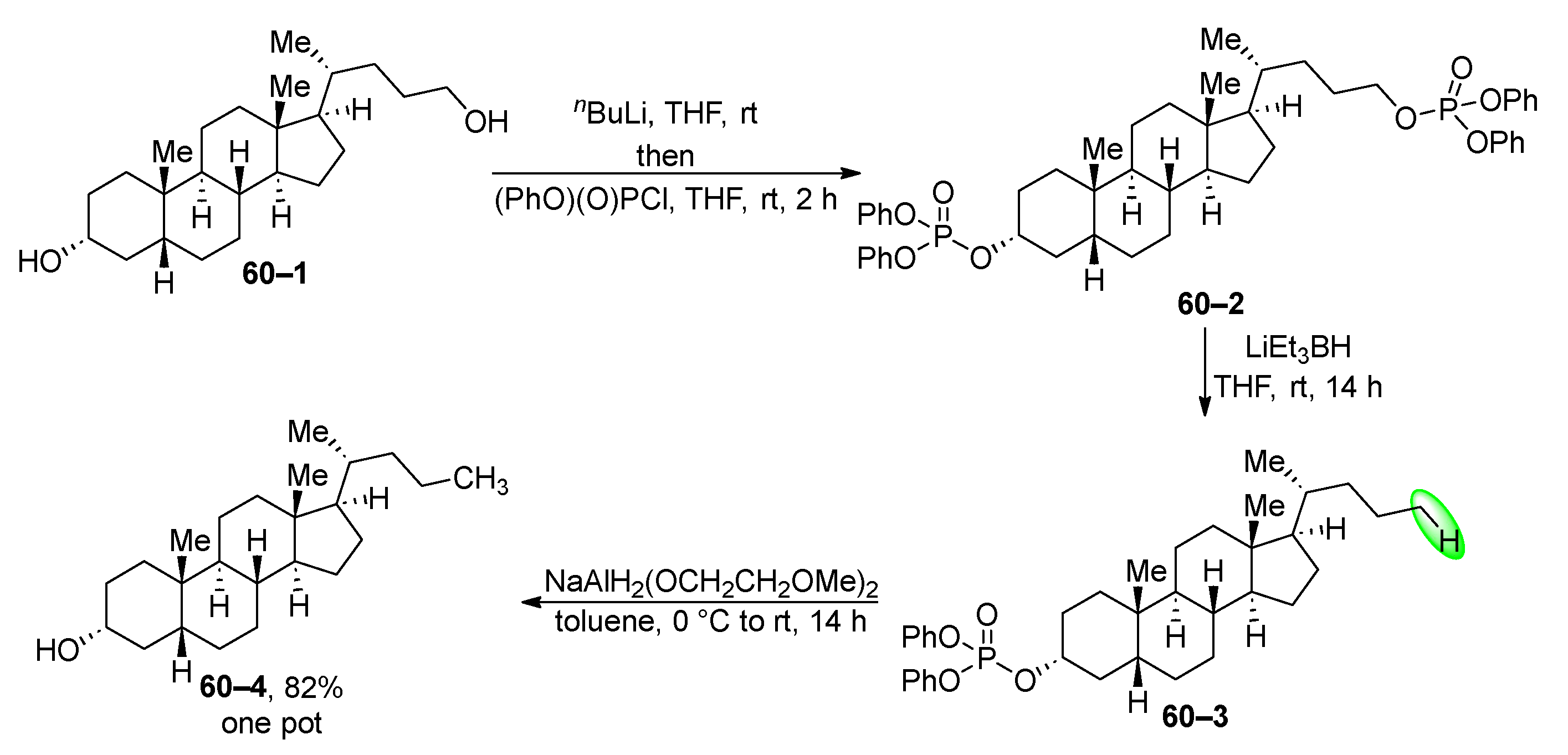
Scheme 61.
Radical deoxygenative coupling of benzyl phosphates with aldehydes and ketones.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Oeser, P.; Tobrman, T. Organophosphates as Versatile Substrates in Organic Synthesis. Molecules 2024, 29, 1593. https://doi.org/10.3390/molecules29071593
AMA Style
Oeser P, Tobrman T. Organophosphates as Versatile Substrates in Organic Synthesis. Molecules. 2024; 29(7):1593. https://doi.org/10.3390/molecules29071593
Chicago/Turabian StyleOeser, Petr, and Tomáš Tobrman. 2024. "Organophosphates as Versatile Substrates in Organic Synthesis" Molecules 29, no. 7: 1593. https://doi.org/10.3390/molecules29071593