

Self-Assembled Matrine-PROTAC Encapsulating Zinc(II) Phthalocyanine with GSH-Depletion-Enhanced ROS Generation for Cancer Therapy
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of LST-1, LST-2, LST-3, LST-4
2.2. LST-1, LST-2, LST-3, and LST-4 Inhibited Tumor Cell Proliferation
2.3. LST-4 Regulated the Levels of p-Akt, p-Erk
2.4. Photophysical Properties of LST-4 and Consumption of Intracellular GSH by LST-4
2.5. Characterization of LST-4 NPs and LST-4@ZnPc NPs
2.6. In Vitro Laser-Triggered Oxidation Regulation by LST-4@ZnPc NPs
2.7. Cellular Uptake of LST-4 NPs and LST-4@ZnPc NPs
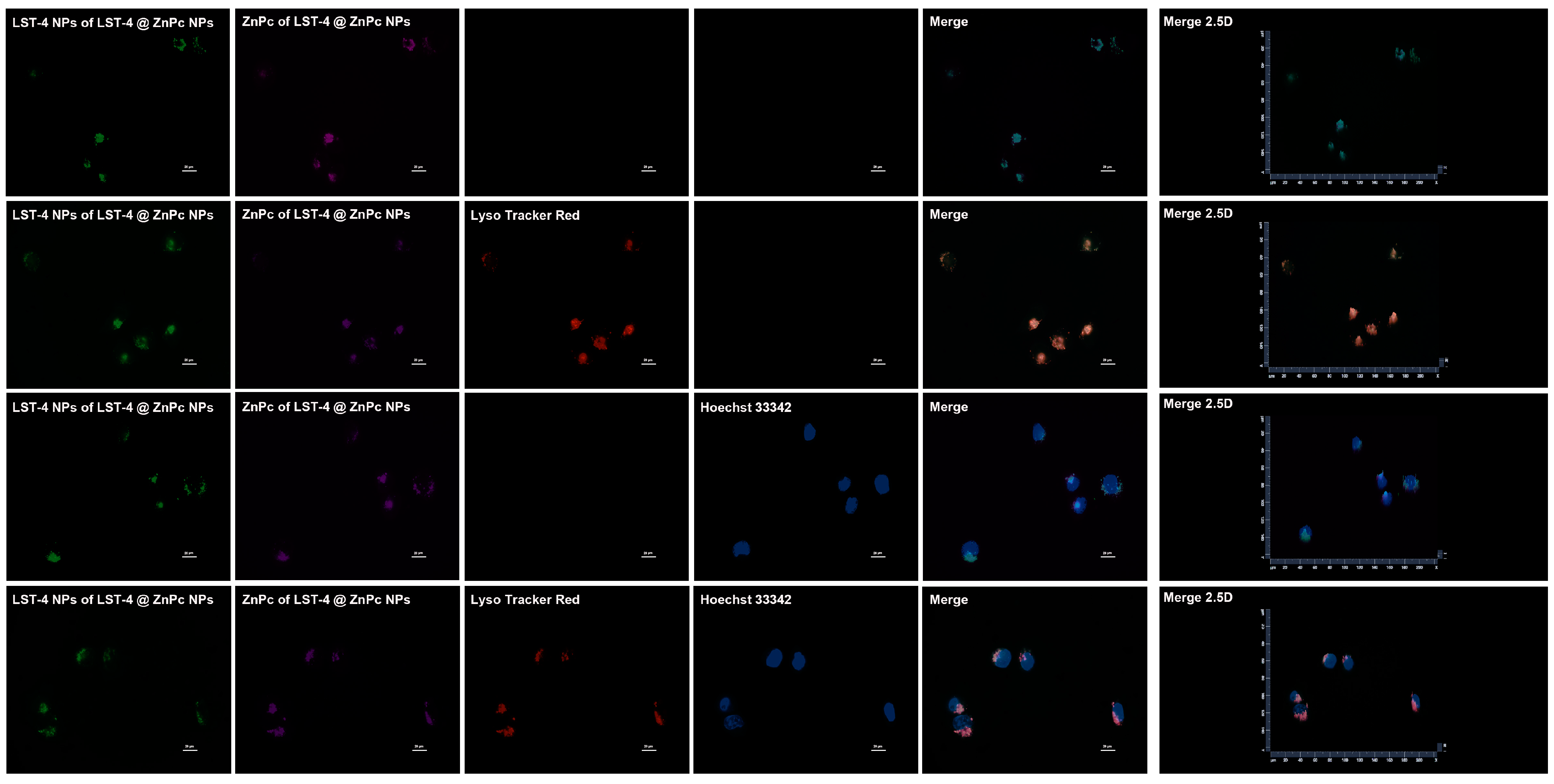
2.8. Subcellular Localization of LST-4 NPs and LST-4@ZnPc NPs
2.9. Intracellular ROS Generation via LST-4@ZnPc NPs and Intracellular GSH Depletion by LST-4 NPs
2.10. In Vitro Inhibition of HepG 2 Cells by LST-4 NPs and LST-4@ZnPc NPs
3. Materials and Methods
3.1. Materials and Agents
3.2. Apparatus
3.3. Synthesis
3.4. UV–Vis Absorption Spectra of LST-4 NPs
3.5. FL Spectra of LST-4 NPs
3.6. Preparation of LST-4 NPs
3.7. Preparation of LST-4@ZnPc NPs
3.8. Characterization
3.9. Loading Efficiency (LE) and In Vitro Release Capacity of LST-4@ZnPc NPs
3.10. In Vitro 1O2 Detection of LST-4@ZnPc NPs
3.11. In Vitro Stability Assay
3.12. Cell Culture
3.13. In Vitro Cytotoxicity Assay
3.14. Western Blot Analysis
3.15. Detection of Intracellular GSH
3.16. In Vitro Cellular Uptake
3.17. Detection of Intracellular ROS
3.18. Subcellular Localization
3.19. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TCM | Traditional Chinese medicine |
| PROTAC | Proteolysis-targeting chimeras |
| GSH | Glutathione |
| EPR | Enhanced permeability and retention |
| ROS | Reactive oxygen species |
| PDT | Photodynamic therapy |
| ZnPc | Zinc phthalocyanine |
| POI | Protein of interest |
| TME | Tumor microenvironment |
| PS | Photosensitizer |
| PEG | Polyethylene glycol |
| PBS | Phosphate buffer solution |
| FBS | Fetal bovine serum |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DPBF | 1,3-diphenylisobenzofuran |
| CCK-8 | Cell counting kit 8 |
| THF | Tetrahydrofuran |
| DMSO | Dimethyl sulfoxide |
| TBS | Tris-buffered saline |
| EDCI | 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride |
| HOBT | 1-hydroxybenzotriazole |
| DIPEA | N,N-diisopropylethylamine |
| LE | Loading efficiency |
| FL | Fluorescence |
References
- Ren, G.; Ding, G.; Zhang, H.; Wang, H.; Jin, Z.; Yang, G.; Han, Y.; Zhang, X.; Li, G.; Li, W. Antiviral activity of sophoridine against enterovirus 71 in vitro. J. Ethnopharmacol. 2019, 236, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Zhou, Q.; Li, X.; Zhou, X. Total alkaloids of Sophora alopecuroides and matrine inhibit auto-inducer 2 in the biofilms of Staphylococcus epidermidis. Microb. Pathog. 2019, 136, 103698. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Tang, X.; Zhuang, X.; Hu, Z.; He, K.; Wu, Y.; Dai, T. Matrine induces apoptosis via targeting CCR7 and enhances the effect of anticancer drugs in non-small cell lung cancer in vitro. Innate. Immun. 2018, 24, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Luo, D.; Yang, L.; Cheng, W.; He, L.J.; Kuang, G.K.; Li, M.M.; Li, Y.L.; Wang, G.C. Matrine-Type Alkaloids from the Roots of Sophora flavescens and Their Antiviral Activities against the Hepatitis B Virus. J. Nat. Prod. 2018, 81, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Chen, N.H.; Wang, W.Z.; Zhang, J.H.; Li, C.J.; Zhuo, X.F.; Tu, Z.C.; Wu, Z.U.; Fan, C.L.; Zhang, H.P. Structurally diverse matrine-based alkaloids with anti-inflammatory effects from sophora alopecuroides. Chin. J. Chem. 2021, 39, 3339–3346. [Google Scholar] [CrossRef]
- Cho, C.H.; Chuang, C.Y.; Chen, C.F. Study of the antipyretic activity of matrine. A lupin alkaloid isolated from Sophora subprostrata. Planta. Med. 1986, 52, 343–345. [Google Scholar] [CrossRef]
- Wang, H.Y.; Li, Y.X.; Dun, L.L.; Xu, T.T.; Hao, Y.J.; Liu, H.Y.; Ma, L.; Jiang, Y.X.; Wang, Y.R.; Yu, J.Q. Antinociceptive effects of matrine on neuropathic pain induced by chronic constriction injury. Pharm. Biol. 2013, 51, 844–850. [Google Scholar]
- Zhang, H.; Chen, L.; Sun, X.; Yang, Q.; Wan, L.; Guo, C. Matrine: A Promising Natural Product With Various Pharmacological Activities. Front. Pharmacol. 2020, 11, 588. [Google Scholar] [CrossRef]
- Sun, X.Y.; Jia, L.Y.; Rong, Z.; Zhou, X.; Cao, L.Q.; Li, A.H.; Guo, M.; Jin, J.; Wang, Y.D.; Huang, L.; et al. Research Advances on Matrine. Front. Chem. 2022, 10, 867318. [Google Scholar] [CrossRef]
- You, L.; Yang, C.; Du, Y.; Liu, Y.; Chen, G.; Sai, N.; Dong, X.; Yin, X.; Ni, J. Matrine Exerts Hepatotoxic Effects via the ROS-Dependent Mitochondrial Apoptosis Pathway and Inhibition of Nrf2-Mediated Antioxidant Response. Oxid. Med. Cell. Longev. 2019, 2019, 1045345. [Google Scholar] [CrossRef]
- Li, X.; Tang, Z.; Wen, L.; Jiang, C.; Feng, Q. Matrine: A review of its pharmacology, pharmacokinetics, toxicity, clinical application and preparation researches. J. Ethnopharmacol. 2021, 269, 113682. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, S.; Yin, T.; Kulkarni, K.H.; Teng, Y.; You, M.; Hu, M. Biopharmaceutical and pharmacokinetic characterization of matrine as determined by a sensitive and robust UPLC-MS/MS method. J. Pharm. Biomed. Anal. 2010, 51, 1120–1127. [Google Scholar] [CrossRef]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Sincere, N.I.; Anand, K.; Ashique, S.; Yang, J.; You, C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules 2023, 28, 4014. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- An, J.; Hu, Y.G.; Cheng, K.; Li, C.; Hou, X.L.; Wang, G.L.; Zhang, X.S.; Liu, B.; Zhao, Y.D.; Zhang, M.Z. ROS-augmented and tumor-microenvironment responsive biodegradable nanoplatform for enhancing chemo-sonodynamic therapy. Biomaterials. 2020, 234, 119761. [Google Scholar] [CrossRef]
- Shi, J.H.; Wang, T.R.; You, Y.Q.; Akhtar, M.L.; Liu, Z.J.; Han, F.; Li, Y.; Wang, Y. Enhancement of ultralow-intensity NIR light-triggered photodynamic therapy based on exo- and endogenous synergistic effects through combined glutathione-depletion chemotherapy. Nanoscale 2019, 11, 13078–13088. [Google Scholar] [CrossRef]
- Huang, F.; Xia, F. A smart photodynamic therapy strategy: Depletion of glutathione in cancer cells. Sci. Bull. 2017, 62, 312–313. [Google Scholar] [CrossRef]
- Deng, Y.; Jia, F.; Chen, S.; Shen, Z.; Jin, Q.; Fu, G.; Ji, J. Nitric oxide as an all-rounder for enhanced photodynamic therapy: Hypoxia relief, glutathione depletion and reactive nitrogen species generation. Biomaterials 2018, 187, 55–65. [Google Scholar] [CrossRef]
- Cao, H.; Zhong, S.; Wang, Q.; Chen, C.; Tian, J.; Zhang, W. Enhanced photodynamic therapy based on an amphiphilic branched copolymer with pendant vinyl groups for simultaneous GSH depletion and Ce6 release. J. Mater. Chem. B 2020, 8, 478–483. [Google Scholar] [CrossRef]
- Lan, J.S.; Liu, L.; Zeng, R.F.; Qin, Y.H.; Zhang, T. Tumor-specific carrier-free nanodrugs with GSH depletion and enhanced ROS generation for endogenous synergistic anti-tumor by a chemotherapy-photodynamic therapy. Chem. Eng. J. 2021, 407, 127212. [Google Scholar] [CrossRef]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef]
- Plekhova, N.; Shevchenko, O.; Korshunova, O.; Stepanyugina, A.; Tananaev, I.; Apanasevich, V. Development of Novel Tetrapyrrole Structure Photosensitizers for Cancer Photodynamic Therapy. Bioengineering 2022, 9, 82. [Google Scholar] [CrossRef]
- Li, Q.; Huang, C.; Liu, L.; Hu, R.; Qu, J. Enhancing Type I Photochemistry in Photodynamic Therapy Under Near Infrared Light by Using Antennae-Fullerene Complexes. Cytom. A 2018, 93, 997–1003. [Google Scholar] [CrossRef]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef]
- Ke, W.; Li, J.; Mohammed, F.; Wang, Y.; Tou, K.; Liu, X.; Wen, P.; Kinoh, H.; Anraku, Y.; Chen, H.; et al. Therapeutic Polymersome Nanoreactors with Tumor-Specific Activable Cascade Reactions for Cooperative Cancer Therapy. ACS Nano 2019, 13, 2357–2369. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, H.; Zhi, K.; Yang, X. Exploration of the Natural Active Small-Molecule Drug-Loading Process and Highly Efficient Synergistic Antitumor Efficacy. ACS Appl. Mater. Interfaces 2020, 12, 6827–6839. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.L.; Zhu, X.; Zhou, Z.; Zhang, S.W.; Yang, D.; Zhao, B.; Zhang, Y.P.; Deng, J.P.; Cheng, Y.X.; Zheng, Y.X.; et al. Frontiers in circularly polarized luminescence: Molecular design, self-assembly, nanomaterials, and applications. Sci. China Chem. 2021, 64, 2060–2104. [Google Scholar] [CrossRef]
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Tsichlis, P.N. AKT signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yang, C.; Yang, L.; Li, T.; Gong, M.; Wang, H.; Zhang, J.; Zhao, H.; Zou, L.; He, X. Matrine induces autophagy in human neuroblastoma cells via blocking the AKT-mTOR pathway. Med. Oncol. 2022, 39, 167. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, C.; Yao, S.; Xie, B. Blocking autophagic flux enhances matrine-induced apoptosis in human hepatoma cells. Int. J. Mol. Sci. 2013, 14, 23212–23230. [Google Scholar] [CrossRef]
- Liang, X.; Ju, J. Matrine inhibits ovarian cancer cell viability and promotes apoptosis by regulating the ERK/JNK signaling pathway via p38MAPK. Oncol. Rep. 2021, 45, 82. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, T.; Lv, F.; Liu, L.; Ying, J.; Wang, S. Self-Assembled Nanomedicines for Anticancer and Antibacterial Applications. Adv. Healthc. Mater. 2018, 7, e1800670. [Google Scholar] [CrossRef]
- Khatoon, S.S.; Chen, Y.; Zhao, H.; Lv, F.; Liu, L.; Wang, S. In situ self-assembly of conjugated polyelectrolytes for cancer targeted imaging and photodynamic therapy. Biomater. Sci. 2020, 8, 2156–2163. [Google Scholar] [CrossRef]
- Tao, Z.; Hong, G.; Shinji, C.; Chen, C.; Diao, S.; Antaris, A.L.; Zhang, B.; Zou, Y.; Dai, H. Biological imaging using nanoparticles of small organic molecules with fluorescence emission at wavelengths longer than 1000 nm. Angew. Chem. Int. Ed. Engl. 2013, 52, 13002–13006. [Google Scholar] [CrossRef]
- Guo, L.; Wang, H.; Wang, Y.; Feng, L. Tailor-Made Polymer Nano-Assembly with Switchable Function and Amplified Anti-Tumor Therapy. Adv. Healthc. Mater. 2020, 9, e1901492. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Wu, C.; Yang, K.; Wang, Q.; Zhou, Y.; Chen, L.; Li, H. Novel PROTACs for degradation of SHP2 protein. Bioorg. Chem. 2021, 110, 104788. [Google Scholar] [CrossRef]
- Magann, N.L.; Westley, E.; Sowden, M.J.; Gardiner, M.G.; Sherburn, M.S. Total Synthesis of Matrine Alkaloids. J. Am. Chem. Soc. 2022, 144, 19695–19699. [Google Scholar] [CrossRef]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar] [CrossRef]
- Yang, M.; Cao, S.; Sun, X.; Su, H.; Li, H.; Liu, G.; Luo, X.; Wu, F. Self-Assembled Naphthalimide Conjugated Porphyrin Nanomaterials with D-A Structure for PDT/PTT Synergistic Therapy. Bioconjug. Chem. 2020, 31, 663–672. [Google Scholar] [CrossRef]
- Lamch, Ł.; Kulbacka, J.; Pietkiewicz, J.; Rossowska, J.; Dubińska-Magiera, M.; Choromańska, A.; Wilk, K.A. Preparation and characterization of new zinc(II) phthalocyanine-Containing poly(l-lactide)-b-poly(ethylene glycol) copolymer micelles for photodynamic therapy. J. Photochem. Photobiol. B 2016, 160, 185–197. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, S.; Wang, B.; Sun, K.; Li, F.; Liu, Q.; Yu, X.-A.; Jiang, L.; Wang, L. Self-Assembled Matrine-PROTAC Encapsulating Zinc(II) Phthalocyanine with GSH-Depletion-Enhanced ROS Generation for Cancer Therapy. Molecules 2024, 29, 1845. https://doi.org/10.3390/molecules29081845
Lai S, Wang B, Sun K, Li F, Liu Q, Yu X-A, Jiang L, Wang L. Self-Assembled Matrine-PROTAC Encapsulating Zinc(II) Phthalocyanine with GSH-Depletion-Enhanced ROS Generation for Cancer Therapy. Molecules. 2024; 29(8):1845. https://doi.org/10.3390/molecules29081845
Chicago/Turabian StyleLai, Sitong, Bing Wang, Kunhui Sun, Fan Li, Qian Liu, Xie-An Yu, Lihe Jiang, and Lisheng Wang. 2024. "Self-Assembled Matrine-PROTAC Encapsulating Zinc(II) Phthalocyanine with GSH-Depletion-Enhanced ROS Generation for Cancer Therapy" Molecules 29, no. 8: 1845. https://doi.org/10.3390/molecules29081845