3.2. Synthesis
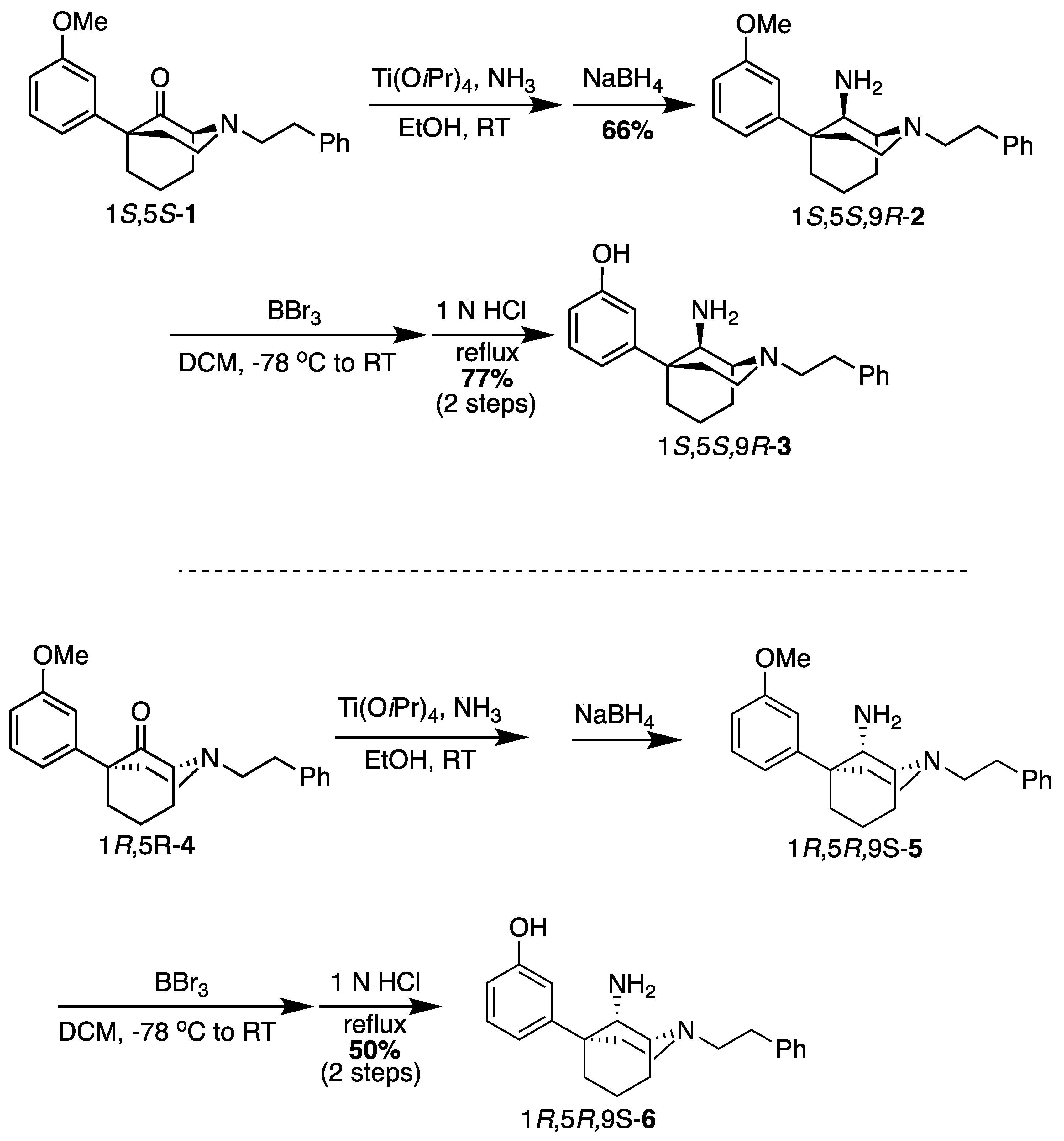
(1S,5S,9R)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (2). In a dry flask, the ketone 1 (1.80 g, 5.15 mmol, 1 equiv), NH4OH (26 mL of a 2 M solution in EtOH, 51.5 mmol, 10 equiv), and Ti(OiPr)4 (3.00 mL, 10.3 mmol, 2 equiv) were combined and stirred overnight at room temperature. The following morning, NaBH4 (292 mg, 7.73 mmol, 1.5 equiv) was added, and the reaction was stirred at room temperature for 1 h before it was quenched with NH4OH and filtered through celite with EtOAc. The filtrate was concentrated, and the residue was purified by flash chromatography with 0 → 10% CMA/CHCl3 to produce 2 as a colorless oil (1.20 g, 66%). 1H NMR (400 MHz; CDCl3): δ 7.27–7.18 (m, 6H), 7.03–6.98 (m, 2H), 6.74–6.72 (m, 1H), 3.76 (s, 3H), 3.56 (s, 1H), 3.05–2.85 (m, 7H), 2.19 (bs, 2H), 1.96–1.51 (m, 8H). 13C NMR (101 MHz; CDCl3): δ 159.77, 150.25, 140.32, 129.42, 128.74, 128.38, 126.07, 118.07, 112.24, 110.77, 57.64, 57.51, 55.32, 55.13, 49.70, 39.98, 39.95, 34.37, 27.77, 21.66, 18.12. HRMS-ESI (m/z): [M+H]+ calcd for C23H31N2O: 351.2436, found: 351.2434, [a]20D +31.7° (c 1.04, CHCl3).
3-((1S,5S,9R)-9-Amino-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (3). In a dry flask, the ether 2 (200 mg, 0.571 mmol, 1 equiv) was dissolved in dichloromethane (13 mL) and cooled to −78 °C before BBr3 (274.6 mL, 2.85 mmol, 5 equiv) was added. After stirring the reaction for 15 min, the cooling bath was removed, and the contents were stirred for an additional 2 h at room temperature. The solvent was removed under vacuum conditions, and the residue was taken up in 1 N HCl and heated to reflux for 1 h. After cooling to room temperature, the pH was adjusted to 8 with NH4OH. The aqueous solution was extracted with dichloromethane (3x). The organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography with 2 → 15% CMA/CHCl3, which produced 3 as a white foam (147 mg, 77%). The HCl salt was prepared by dissolving the solid (40 mg) in 400 μL of dichloromethane and adding 2 N HCl (4 equiv) in Et2O, resulting in a white solid (24 mg, 50% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.29–6.82 (m, 6H), 6.79–6.70 (m, 2H), 6.69–6.68 (m, 1H), 3.50 (s, 1H), 3.12–2.95 (m, 3H), 2.85–2.74 (m, 4H), 2.33–2.21 (m, 2H), 2.10 (dd, J = 13.6, 4.1 Hz, 1H), 1.96–1.84 (m, 2H), 1.77–1.62 (m, 2H), 1.51–1.48 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 158.31, 149.57, 140.62, 130.39, 128.84, 128.46, 126.06, 116.70, 114.47, 111.41, 56.77, 56.27, 55.11, 48.50, 42.36, 40.87, 34.66, 29.04, 26.12, 23.31. HRMS-ESI (m/z): [M+H]+ calcd for C22H29N2O: calc: 337.2280, found: 337.2285, mp 70–73 °C, [α]20D +8.74° (c 1.12, CHCl3), CHN calc. for C22H30Cl2N2O · 1 H2O: C, 61.82%; H, 7.55%; N, 6.55%; found: C, 61.50%; H, 7.24%; N, 6.30%.
(1R,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (5). Using the ketone 1R,4R-4, the procedure followed the preparation of 2 (on a 473 mg scale) to obtain 5 as a yellow oil (235 mg, 50%). 1H-NMR (400 MHz; CDCl3): δ 7.28–7.14 (m, 6H), 6.95 (d, J = 7.9 Hz, 1H), 6.90 (t, J = 1.9 Hz, 1H), 6.73 (dd, J = 8.1, 2.3 Hz, 1H), 3.79 (s, 3H), 3.25 (d, J = 1.9 Hz, 1H), 3.06–2.84 (m, 3H), 2.84–2.76 (m, 4H), 2.36–2.25 (m, 2H), 1.98 (dd, J = 13.6, 4.9 Hz, 1H), 1.92–1.70 (m, 3H), 1.65–1.53 (m, 4H). 13C-NMR (101 MHz; CDCl3): δ 159.83, 150.97, 140.83, 129.48, 128.76, 128.28, 125.89, 117.99, 112.29, 110.52, 57.41, 56.81, 55.36, 55.19, 48.55, 41.49, 41.29, 34.64, 28.91, 25.76, 23.21. HRMS-ESI (m/z): [M+H]+ calcd for C23H30N2O: 351.2436, found: 251.2441, [α]20D −31.3° (c 1.10, CHCl3).
3-((1R,5R,9S)-9-Amino-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (6). Using the ether 5, the procedure followed the preparation of 3 on a 353 mg scale, obtaining the phenol as a white foam (233 mg, 66%). Salt formation to a hydrochloride produced a white solid (223 mg, 86% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.29–6.82 (m, 6H), 6.79–6.70 (m, 2H), 6.69–6.68 (m, 1H), 3.50 (s, 1H), 3.12–2.95 (m, 3H), 2.85–2.74 (m, 4H), 2.33–2.21 (m, 2H), 2.10 (dd, J = 13.6, 4.1 Hz, 1H), 1.96–1.84 (m, 2H), 1.77–1.62 (m, 2H), 1.51–1.48 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 158.31, 149.57, 140.62, 130.39, 128.84, 128.46, 126.06, 116.70, 114.47, 111.41, 56.77, 56.27, 55.11, 48.50, 42.36, 40.87, 34.66, 29.04, 26.12, 23.31. HRMS-ESI (m/z): [M+H]+ calcd for C22H29N2O: 337.2280, found: 337.2284, mp 71–73 °C, [α]20D −10.6° (c 1.08, CHCl3), CHN calc. for C22H30Cl2N2O · 0.25 H2O: C, 63.84%; H, 7.43%; N, 6.77%; found: C, 63.77%; H, 7.22%; N, 6.63%.
(1S,5S,9R)-5-(3-Methoxyphenyl)-N-methyl-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (7). In a pressure flask, the oxide 1 (300 mg, 0.858 mmol, 1 equiv), MeNH2·HCl (232 mg, 3.42 mmol, 4 equiv), triethylamine (480 mL, 3.43 mmol, 4 equiv), Ti(OiPr)4 (1.01 mL, 3.42 mmol, 4 equiv), and EtOH (3 mL) were combined and heated to 50 °C overnight. The reaction was then cooled to room temperature, and NaBH4 (49 mg, 1.29 mmol, 1.5 equiv) was added. The mixture was stirred for 1 h at room temperature. The reaction was quenched with NH4OH and filtered through celite with EtOAc, and the filtrate was concentrated. The residue was purified by chromatography with 0 → 15% CMA/CHCl3 to produce 7 as a yellow oil (245 mg, 78%). 1H NMR (400 MHz; CDCl3): δ 7.27–7.14 (m, 6H), 6.92 (d, J = 7.9 Hz, 1H), 6.87 (t, J = 2.0 Hz, 1H), 6.71–6.68 (m, 1H), 3.78 (s, 3H), 3.12–3.03 (m, 3H), 2.98–2.75 (m, 5H), 2.35–2.24 (m, 2H), 2.20 (s, 3H), 2.04–1.99 (m, 1H), 1.91–1.69 (m, 3H), 1.65–1.58 (m, 1H), 1.48–1.44 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 159.51, 151.57, 140.85, 129.12, 128.65, 128.20, 125.81, 117.71, 111.97, 110.16, 63.84, 56.57, 55.08, 52.47, 48.67, 42.42, 40.49, 34.89, 34.31, 29.81, 25.92, 23.19. HRMS-ESI (m/z): [M+H]+ calcd for C24H33N2O: 365.2593, found: 365.2588, [α]20D +15.1° (c 1.01, CHCl3).
3-((1S,5S,9R)-9-(Methylamino)-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (8). In a dry flask, ether 7 (245 mg, 0.672 mmol, 1 equiv) was dissolved in dichloromethane (15 mL) and cooled to −78 °C before BBr3 (320 uL, 3.36 mmol, 5 equiv) was added dropwise. After the reaction was stirred at −78 °C for 15 min, the cooling bath was removed, and the contents were allowed to stir at room temperature for 2 h. The solvent was removed in vacuo, and the residue was taken up in 1 N HCl (10 mL) and heated to reflux for 1 h. After cooling to room temperature, the pH was adjusted to 8 with NH4OH, and the aqueous mixture was extracted with CHCl3 (3x). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash chromatography with 2 → 20% CMA/CHCl3 to produce 8 as a yellow oil, which solidified upon drying (89 mg, 38%). The HCl salt was prepared by dissolving the free base (80 mg) in a minimal amount of dichloromethane and adding 2 M HCl (4 equiv) in Et2O, resulting in a pink solid (77 mg, 80% recovery). 1H NMR (400 MHz; CDCl3): δ 7.29–7.15 (m, 5H), 6.98 (t, J = 7.9 Hz, 1H), 6.73 (d, J = 7.7 Hz, 1H), 6.53 (s, 1H), 6.20–6.18 (m, 1H), 3.09–3.04 (m, 3H), 2.89–2.79 (m, 5H), 2.38–2.31 (m, 2H), 2.14 (s, 3H), 2.01–1.97 (m, 1H), 1.88–1.71 (m, 3H), 1.61–1.58 (m, 1H), 1.49–1.44 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.28, 149.90, 140.79, 128.99, 128.74, 128.37, 126.06, 115.68, 114.04, 113.28, 64.61, 56.63, 53.19, 48.14, 42.45, 39.86, 34.41, 34.23, 29.49, 26.06, 23.09. HRMS-ESI (m/z): [M+H]+ calcd for C23H31N2O: 351.2436, found: 351.2430, mp 60–65 °C, [α]20D +3.79° (c 1.12, CHCl3), CHN calc. for C23H32Cl2N2O · 1.38 H2O: C, 61.62%; H, 7.82%; N, 6.25%; found: C, 61.81%; H, 7.73%; N, 6.04%.
(1R,5R,9S)-5-(3-Methoxyphenyl)-N-methyl-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (9). Using the oxide 4, the procedure followed the preparation of 7 on a 300 mg scale, obtaining 9 as a colorless oil (182 mg, 58%). 1H NMR (400 MHz; CDCl3): δ 7.27–7.14 (m, 6H), 6.92 (d, J = 7.9 Hz, 1H), 6.87 (t, J = 2.0 Hz, 1H), 6.71–6.68 (m, 1H), 3.78 (s, 3H), 3.12–3.03 (m, 3H), 2.98–2.75 (m, 5H), 2.35–2.24 (m, 2H), 2.20 (s, 3H), 2.04–1.99 (m, 1H), 1.91–1.69 (m, 3H), 1.65–1.58 (m, 1H), 1.48–1.44 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 159.51, 151.57, 140.85, 129.12, 128.65, 128.20, 125.81, 117.71, 111.97, 110.16, 63.84, 56.57, 55.08, 52.47, 48.67, 42.42, 40.49, 34.89, 34.31, 29.81, 25.92, 23.19. HRMS-ESI (m/z): [M+H]+ calcd for C24H33N2O: 365.2593, found: 365.2587, [α]20D −19.1° (c 0.99, CHCl3).
3-((1R,5R,9S)-9-(Methylamino)-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (10). Using the ether 9, the procedure followed the preparation of 8 on a 148 mg scale, obtaining an off-white foam (54 mg, 38%). Hydrochloride salt formation produced a white solid (55 mg, 85% recovery). 1H NMR (400 MHz; CDCl3): δ 7.29–7.15 (m, 5H), 6.98 (t, J = 7.9 Hz, 1H), 6.73 (d, J = 7.7 Hz, 1H), 6.53 (s, 1H), 6.20–6.18 (m, 1H), 3.09–3.04 (m, 3H), 2.89–2.79 (m, 5H), 2.38–2.31 (m, 2H), 2.14 (s, 3H), 2.01–1.97 (m, 1H), 1.88–1.71 (m, 3H), 1.61–1.58 (m, 1H), 1.49–1.44 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.28, 149.90, 140.79, 128.99, 128.74, 128.37, 126.06, 115.68, 114.04, 113.28, 64.61, 56.63, 53.19, 48.14, 42.45, 39.86, 34.41, 34.23, 29.49, 26.06, 23.09. HRMS-ESI (m/z): [M+H]+ calcd for C23H31N2O: 351.2436, found: 351.2433, mp 62–65 °C, [α]20D −1.86° (c 1.01, CHCl3), CHN calc. for C23H32Cl2N2O · 0.8 H2O: C, 63.09%; H, 7.74%; N, 6.40%; found: C, 63.09%; H, 7.55%; N, 6.22%.
(1S,5S)-9-Hydrazineyl-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonane (11). The combined oxide 1 (100 mg, 0.286 mmol, 1 equiv), hydrazine hydrate (28 mL, 0.572 mmol, 2 equiv), acetic acid (8.2 mL, 0.143 mmol, 0.5 eq) and benzene (4 mL) were combined in a flask. The flask was heated to reflux under Dean–Stark conditions overnight. The following day, TLC showed no starting material. After cooling to room temperature, the reaction was concentrated; the residue was taken up in EtOH (4 mL); and NaBH4 was added (11 mg, 0.286 mmol, 1 equiv). Upon the addition of the reductant, hydrogen gas evolution was observed, and once this ceased, the reaction was allowed to stir for 30 min longer to ensure the complete reduction of the hydrazone. The reaction was quenched with NH4OH and filtered through celite with EtOH, and the filtrate was concentrated into a yellow oil. It was used directly in the next step without purification. Hydrazine 11 did not react with titanium trichloride in basic media. 1H-NMR (400 MHz; CDCl3): δ 7.31–7.12 (m, 5H), 6.98–6.94 (m, 2H), 6.61 (dd, J = 8.1, 2.4 Hz, 1H), 4.24 (t, J = 3.3 Hz, 1H), 3.66 (s, 3H), 3.19–3.14 (m, 1H), 2.76–2.72 (m, 3H), 2.63–2.58 (m, 2H), 2.52–2.45 (m, 1H), 2.22–2.13 (m, 5H), 1.99 (d, J = 13.7 Hz, 1H), 1.67–1.63 (m, 1H), 1.48–1.43 (m, 1H).
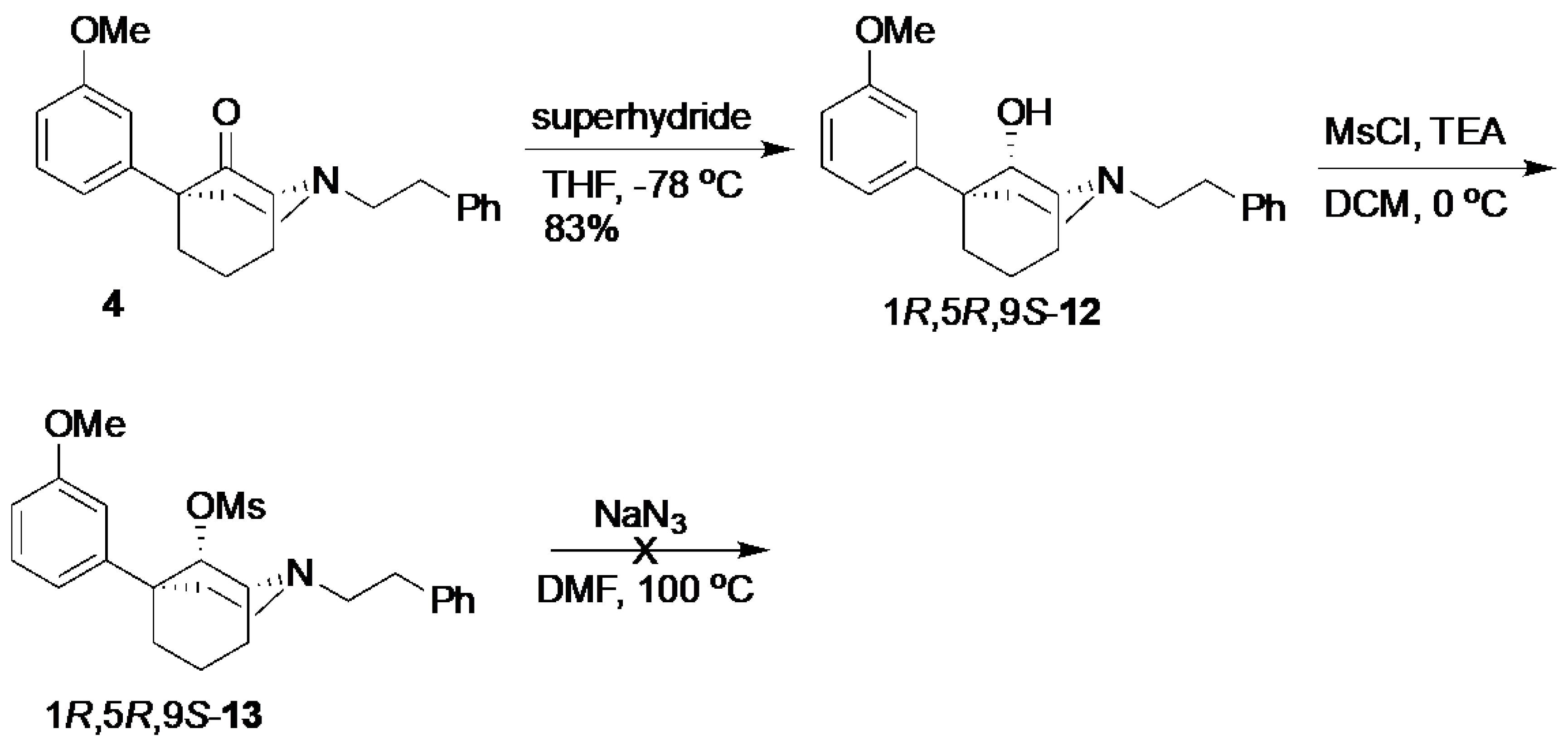
(1R,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-ol (
12) [
3]. The compound
4 (500 mg, 1.43 mmol, 1 equiv) was dissolved in anhydrous THF (12.5 mL) in a flame-dried flask. After cooling to −78 °C, superhydride (2.16 mL of a 1 M solution, 2.16 mmol, 1.51 equiv) was added dropwise. After stirring for 1 h, TLC showed no remaining starting material. The mixture was quenched with MeOH, concentrated, and purified by chromatography with 0 to 2% MeOH/dichloromethane, yielding the alcohol
12 as a yellow oil (416 mg, 83%). Characterization data agreed with the literature. The product was used in the next step without further purification.
(1R,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl methanesulfonate (13). We dissolved 12 in dichloromethane (4 mL) in a dry flask and cooled the contents to 0 °C before adding TEA (181 mL, 1.30 mmol, 1.1 equiv), followed by MsCl (138 mL, 1.78 mmol, 1.5 equiv). After stirring for 1 h at room temperature, TLC showed no starting material. The reaction was quenched with H2O; the aqueous layer was extracted with dichloromethane (3x); and the combined organic layers were washed with NaHCO3 and dried over Na2SO4, filtered, and concentrated into an orange oil. The product 13 was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 7.29–7.16 (m, 6H), 7.00 (d, J = 7.9 Hz, 1H), 6.95 (t, J = 1.8 Hz, 1H), 6.75 (dd, J = 8.1, 2.3 Hz, 1H), 5.12 (d, J = 2.5 Hz, 1H), 3.81 (s, 3H), 3.46 (d, J = 8.1, 2.4 Hz, 1H), 3.16–3.13 (m, 2H), 2.89–2.78 (m, 4H), 2.57–2.49 (m, 1H), 2.41 (s, 3H), 2.36–2.30 (m, 1H), 2.11–2.08 (m, 1H), 1.98–1.85 (m, 2H), 1.67–1.60 (m, 3H). 13C NMR (101 MHz; CDCl3): δ 159.61, 148.41, 140.56, 129.29, 128.78, 128.29, 125.94, 117.82, 111.98, 111.28, 84.68, 56.81, 56.11, 55.23, 48.20, 41.18, 39.80, 38.59, 34.12, 29.54, 25.69, 21.91.
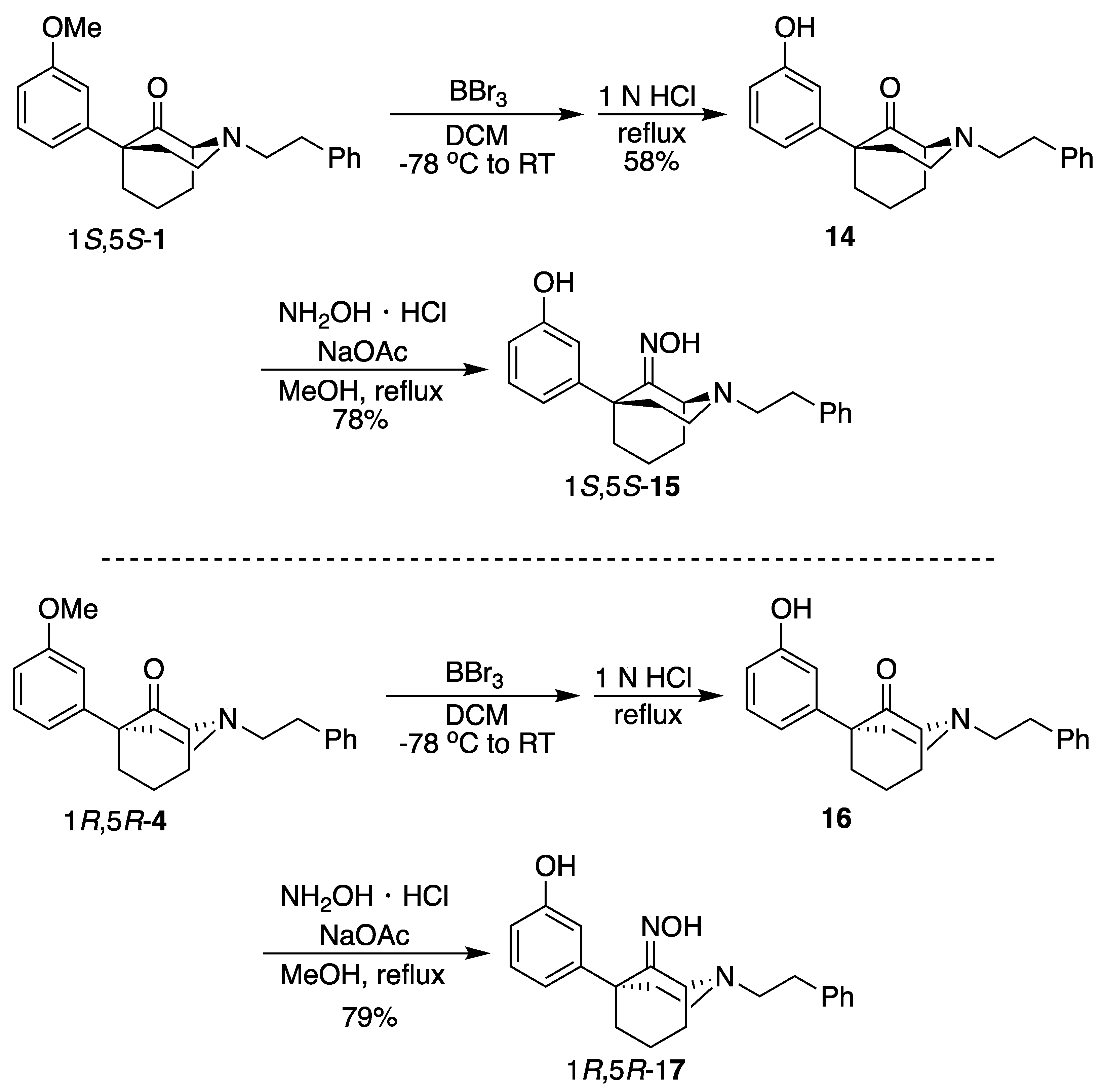
(1S,5S)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one (
14). In a dry flask, ketone
1 (250 mg, 0.743 mmol, 1 equiv) was dissolved in dichloromethane (17 mL), and the solution was cooled to –78 °C. To this mixture was added BBr
3 (360 mL, 3.71 mmol, 5 equiv), and the reaction was stirred for 15 min before the cooling bath was removed. After 2 h at room temperature, the reaction was analyzed by TLC (5% CMA/CHCl
3), and no starting material was present. The solvent was removed in vacuo, and the residue was taken up in 1 N HCl (10 mL) and heated to reflux for 1 h. After cooling to room temperature, the pH was adjusted to 8 with NH
4OH, and the aqueous solution was extracted with CHCl
3 (3x). The combined organic layers were dried over Na
2SO
4, filtered, and concentrated. The crude product was purified by flash chromatography with 0 → 5% CMA/CHCl
3 to produce
14 as a yellow solid (145 mg, 58%). Characterization data agreed with the literature [
4].
(1S,5S)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one oxime (15). In a flask, 14 (145 mg, 0.432 mmol, 1 equiv), NaOAc (71.0 mg, 0.519 mmol, 1.2 equiv), hydroxylamine hydrochloride (36.0 mg, 0.519 mmol, 1.2 equiv), and MeOH (4.3 mL) were combined and heated to reflux for 2 h. After cooling to room temperature, the reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc (3x). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product 15 was purified by flash chromatography with 2 → 15% CMA/CHCl3 to produce a white solid (119 mg, 79%). The HCl salt was prepared by dissolving the free base (90 mg) in minimal EtOAc and adding 2 M HCl in Et2O (2 equiv), resulting in a white solid (70 mg, 70% recovery). 1H NMR (400 MHz; DMSO-d6): δ 10.57 (s, 1H), 9.12 (s, 1H), 7.30–7.24 (m, 4H), 7.20–7.16 (m, 1H), 7.06–7.02 (m, 1H), 6.75–6.74 (m, 2H), 6.56–6.54 (m, 1H), 4.42–4.41 (m, 1H), 3.12–2.83 (m, 1H), 2.83–2.64 (m, 5H), 2.29–2.23 (m, 1H), 2.18–2.11 (m, 1H), 2.09–1.98 (m, 3H), 1.97–1.89 (m, 1H), 1.59–1.48 (m, 1H), 1.41–1.37 (m, 1H). 13C NMR (101 MHz; DMSO-d6): δ 162.20, 156.88, 148.58, 140.85, 129.09, 128.62, 128.48, 126.23, 118.23, 115.38, 112.92, 58.66, 56.47, 51.36, 48.44, 43.90, 38.89, 34.22, 30.55, 19.77. HRMS-ESI (m/z): [M+H]+ calcd for C22H27N2O2: 351.2073, found: 351.2075, mp 206–208 °C, [α]20D +26.2° (c 1.02, MeOH). CHN calc. for C22H27ClN2O2 · 0.7 H2O: C, 66.14%; H, 7.16%; N, 7.01%; found C, 66.25%; H, 6.97%; N, 6.76%.
(1R,5R)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one (
16). Using
4, the procedure followed that of
14 on a 2.01 g scale to obtain
16 as a yellow oil (664 mg, 34%). Characterization data agreed with the literature [
3].
(1R,5R)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one oxime (17). Using 16, the procedure followed that of 14 on a 664 mg scale to obtain 17 as a white solid (549 mg, 79%). The HCl salt was prepared by dissolving the free base (200 mg) in minimal EtOAc and adding 2 M HCl in Et2O (2 equiv), resulting in a white solid (121 mg, 55% recovery). 1H NMR (400 MHz; DMSO-d6): δ 10.57 (s, 1H), 9.12 (s, 1H), 7.30–7.24 (m, 4H), 7.20–7.16 (m, 1H), 7.06–7.02 (m, 1H), 6.75–6.74 (m, 2H), 6.56–6.54 (m, 1H), 4.42–4.41 (m, 1H), 3.12–2.83 (m, 1H), 2.83–2.64 (m, 5H), 2.29–2.23 (m, 1H), 2.18–2.11 (m, 1H), 2.09–1.98 (m, 3H), 1.97–1.89 (m, 1H), 1.59–1.48 (m, 1H), 1.41–1.37 (m, 1H). 13C NMR (101 MHz; DMSO-d6): δ 162.20, 156.88, 148.58, 140.85, 129.09, 128.62, 128.48, 126.23, 118.23, 115.38, 112.92, 58.66, 56.47, 51.36, 48.44, 43.90, 38.89, 34.22, 30.55, 19.77. HRMS-ESI (m/z): [M+H]+ calcd for C22H27N2O2: 351.2073, found: 351.2076, mp 208–210 °C. CHN calc. for C22H27ClN2O2 · 0.25 H2O: C, 67.51%; H, 7.08%; N, 7.16%; found: C, 67.65%; H, 7.05%; N, 6.98%.
(1S,5S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one oxime (18). Using ketone 1, the procedure followed that of compound 19 on a 1.34 g scale to obtain 18 as a white solid (1.18 g, 84%). 1H NMR (400 MHz; CDCl3): δ 7.89 (bs, 1H), 7.31–7.27 (m, 2H), 7.24–7.18 (m, 4H), 6.93 (d, J = 7.9 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.1, 2.4 Hz, 1H), 4.54 (bs, 1H), 3.74 (s, 3H), 3.20 (bs, 1H), 2.86–2.74 (m, 5H), 2.45–2.39 (m, 1H), 2.23–2.02 (m, 5H), 1.69–1.66 (m, 1H), 1.60–1.55 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 159.01, 147.31, 140.32, 128.78, 128.62, 128.34, 126.01, 119.84, 113.82, 110.95, 58.66, 55.13, 50.92, 48.62, 44.05, 39.27, 38.50, 34.30, 30.37, 19.64. HRMS-ESI (m/z): [M+H]+ calcd for C23H29N2O2: 365.2229, found: 365.2231, mp 122.9 °C, [α]20D +31.6° (c 1.03, CHCl3).
(1R,5R)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-one oxime (19). Ketone 4 (361 mg, 1.03 mmol, 1 equiv), hydroxylamine hydrochloride (86.1 mg, 1.24 mmol, 1.2 equiv), and sodium acetate trihydrate (169 mg, 1.24 mmol, 1.2 equiv) were placed in a flask and suspended in MeOH (10 mL). The contents were heated to reflux, during which time all of the solids dissolved. After heating for 2 h, no starting material remained as seen through TLC. The reaction was cooled to room temperature and quenched with saturated aqueous NaHCO3, and the aqueous layer was extracted with dichloromethane (3x). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was purified by flash chromatography with 40% EtOAc/hexane to produce 19 as a white solid (307 mg, 82%). 1H NMR (400 MHz; CDCl3): δ 8.35 (bs, 1H), 7.32–7.28 (m, 2H), 7.24–7.18 (m, 4H), 6.93 (d, J = 7.9 Hz, 1H), 6.89 (t, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.1, 2.4 Hz, 1H), 4.51 (bs, 1H), 3.74 (s, 3H), 3.18–3.17 (m, 1H), 2.88–2.83 (m, 4H), 2.79–2.73 (m, 1H), 2.45–2.38 (m, 1H), 2.22–2.08 (m, 4H), 2.04–2.00 (m, 1H), 1.68–1.64 (m, 1H), 1.59–1.52 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 164.60, 159.03, 147.33, 140.33, 128.79, 128.62, 128.34, 125.99, 119.83, 113.81, 110.97, 58.68, 55.13, 50.89, 48.61, 44.06, 39.27, 38.44, 34.32, 30.40, 19.63. HRMS-ESI (m/z): [M+H]+ calcd for C23H29N2O2: 365.2229, found: 365.2225, mp 119–121 °C, [α]20D –30.7° (c 1.08, CHCl3).
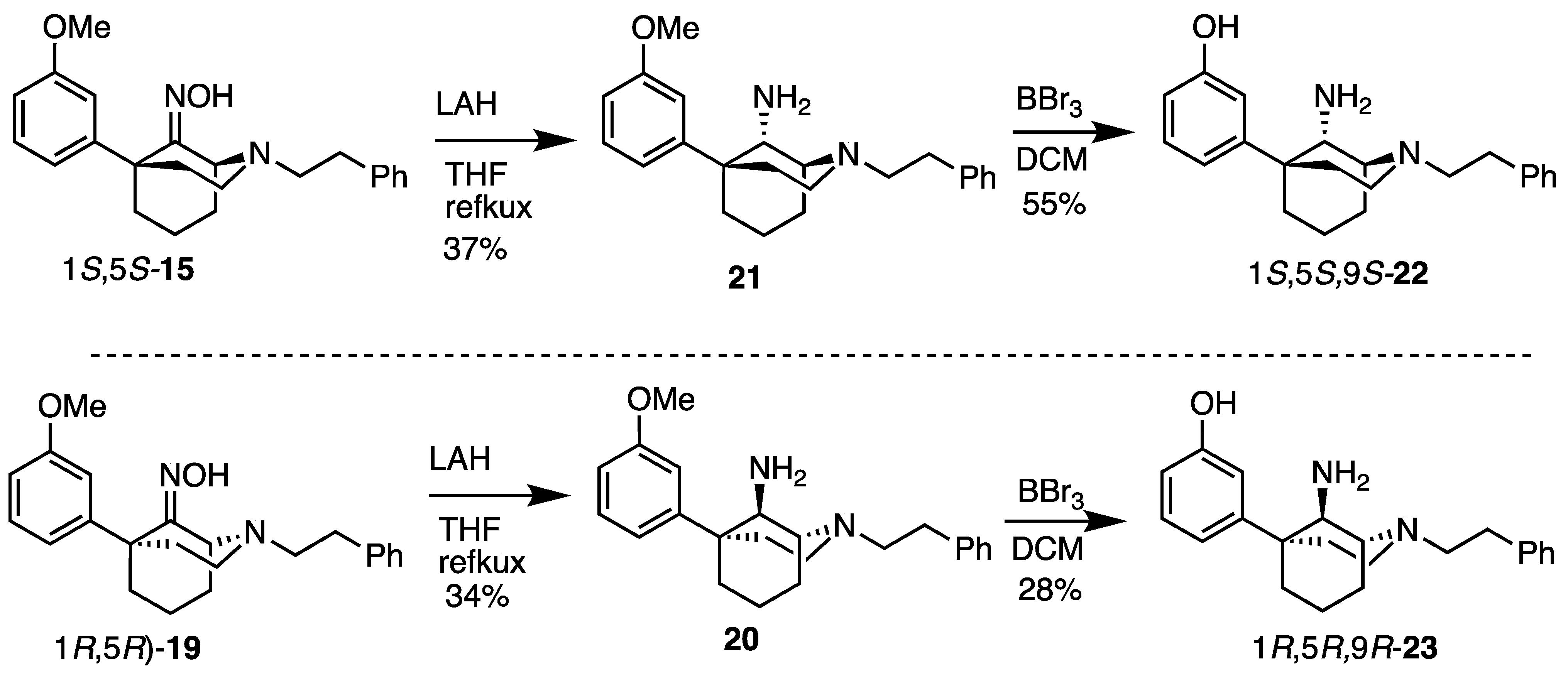
(1R,5R,9R)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (20). Using 19, the procedure followed the preparation of 21 on a 1.09 g scale; the only change was in the reaction workup procedure. The reaction was cooled to 0 °C and diluted with Et2O, followed by the addition of H2O (460 mL), 15% NaOH (460 uL), and an additional portion of H2O (1.40 mL). After stirring for 15 min, 2.30 g of Na2SO4 · 10 H2O was added, and the contents were stirred for an additional 15 min. The solids were filtered off and washed with Et2O, and the filtrate was concentrated and purified as described previously to obtain 20 as a yellow oil (361 mg, 34%). 1H-NMR (400 MHz; CDCl3): δ 7.33–7.20 (m, 6H), 7.06–6.99 (m, 2H), 6.77–6.75 (m, 1H), 3.81 (s, 3H), 3.66 (bs, 1H), 3.13 (bs, 1H), 3.13–3.05 (m, 2H), 3.05–2.91 (m, 4H), 2.28–2.22 (m, 2H), 2.02–1.77 (m, 6H). 13C-NMR (101 MHz; CDCl3): δ 159.76, 150.36, 140.46, 129.41, 128.73, 128.36, 126.03, 118.08, 112.24, 110.74, 57.69, 57.46, 55.46, 55.15, 49.71, 40.09, 40.02, 34.50, 27.81, 21.70, 18.17. HRMS-ESI (m/z): [M+H]+ calcd for C23H31N2O: 351.2436, found: 351.2435, [α]20D –37.3° (c 1.02, CHCl3).
(1S,5S,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-amine (21). We dissolved the oxime 15 (292 mg, 0.801 mmol, 1 equiv) in anhydrous THF (15 mL) and added LAH (122 mg, 3.20 mmol, 4 equiv) gradually to avoid excessive foaming. The contents were heated to reflux overnight then cooled to room temperature the following day and quenched slowly with 1 N HCl to pH 1. The pH was then adjusted to 9 by the addition of NH4OH, and the suspension was filtered through celite with EtOAc. The filtrate was concentrated and purified by flash chromatography with a gradual gradient of 1 → 8% CMA/CHCl3 (the slower the gradient and the longer the column, the better the separation of the diastereomers) to produce 21 as a pale-yellow oil (105 mg, 37%).1H-NMR (400 MHz; CDCl3): δ 7.33–7.20 (m, 6H), 7.06–6.99 (m, 2H), 6.77–6.75 (m, 1H), 3.81 (s, 3H), 3.66 (bs, 1H), 3.13 (bs, 1H), 3.13–3.05 (m, 2H), 3.05–2.91 (m, 4H), 2.28–2.22 (m, 2H), 2.02–1.77 (m, 6H). 13C-NMR (101 MHz; CDCl3): δ 159.76, 150.36, 140.46, 129.41, 128.73, 128.36, 126.03, 118.08, 112.24, 110.74, 57.69, 57.46, 55.46, 55.15, 49.71, 40.09, 40.02, 34.50, 27.81, 21.70, 18.17. HRMS-ESI (m/z): [M+H]+ calcd for C23H31N2O: 351.2436, found: 351.2433, [α]20D –32.4° (c 0.98, CHCl3).
3-((1S,5S,9S)-9-Amino-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (22). Using the ether 21, the procedure followed the preparation of 23 on a 361 mg scale. The phenol 22 was obtained as a white foam (189 mg, 55%). Hydrochloride salt formation produced a white powder (208 mg, 90% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.28–7.15 (m, 6H), 6.89–6.87 (m, 2H), 6.69–6.67 (m, 1H), 3.58 (d, J = 2.9 Hz, 1H), 3.14 (s, 1H), 3.02–2.99 (m, 2H), 2.89–2.80 (m, 4H), 2.25–2.11 (m, 2H), 1.98–1.74 (m, 5H), 1.68–1.62 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 157.33, 149.26, 139.79, 129.76, 128.72, 128.41, 126.15, 117.29, 114.33, 112.61, 57.51, 57.04, 55.00, 49.56, 39.53, 39.44, 33.78, 27.57, 21.28, 18.05. HRMS-ESI (m/z): [M+H]+ calcd for C22H29N2O: 337.2280, found: 337.2281, mp 72–73 °C, [α]20D −21.4° (c 1.00, CHCl3).
3-((1R,5R,9R)-9-Amino-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (23). We dissolved the ether 20 (538 mg, 1.53 mmol, 1 equiv) in dichloromethane (9 mL) and cooled the solution to −78 °C before BBr3 (739 mL, 7.67 mmol, 5 equiv) was added. After 15 min, the cooling bath was removed, and the reaction was allowed to stir at room temperature for 2 h, at which point TLC showed no starting material. The reaction was quenched with MeOH, concentrated, and refluxed in 9 mL of 1 N HCl for 1 h to break up boron complexes. After cooling to room temperature, the pH was adjusted to 9 with NH4OH, and the aqueous layer was extracted with dichloromethane (3x). The organic layers were combined, dried over Na2SO4, filtered, and concentrated. Purification by flash chromatography with 2 → 15% CMA/CHCl3 afforded 142 mg (28% yield) of 23 as a white foam. A salt was made by dissolving 142 mg of the free base in a minimal amount of MeOH and adding concentrated HCl (2 equiv per amine, 4 equiv total). After the solution was stirred for 1 h, the solvent was removed under vacuum conditions, and the pasty material recrystallized from EtOH to yield 23 as a fluffy white solid (163 mg, 94% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.28–7.15 (m, 6H), 6.89–6.87 (m, 2H), 6.69–6.67 (m, 1H), 3.58 (d, J = 2.9 Hz, 1H), 3.14 (s, 1H), 3.02–2.99 (m, 2H), 2.89–2.80 (m, 4H), 2.25–2.11 (m, 2H), 1.98–1.74 (m, 5H), 1.68–1.62 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 157.33, 149.26, 139.79, 129.76, 128.72, 128.41, 126.15, 117.29, 114.33, 112.61, 57.51, 57.04, 55.00, 49.56, 39.53, 39.44, 33.78, 27.57, 21.28, 18.05. HRMS-ESI (m/z): [M+H]+ calcd for C22H29N2O: 337.2280, found: 337.2280, mp 79.6–81.5 °C, [α]20D +20.9° (c 1.01, CHCl3). CHN calc. for C22H30Cl2N2O · 3.8 H2O: C, 55.3%; H, 7.93%; N, 5.86%; found: C, 55.24%; H, 7.80%; N, 5.75%.
(1S,5S)-5-(3-Methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-one (
25). In a flask, the secondary amine
24 [
4] (3.32 g, 13.5 mmol, 1 equiv) and DMAP (165 mg, 1.35 mmol, 0.100 equiv) were dissolved in dichloromethane (34 mL). The contents were cooled to 0 °C, and TEA (2.83 mL, 20.3 mmol, 1.5 equiv) was added, followed by Boc
2O (4.66 mL, 20.3 mmol, 1.5 equiv). After stirring overnight, a spatula tip’s worth of imidazole was added, and the solution was stirred for another 1 h to quench excess Boc
2O. The contents were transferred to a separatory funnel and washed with dilute HCl and H
2O; the aqueous layer was back extracted with dichloromethane (2x); and the organic layers dried over Na
2SO
4. The removal of the solvent in vacuo, followed by purification by flash chromatography with 0 to 40% EtOAc/hexane, produced
25 as a viscous yellow oil (3.69 g, 79%).
1H-NMR (400 MHz; CDCl
3): δ 7.30–7.26 (m, 1H), 6.82–6.76 (m, 3H), 4.31 (bs, 1H), 4.24–4.19 (m, 1H), 3.80 (s, 3H), 3.22–3.15 (m, 1H), 2.56–2.48 (m, 2H), 2.39–2.31 (m, 2H), 2.21–2.18 (m, 2H), 1.80–1.72 (m, 2H), 1.49 (s, 9H).
13C-NMR (101 MHz; CDCl
3): δ 211.81, 159.25, 154.62, 145.59, 129.05, 119.36, 113.52, 111.31, 80.30, 63.62, 55.09, 52.81, 40.98, 40.64, 38.09, 35.54, 28.39, 17.56. HRMS-ESI (
m/z): [M+Na]
+ calcd for C
20H
27NO
4Na: 368.1838, found: 368.1833 [α]
20D −37.1° (
c 1.08, CHCl
3).
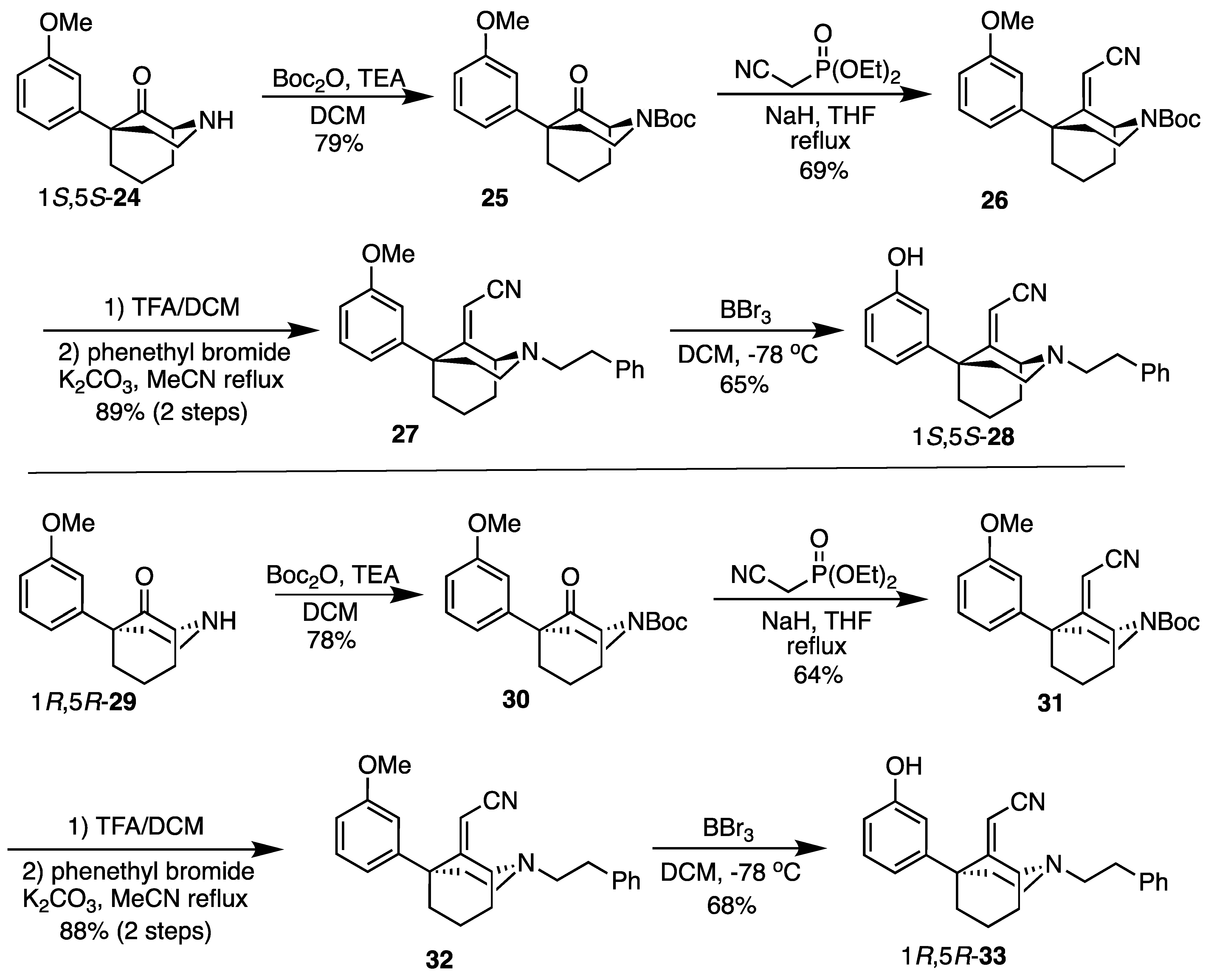
tert-Butyl (1S,5S)-9-(cyanomethylene)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonane-2-carboxylate (26). To a flame-dried flask was added NaH (452 mg, 11.3 mmol, 3 equiv, 60% dispersion in mineral oil). The solid was washed with hexane 3 times under nitrogen conditions before THF (10 mL) was added. To this suspension, diethyl (cyanomethyl)phosphonate (1.83 mL, 11.3 mmol, 3 equiv) was added dropwise. Once gas evolution ceased and a clear solution formed, N-Boc ketone 25 (1.3 g, 3.76 mmol, 1 equiv) as a solution in THF (7 mL) was added, and the reaction mixture was heated to reflux overnight. After cooling to room temperature, EtOH was added dropwise to quench excess NaH, and the contents were concentrated in vacuo. The residual oil was purified by chromatography with 0 to 20% EtOAc/hexane to yield 26 as a white solid (961 mg, 691H-NMR (400 MHz; CDCl3): δ 7.23 (t, J = 7.9 Hz, 1H), 6.78–6.74 (m, 3H), 5.11–5.05 (m, 1H), 4.55 (s, 1H), 4.03–3.94 (m, 1H), 3.74 (s, 3H), 3.14–3.08 (m, 1H), 2.37–2.34 (m, 1H), 2.24–2.20 (m, 2H), 2.06–1.97 (m, 3H), 1.67–1.66 (m, 1H), 1.46 (s, 10H). 13C-NMR (101 MHz; CDCl3): δ 171.58, 159.77, 155.04, 147.40, 129.75, 119.59, 116.06, 113.94, 111.56, 94.92, 80.61, 55.64, 55.38, 45.79, 40.55, 39.89, 38.45, 34.22, 28.59, 18.06. HRMS-ESI (m/z): [M+Na]+ calcd for C22H28N2O3Na: 391.1998, found: 391.1997, mp 124–126 °C, [α]20D –44.3° (c 1.10, CHCl3).
2-((1S,5S)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile (27). A solution of 26 (500 mg, 1.36 mmol, 1 equiv) in dichloromethane (4.6 mL) was prepared and cooled to 0 °C before TFA (2.09 mL, 27.1 mmol, 20 equiv) was added dropwise. After 30 min, TLC showed no evidence of starting material. The pH was adjusted to 9 with NH4OH, and the aqueous layer was extracted with CHCl3 (3x). The combined organic layers were dried over Na2SO4, filtered, and concentrated to provide 2-((1S,5S)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile as a golden oil. This secondary amine was used directly in the next step without purification. 1H-NMR (400 MHz; CDCl3): δ 7.27–7.23 (m, 1H), 6.84 (d, J = 7.9 Hz, 1H), 6.80 (t, J = 2.0 Hz, 1H), 6.76 (dd, J = 8.1, 2.3 Hz, 1H), 4.58 (s, 1H), 4.29 (bs, 1H), 3.76 (s, 3H), 3.45–3.38 (m, 1H), 2.93 (dt, J = 13.1, 6.3 Hz, 1H), 2.37–2.22 (m, 3H), 2.15–2.07 (m, 4H), 1.88–1.78 (m, 1H), 1.75–1.69 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 175.31, 159.51, 147.07, 129.36, 119.51, 116.54, 113.80, 111.26, 92.18, 55.22, 53.91, 46.09, 41.81, 41.51, 39.51, 34.61, 20.04. HRMS-ESI (m/z): [M+H]+ calcd for C17H21N2O: 269.1654, found: 269.1653, [α]20D −69.2° (c 1.11, CHCl3).
To a flask were added K2CO3 (375 mg, 2.71 mmol, 2 equiv), the secondary amine obtained above (364 mg, 1.36 mmol, 1 equiv), phenethyl bromide (278 mL, 2.03 mmol, 1.5 equiv), and MeCN (3.2 mL). The suspension was heated to reflux overnight, and the solids were filtered off and washed with MeCN. The filtrate was concentrated and purified by chromatography with 0 to 20% EtOAc/hexane to obtain the tertiary amine 27 as a colorless oil (449 mg, 891H-NMR (400 MHz; CDCl3): δ 7.31–7.18 (m, 6H), 6.89–6.79 (m, 3H), 4.62 (s, 1H), 4.11–4.09 (m, 1H), 3.80 (s, 3H), 3.16–3.10 (m, 1H), 2.93–2.84 (m, 4H), 2.78–2.72 (m, 1H), 2.39–2.35 (m, 1H), 2.24–2.05 (m, 5H), 1.70–1.66 (m, 1H), 1.65–1.54 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 173.79, 159.51, 147.46, 140.15, 129.35, 128.77, 128.36, 126.03, 119.68, 116.75, 113.92, 111.27, 93.40, 60.44, 58.82, 55.24, 48.28, 45.79, 39.58, 38.77, 34.49, 32.58, 19.49. HRMS-ESI (m/z): [M+H]+ calcd for C25H29N2O: 373.2280, found: 373.2281, [α]20D –11.6° (c 1.00, CHCl3).
2-((1S,5S)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile (28). A solution of 27 (782 mg, 2.10 mmol, 1 equiv) in dichloromethane (47 mL) was prepared and cooled to −78 °C before BBr3 (1.01 mL, 10.5 mmol, 5 equiv) was added dropwise. After 15 min, the cooling bath was removed, and the solution was allowed to stir at room temperature for an additional 2 h. The solvent was removed in vacuo, and the residue was taken up in H2O. The pH was adjusted to 9 with NH4OH, and the contents were stirred for 30 min to break up borane complexes. The aqueous layer was extracted with CHCl3 (3x), and the combined organic layers were dried over Na2SO4. The solids were filtered off, and the filtrate was concentrated then purified by chromatography using 0 to 20% EtOAc/hexane to produce a white foam (487 mg, 65%). Crystallization was obtained by dissolving 100 mg of product in minimal aq MeOH then adding 2 N HCl in Et2O (2 equiv) to produce 28 as a white solid (65 mg, 63% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.32–7.19 (m, 6H), 6.86–6.84 (m, 1H), 6.77 (t, J = 2.1 Hz, 1H), 6.74 (dd, J = 7.8, 2.2 Hz, 1H), 4.65 (s, 1H), 4.13–4.12 (m, 1H), 3.19–3.13 (m, 1H), 2.94–2.85 (m, 4H), 2.81–2.74 (m1H), 2.42–2.36 (m, 1H), 2.24–2.08 (m, 5H), 1.72–1.63 (m, 1H), 1.61–1.57 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 173.76, 155.83, 147.79, 140.15, 129.72, 128.91, 128.54, 126.24, 119.66, 116.85, 114.65, 114.08, 93.65, 60.51, 58.89, 48.39, 45.86, 39.70, 38.71, 34.54, 32.47, 19.74. HRMS-ESI (m/z): [M+H]+ calcd for C24H27N2O: 359.2123, found: 359.2122, mp 152–153 °C, [α]20D –11.0° (c 0.99, CHCl3). CHN calc. for C24H27ClN2O · 0.15 H2O: C, 72.49%; H, 6.92%; N, 7.04%; found C, 72.48%; H, 6.90%; N, 7.07%.
tert-Butyl (1R,5R)-5-(3-methoxyphenyl)-9-oxo-2-azabicyclo[3.3.1]nonane-2-carboxylate (30). The procedure followed that of 25 on a 3.88 g scale to obtain 30 as a yellow oil (4.26 g, 78%). 1H-NMR (400 MHz; CDCl3): δ 7.30–7.26 (m, 1H), 6.82–6.76 (m, 3H), 4.31 (bs, 1H), 4.24–4.19 (m, 1H), 3.80 (s, 3H), 3.22–3.15 (m, 1H), 2.56–2.48 (m, 2H), 2.39–2.31 (m, 2H), 2.21–2.18 (m, 2H), 1.80–1.72 (m, 2H), 1.49 (s, 9H). 13C-NMR (101 MHz; CDCl3): δ 211.81, 159.25, 154.62, 145.59, 129.05, 119.36, 113.52, 111.31, 80.30, 63.62, 55.09, 52.81, 40.98, 40.64, 38.09, 35.54, 28.39, 17.56. HRMS-ESI (m/z): [M+Na]+ calcd for C20H27NO4Na: 368.1838, found: 368.1833 [α]20D +42.9° (c 1.13, CHCl3).
tert-Butyl (1R,5R)-9-(cyanomethylene)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonane-2-carboxylate (31). The procedure followed that of 26 on a 4.26 g scale to obtain 31 as a white solid (2.91 g, 64%). 1H-NMR (400 MHz; CDCl3): δ 7.23 (t, J = 7.9 Hz, 1H), 6.78–6.74 (m, 3H), 5.11–5.05 (m, 1H), 4.55 (s, 1H), 4.03–3.94 (m, 1H), 3.74 (s, 3H), 3.14–3.08 (m, 1H), 2.37–2.34 (m, 1H), 2.24–2.20 (m, 2H), 2.06–1.97 (m, 3H), 1.67–1.66 (m, 1H), 1.46 (s, 10H). 13C-NMR (101 MHz; CDCl3): δ 171.58, 159.77, 155.04, 147.40, 129.75, 119.59, 116.06, 113.94, 111.56, 94.92, 80.61, 55.64, 55.38, 45.79, 40.55, 39.89, 38.45, 34.22, 28.59, 18.06. HRMS-ESI (m/z): [M+Na]+ calcd for C22H28N2O3Na: 391.1998, found: 391.1998, mp 123–126 °C, [α]20D +44.0° (c 1.02, CHCl3).
2-((1R,5R)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile (32). Using the N-Boc 31 to obtain the secondary amine (1R,5R)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile on a 2.50 g scale, the procedure followed that of 26 to give rise to the secondary amine, leading to 27. The secondary amine 2-((1R,5R)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile was obtained as a yellow oil and was used directly in the next step without purification. 1H-NMR (400 MHz; CDCl3): δ 7.27–7.23 (m, 1H), 6.84 (d, J = 7.9 Hz, 1H), 6.80 (t, J = 2.0 Hz, 1H), 6.76 (dd, J = 8.1, 2.3 Hz, 1H), 4.58 (s, 1H), 4.29 (bs, 1H), 3.76 (s, 3H), 3.45–3.38 (m, 1H), 2.93 (dt, J = 13.1, 6.3 Hz, 1H), 2.37–2.22 (m, 3H), 2.15–2.07 (m, 4H), 1.88–1.78 (m, 1H), 1.75–1.69 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 175.31, 159.51, 147.07, 129.36, 119.51, 116.54, 113.80, 111.26, 92.18, 55.22, 53.91, 46.09, 41.81, 41.51, 39.51, 34.61, 20.04. HRMS-ESI (m/z): [M+H]+ calcd for C17H21N2O: 269.1654, found: 269.1654, [α]20D +66.0° (c 0.98, CHCl3).
Using the 1R,5R secondary amine above, the procedure for the tertiary amine 32 followed that of the preparation of 27 on a 1.89 g scale. A colorless oil (32, 2.32 g, 88%) was obtained. 1H-NMR (400 MHz; CDCl3): δ 7.31–7.18 (m, 6H), 6.89–6.79 (m, 3H), 4.62 (s, 1H), 4.11–4.09 (m, 1H), 3.80 (s, 3H), 3.16–3.10 (m, 1H), 2.93–2.84 (m, 4H), 2.78–2.72 (m, 1H), 2.39–2.35 (m, 1H), 2.24–2.05 (m, 5H), 1.70–1.66 (m, 1H), 1.65–1.54 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 173.79, 159.51, 147.46, 140.15, 129.35, 128.77, 128.36, 126.03, 119.68, 116.75, 113.92, 111.27, 93.40, 60.44, 58.82, 55.24, 48.28, 45.79, 39.58, 38.77, 34.49, 32.58, 19.49. HRMS-ESI (m/z): [M+H]+ calcd for C25H29N2O: 373.2280, found: 373.2275, [α]20D +12.3° (c 1.09, CHCl3).
2-((1R,5R)-5-(3-hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-ylidene)acetonitrile (33). Using 32, the procedure followed that of the preparation of 28 on a 500 mg scale. A white foam (292 g, 61%) was obtained. Crystallization produced 33 as a white solid (76 mg, 68% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.30–7.17 (m, 6H), 6.84–6.82 (m, 1H), 6.76 (t, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.0, 1.9 Hz, 1H), 4.64 (s, 1H), 4.13–4.11 (m, 1H), 3.19–3.13 (m, 1H), 2.91–2.83 (m, 4H), 2.80–2.74 (m, 1H), 2.41–2.34 (m, 1H), 2.23–2.07 (m, 5H), 1.71–1.66 (m, 1H), 1.63–1.55 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 173.58, 155.79, 147.52, 139.93, 129.55, 128.74, 128.39, 126.1, 119.39, 116.69, 114.54, 113.98, 93.49, 60.29, 58.67, 48.22, 45.69, 39.54, 38.47, 34.33, 32.17, 19.64. HRMS-ESI (m/z): [M+H]+ calcd for C24H27N2O: 359.2123, found: 359.2122, mp 154 °C, [α]20D +11.5° (c 1.02, CHCl3). CHN calc. for C24H27ClN2O · 1.45 H2O: C, 68.46%; H, 7.16%; N, 6.65%; found C, 68.19%; H, 6.87%; N, 6.44%.
2-((1S,5R,9S)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (34). We dissolved the unsaturated nitrile 28 (170 mg, 0.474 mmol, 1 equiv) in EtOH (33 mL) and passed it through the H-cube flow reactor under the following conditions: 70 °C, 50 bar H2, 10% Pd/C, 1.2 mL/min flow rate. The reaction mixture was concentrated and purified by chromatography with 0 to 8% CMA/CHCl3 to give rise to a white foam (30 mg, 18%). Crystallization was performed by dissolving 30 mg of product in minimal aq MeOH, then adding 2 N HCl in Et2O (2 equiv) to produce 34 as a white solid (23 mg, 70% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.31–7.16 (m, 6H), 6.92 (bs, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.75 (dd, J = 8.0, 2.2 Hz, 1H), 3.34 (d, J = 1.3 Hz, 1H), 3.19–3.17 (m, 1H), 3.17–3.01 (m, 1H), 2.99–2.89 (m, 5H), 2.41–2.39 (m, 1H), 2.39–2.38 (m, 1H), 2.13–1.61 (m, 7H), 1.55–1.49 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 156.49, 149.10, 139.63, 129.89, 128.72, 128.48, 126.25, 118.73, 117.20, 113.83, 112.86, 57.82, 54.76, 49.46, 42.09, 40.25, 37.62, 33.90, 27.93, 21.09, 18.11, 16.52. HRMS-ESI (m/z): [M+H]+ calcd for C24H29N2O: 361.2280, found: 361.2282, mp 63–66 °C, [α]20D −2.43° (c 1.30, CHCl3). CHN calc. for C24H29ClN2O: C, 72.62%; H, 7.36%; N, 7.06%; found C, 72.69%; H, 7.24%; N, 7.11%.
2-((1R,5S,9R)-5-(3-hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (35). Using 33, the saturated cyanomethyl compound was prepared analogously to 34 on a 479 mg scale to obtain 35 as a white foam (205 mg, 43%). Hydrochloride salt formation produced the 1R,5S,9R-saturated nitrile 35 as a white solid (180 mg, 80% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.31–7.16 (m, 6H), 6.92 (bs, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.75 (dd, J = 8.0, 2.2 Hz, 1H), 3.34 (d, J = 1.3 Hz, 1H), 3.19–3.17 (m, 1H), 3.17–3.01 (m, 1H), 2.99–2.89 (m, 5H), 2.41–2.39 (m, 1H), 2.39–2.38 (m, 1H), 2.13–1.61 (m, 7H), 1.55–1.49 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 156.49, 149.10, 139.63, 129.89, 128.72, 128.48, 126.25, 118.73, 117.20, 113.83, 112.86, 57.82, 54.76, 49.46, 42.09, 40.25, 37.62, 33.90, 27.93, 21.09, 18.11, 16.52. HRMS-ESI (m/z): [M+H]+ calcd for C24H29N2O: 361.2280, found: 361.2280, mp 63–66 °C, [α]20D +3.75° (c 0.99, CHCl3). CHN calc. for C24H29ClN2O · 0.1 H2O: C, 72.14%; H, 7.20%; N, 6.99%; found C, 72.29%; H, 7.38%; N, 7.03%.
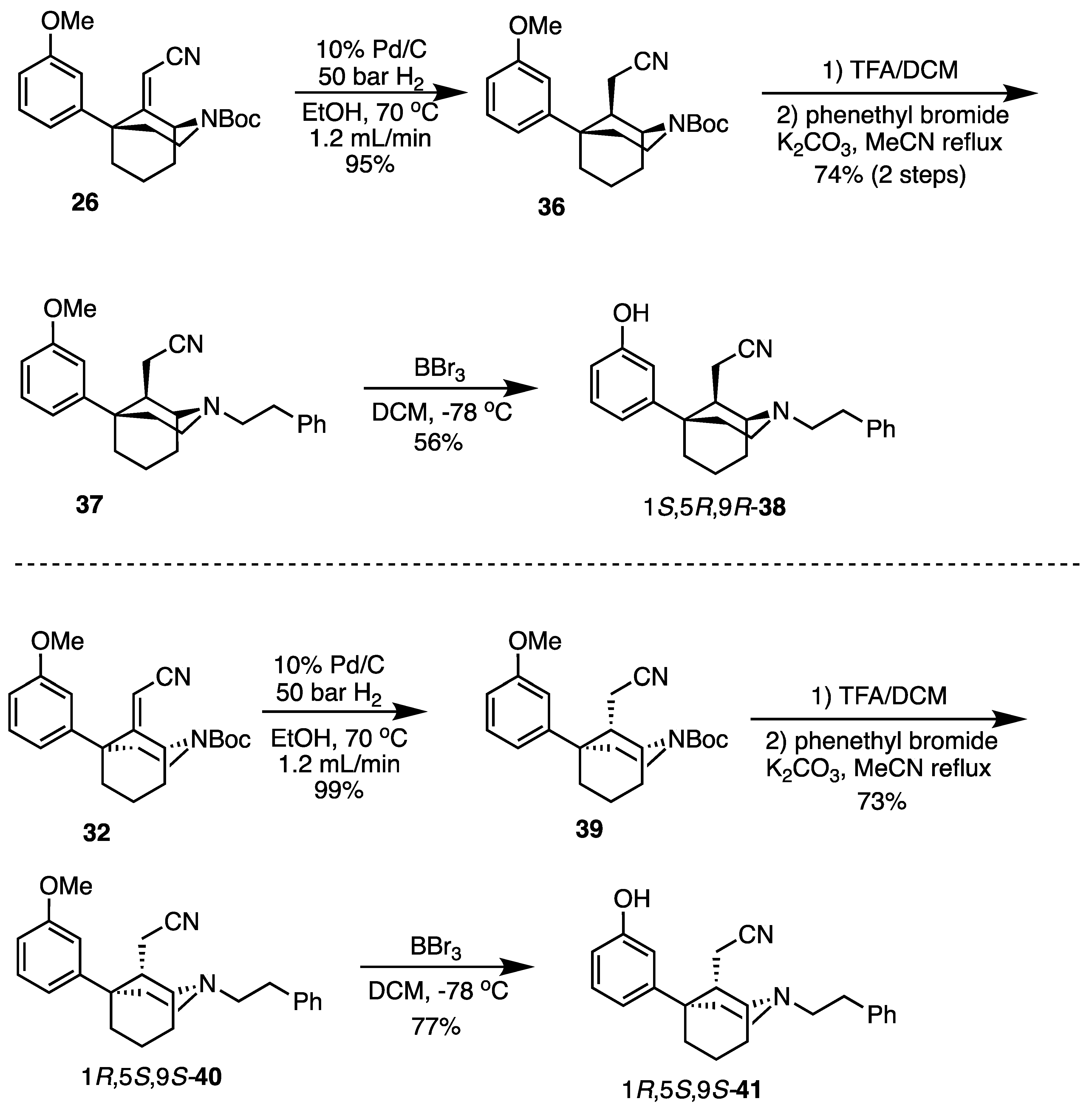
tert-Butyl (1S,5R,9R)-9-(cyanomethyl)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonane-2-carboxylate (36). The procedure followed that of the preparation of 34 on a 475 mg scale to obtain 36 as a white solid (453 mg, 95%). 1H-NMR (400 MHz; CDCl3): δ 7.29–7.24 (m, 1H), 6.84 (t, J = 6.7 Hz, 1H), 6.80–6.74 (m, 2H), 4.56 (d, J = 2.6 Hz, 1H), 4.15–4.09 (m, 1H), 3.79 (s, 3H), 3.58–3.49 (m, 1H), 2.43 (d, J = 11.7 Hz, 1H), 2.28–2.21 (m, 1H), 2.11–1.69 (m, 9H), 1.47 (s, 9H). 13C-NMR (101 MHz; CDCl3): δ 159.93, 155.77, 148.94, 129.87, 119.19, 117.14, 111.92, 110.79, 80.21, 55.19, 48.96, 42.46, 41.76, 40.54, 38.22, 31.17, 28.42, 27.34, 21.69, 16.64. HRMS-ESI (m/z): [M+Na]+ calcd for C22H30N2O3Na: 393.2154, found: 393.2152, mp 143–146 °C, [α]20D −63.8° (c 1.09, CHCl3).
2-((1S,5R,9R)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (37). We combined 36 (453 mg, 1.22 mmol, 1 equiv), dichloromethane (4.5 mL), and TFA (1.88 mL, 24.5 mmol, 20 equiv) in a flask and stirred them at room temperature. After 30 min, TLC showed no starting material. The reaction was quenched with NaHCO3; the aqueous layer was extracted with dichloromethane 3 times; and the combined organic layers were dried over Na2SO4. The drying agent was filtered off, and the filtrate was concentrated to obtain the secondary amine, 2-((1S,5R,9R)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile, as a golden oil, which was used directly in the next step without purification. 1H-NMR (400 MHz; CDCl3): δ 7.24 (t, J = 8.0 Hz, 1H), 6.86 (dd, J = 7.6, 1.3 Hz, 1H), 6.79 (t, J = 2.1 Hz, 1H), 6.72 (dd, J = 8.1, 2.4 Hz, 1H), 3.78 (s, 3H), 3.63 (td, J = 12.5, 5.5 Hz, 1H), 3.32 (d, J = 3.1 Hz, 1H), 3.04 (dd, J = 12.4, 7.6 Hz, 1H), 2.76 (dd, J = 16.7, 11.6 Hz, 1H), 2.35 (d, J = 11.5 Hz, 1H), 2.17–1.72 (m, 10H). 13C-NMR (101 MHz; CDCl3): δ 159.82, 150.24, 129.66, 120.12, 117.28, 111.89, 110.55, 55.18, 48.97, 42.46, 41.98, 41.67, 38.53, 34.04, 28.85, 22.95, 16.72. HRMS-ESI (m/z): [M+H]+ calcd for C17H23N2O: 271.1810, found: 271.1806, [α]20D −8.6° (c 1.03, CHCl3).
The conversion of the secondary amine to the tertiary amine followed the preparation of 27 on a 331 mg scale to obtain 37 as a yellow oil (345 mg, 75%). 1H-NMR (400 MHz; CDCl3): δ 7.29–7.15 (m, 6H), 6.87–6.85 (m, 1H), 6.80 (s, 1H), 6.74 (d, J = 8.0 Hz, 1H), 3.79 (s, 3H), 3.15 (s, 1H), 3.09–2.96 (m, 2H), 2.86–2.72 (m, 5H), 2.40 (d, J = 10.8 Hz, 1H), 2.34–2.30 (m, 1H), 2.08–1.66 (m, 7H), 1.56–1.49 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 159.83, 150.13, 140.56, 129.66, 128.67, 128.29, 125.91, 120.48, 117.44, 112.00, 110.56, 56.48, 55.19, 54.13, 48.25, 43.85, 42.29, 38.45, 34.35, 29.17, 25.57, 23.06, 16.37. HRMS-ESI (m/z): [M+H]+ calcd for C25H31N2O: 375.2436, found: 375.2437 [α]20D −9.5° (c 1.05, CHCl3).
2-((1S,5R,9R)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (38). Using 37, the procedure followed that of the preparation of 8 on a 345 mg scale to obtain 38 as a white foam (156 mg, 47%). Crystallization was performed by dissolving 100 mg of product in minimal aqueous MeOH and then adding 2 N HCl in Et2O (2 equiv) to produce a white solid (82 mg, 74% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.32–7.19 (m, 6H), 6.88 (d, J = 7.9 Hz, 1H), 6.82 (s, 1H), 6.71 (dd, J = 7.9, 2.2 Hz, 1H), 3.31 (s, 1H), 3.08–3.04 (m, 2H), 2.94–2.82 (m, 4H), 2.73–2.69 (m, 1H), 2.29–2.22 (m, 2H), 2.14–1.77 (m, 7H), 1.50–1.49 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 156.83, 149.29, 139.88, 130.03, 128.74, 128.50, 126.25, 118.95, 117.03, 114.09, 113.39, 57.92, 54.59, 49.41, 42.27, 40.43, 37.68, 33.99, 27.99, 21.29, 18.11, 16.58.HRMS-ESI (m/z): [M+H]+ calcd for C24H29N2O: 361.2280, found: 361.2282, mp 73–76 °C, [α]20D +14.0° (c 1.01, CHCl3). CHN calc. for C24H29ClN2O · 0.5 H2O: C, 71.01%; H, 7.45%; N, 6.90%; found C, 71.10%; H, 7.33%; N, 6.76%.
tert-Butyl (1R,5S,9S)-9-(cyanomethyl)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonane-2-carboxylate (39). The procedure followed that of the preparation of 34 on a 155 mg scale to obtain 39 as a white foam (156 mg, 99%). 1H-NMR (400 MHz; CDCl3): δ 7.28–7.24 (m, 1H), 6.84 (t, J = 6.6 Hz, 1H), 6.79–6.73 (m, 2H), 4.56 (d, J = 2.5 Hz, 1H), 4.14–4.09 (m, 1H), 3.78 (s, 3H), 3.57–3.49 (m, 1H), 2.43 (d, J = 11.6 Hz, 1H), 2.27–2.20 (m, 1H), 2.19–1.69 (m, 9H), 1.47 (s, 9H). 13C-NMR (101 MHz; CDCl3): δ 159.93, 155.78, 148.95, 129.87, 119.20, 117.14, 111.92, 110.79, 80.22, 55.19, 48.97, 42.47, 41.77, 40.55, 38.23, 31.17, 28.42, 27.34, 21.69, 16.65. HRMS-ESI (m/z): [M+Na]+ calcd for C22H30N2O3Na: 393.2154, found: 393.2151, mp 144–146 °C, [α]20D +64.6° (c 0.99, CHCl3).
2-((1R,5S,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (40). The procedure to obtain the intermediate secondary amine, 2-((1R,5S,9S)-5-(3-methoxyphenyl)-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile, followed that of the preparation of 37 on a 156 mg scale to obtain a golden oil, which was used directly in the next step without purification. 1H-NMR (400 MHz; CDCl3): δ 7.24 (t, J = 8.0 Hz, 1H), 6.86 (dd, J = 7.6, 1.3 Hz, 1H), 6.79 (t, J = 2.1 Hz, 1H), 6.72 (dd, J = 8.1, 2.4 Hz, 1H), 3.78 (s, 3H), 3.63 (td, J = 12.5, 5.5 Hz, 1H), 3.32 (d, J = 3.1 Hz, 1H), 3.04 (dd, J = 12.4, 7.6 Hz, 1H), 2.76 (dd, J = 16.7, 11.6 Hz, 1H), 2.35 (d, J = 11.5 Hz, 1H), 2.17–1.72 (m, 10H). 13C-NMR (101 MHz; CDCl3): δ 159.82, 150.24, 129.66, 120.12, 117.28, 111.89, 110.55, 55.18, 48.97, 42.46, 41.98, 41.67, 38.53, 34.04, 28.85, 22.95, 16.72. HRMS-ESI (m/z): [M+H]+ calcd for C17H23N2O: 271.1810, found: 271.1810 [α]20D +9.81° (c 1.03, CHCl3).
The procedure to convert the secondary amine to the tertiary amine 40 followed that of the preparation of 37 on a 114 mg scale to obtain the tertiary amine 40 as a yellow oil (116 mg, 73%). 1H-NMR (400 MHz; CDCl3): δ 7.32–7.18 (m, 6H), 6.88 (d, J = 7.9 Hz, 1H), 6.83 (s, 1H), 6.76 (dd, J = 8.1, 2.1 Hz, 1H), 3.81 (s, 3H), 3.18 (s, 1H), 3.13–2.99 (m, 2H), 2.89–2.74 (m, 5H), 2.43 (m, J = 11.1 Hz, 1H), 2.34 (dd, J = 14.0, 3.4 Hz, 1H), 2.10–1.69 (m, 7H), 1.59–1.52 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 159.84, 150.13, 140.56, 129.67, 128.68, 128.29, 125.92, 120.47, 117.44, 112.00, 110.58, 56.47, 55.20, 54.13, 48.26, 43.85, 42.28, 38.45, 34.35, 29.17, 25.56, 23.06, 16.38. HRMS-ESI (m/z): [M+H]+ calcd for C25H31N2O: 375.2436, found: 375.2436, [α]20D +8.8° (c 1.07, CHCl3).
2-((1R,5S,9S)-5-(3-Hydroxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)acetonitrile (41). Using 40, the procedure followed that of the preparation of 8 on a 116 mg scale to obtain the phenol 41 as a white foam (53 mg, 77%). Crystallization was performed by dissolving 63 mg of product in minimal aqueous MeOH and then adding 2 N HCl in Et2O (2 equiv) to afford a white solid (65 mg, 63% recovery). 1H-NMR (400 MHz; CDCl3): δ 7.32–7.19 (m, 6H), 6.88 (d, J = 7.9 Hz, 1H), 6.82 (s, 1H), 6.71 (dd, J = 7.9, 2.2 Hz, 1H), 3.31 (s, 1H), 3.08–3.04 (m, 2H), 2.94–2.82 (m, 4H), 2.73–2.69 (m1H), 2.29–2.22 (m, 2H), 2.14–1.77 (m, 7H), 1.50–1.49 (m, 1H). 13C-NMR (101 MHz; CDCl3): δ 156.83, 149.29, 139.88, 130.03, 128.74, 128.50, 126.25, 118.95, 117.03, 114.09, 113.39, 57.92, 54.59, 49.41, 42.27, 40.43, 37.68, 33.99, 27.99, 21.29, 18.11, 16.58. HRMS-ESI (m/z): [M+H]+ calcd for C24H29N2O: 361.2280, found: 361.2283, mp 73–76 °C, [α]20D −10.5° (c 0.93, CHCl3). CHN calc. for C24H29ClN2O · 0.35 H2O: C, 71.48%; H, 7.42%; N, 6.95%; found C, 71.49%; H, 7.28%; N, 6.83%.
1-(1S,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)-N-methylmethanamine (44) and 1-(1S,5R,9R)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)-N-methylmethanamine (45). Enol ether 42 (1.00 g, 1 equiv, 2.65 mmol) was dissolved in tetrahydrofuran (7.5 mL) and 6 M HCl (10 mL). The reaction was stirred under argon at room temperature overnight. The reaction was cooled down to 0 °C and basified with 7 N NH4OH in methanol (pH 8.5). The aqueous solution was then extracted with CHCl3 (5 × 20 mL). The organic layers were combined and washed with water (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, and filtered. The solvent was removed under vacuum conditions to give rise to the mixture of aldehydes (43) as a bluish green oil. The aldehydes were dried under high vacuum for 30 min and used immediately in the next step without purification. The mixture of aldehydes was dissolved in ethanol (10 mL) and transferred to a pressure flask. Titanium(IV) isopropoxide (1.51 g, 1.61 mL, 2 equiv, 5.30 mmol) and 33% methylamine solution in ethanol (2.49 g, 3.30 mL, 10 equiv, 26.5 mmol) were added to the flask. The pressure flask was flushed with argon, sealed, and stirred at 50 °C overnight. The reaction was cooled to 0 °C. Sodium borohydride (200 mg, 2 equiv, 5.30 mmol) was then added portionwise, and the reaction was stirred for 2 h. Water was added to quench the reaction, and the pH was adjusted to 9 by adding NH4OH solution. The reaction mixture was filtered through celite and washed several times with ethyl acetate. The filtrate was transferred to a separatory funnel. The aqueous phase was extracted with ethyl acetate (3 × 20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4, and filtered. The solvent was removed under vacuum conditions to yield a dark-yellow residue. The residue was separated and purified by flash chromatography (slow gradient 0–20% CHCl3/MeOH/NH4OH) to give rise to the diastereomers 1S,5R,9S-44 and 1S,5R,9R-45 in a 75% yield for the combined diastereomers.
For 1S,5R,9S-44, the product was isolated as a pale-yellow oil (437.5 mg). 1H NMR (400 MHz; CDCl3) δ: 7.27–7.24 (m, 3H), 7.20 (dd, J = 7.1, 1.9 Hz, 2H), 7.15 (t, J = 7.1 Hz, 1H), 6.92 (d, J = 7.9 Hz, 1H), 6.86 (t, J = 1.9 Hz, 1H), 6.70 (dd, J = 8.1, 2.3 Hz, 1H), 3.79 (s, 3H), 3.05 (dd, J = 11.4, 4.8 Hz, 2H), 2.98 (d, J = 2.8 Hz, 1H), 2.85–2.66 (m, 5H), 2.30–2.25 (m, 2H), 2.10 (s, 3H), 2.08 (d, J = 3.8 Hz, 1H), 1.95 (dt, J = 11.6, 4.4 Hz, 2H), 1.87 (dt, J = 12.5, 6.1 Hz, 1H), 1.82–1.77 (m, 1H), 1.70–1.62 (m, 3H), 1.49–1.42 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 159.46, 151.76, 141.96, 129.02, 128.67, 128.10, 125.76, 117.86, 112.03, 109.94, 56.69, 55.11, 52.96, 49.37, 48.81, 45.83, 42.86, 38.21, 36.87, 34.04, 30.14, 25.81, 23.31. HRMS-ESI (m/z): [M+H]+ calc for C25H35N2O: 379.2749; found, 379.2747.
For 1S,5R,9R-45, the product was obtained as a pale-yellow oil (314.5 mg). 1H NMR (400 MHz; CDCl3) δ: 7.30–7.17 (m, 6H), 7.01 (d, J = 8.0 Hz, 1H), 6.95 (s, 1H), 6.72 (dd, J = 8.1, 2.2 Hz, 1H), 3.79 (s, 3H), 3.20 (bs, 1H), 3.01 (dd, J = 10.1, 3.8 Hz, 2H), 2.88–2.77 (m, 4H), 2.49 (t, J = 11.1 Hz, 1H), 2.39 (d, J = 10.6 Hz, 1H), 2.26 (s, 3H), 2.24–2.20 (m, 2H), 2.01–1.91 (m, 4H), 1.81–1.71 (m, 2H), 1.55–1.49 (m, 2H). 13C NMR (101 MHz; CDCl3): δ 159.51, 151.56, 140.69, 129.09, 128.73, 128.31, 125.91, 117.99, 112.08, 110.31, 58.26, 55.15, 53.57, 50.00, 49.76, 45.48, 41.57, 37.84, 36.70, 34.65, 29.32, 21.72, 18.58. HRMS-ESI (m/z): [M+H]+ calc for C25H35N2O: 379.2749; found, 379.2747.
1-((1S,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)-N-methylmethanamine (46). The ether 44 (320 mg, 1 equiv, 845 µmol) was dissolved in anhydrous dichloromethane (20 mL), and the solution was cooled to −78 °C. To this cooled solution was added boron tribromide (1.06 mg, 400 µL, 5 equiv, 4.23 mmol) dropwise. The reaction was stirred at −78 °C for 15 min and then warmed up to room temperature for 2 h. The reaction was cooled to 0 °C, quenched with MeOH, and stirred for 30 min. Then, 2 N HCl (10 mL) was added and distilled at 100 °C (to remove the volatile solvents) under nitrogen conditions for 1 h. The reaction was allowed to cool to room temperature and basified to pH 10 using aqueous NH4OH. The aqueous phase was extracted with CHCl3:MeOH (9:1) (3 × 20 mL). The organic layers were combined and washed with brine (20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The solvent was removed in vacuo to give rise to a pale-yellow oil. The crude sample was purified using flash chromatography (0–20% CHCl3/MeOH/NH4OH), and 46 was obtained as a pale-yellow oil (200 mg, 65%). 1H NMR (400 MHz; CDCl3): δ 7.27–7.10 (m, 6H), 6.78–6.76 (m, 2H), 6.58 (dd, J = 8.1, 1.5 Hz, 1H), 3.47 (bs, 1H), 3.10–3.02 (m, 2H), 2.93 (s, 1H), 2.84–2.73 (m, 4H), 2.67 (dd, J = 11.7, 9.0 Hz, 1H), 2.27–2.20 (m, 3H), 2.13 (dd, J = 11.8, 4.0 Hz, 1H), 2.06 (s, 3H), 1.97–1.88 (m, 1H), 1.87–1.85 (m, 2H), 1.84–1.72 (m, 1H), 1.70–1.61 (m, 1H), 1.45–1.38 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.32, 151.11, 140.82, 129.26, 128.63, 128.17, 125.85, 116.32, 113.51, 112.78, 56.58, 53.58, 49.70, 48.74, 45.04, 42.86, 38.07, 36.30, 33.92, 29.98, 25.73, 23.28. HRMS-ESI (m/z): [M+H]+ calc for C24H33N2O: 365.2593; found, 365.2593. [α]20D +8.8 (c 1.1, CHCl3), mp (dioxalate salt): 143.4–145.1 °C. CHN calcd for C28H36N2O9 • 0.7H2O: C, 60.36; H, 6.77; N, 5.03. Found: C, 60.29; H, 6.72; N, 5.12.
3-((1S,5R,9R)-9-((Methylamino)methyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (47). The ether 45 (180 mg, 1 equiv, 475 µmol) was dissolved in anhydrous dichloromethane (15 mL), and the solution was cooled to −78 °C. To this cooled solution was added boron tribromide (596 mg, 225 µL, 5 equiv, 2.38 mmol) dropwise. The reaction was stirred at −78 °C for 15 min and then warmed up to room temperature for 2 h. The reaction was cooled to 0 °C, quenched with MeOH, and stirred for 30 min. Then, 2 N HCl (10 mL) was added and distilled at 100 °C (to remove the volatile solvents) under nitrogen conditions for 1 h. The reaction was allowed to cool to room temperature and basified to pH 10 using aqueous NH4OH. The aqueous phase was extracted with CHCl3:MeOH (9:1) (3 × 20 mL). The organic layers were combined and washed with brine (20 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The solvent was removed in vacuo to give rise to a pale-yellow foam. The crude sample was purified using flash chromatography (0–20% CHCl3/MeOH/NH4OH), and 47 was obtained as an off-white foam (140 mg, 81%). 1H NMR (400 MHz; CD3OD): δ 7.28–7.26 (m, 1H), 7.24–7.22 (m, 3H), 7.18–7.14 (m, 1H), 7.14–7.10 (m, 1H), 6.88 (d, J = 8.0 Hz, 1H), 6.84 (t, J = 2.0 Hz, 1H), 6.60 (dd, J = 7.9, 2.2 Hz, 1H), 3.29 (s, 1H), 3.06 (td, J = 12.0, 5.0 Hz, 1H), 2.99–2.95 (m, 1H), 2.90–2.78 (m, 4H), 2.58 (dd, J = 12.1, 10.6 Hz, 1H), 2.43 (dd, J = 10.4, 2.7 Hz, 1H), 2.25–2.17 (m, 5H), 2.04–1.95 (m, 4), 1.84–1.79 (m, 1H), 1.72 (ddd, J = 14.1, 4.9, 2.0 Hz, 1H), 1.59–1.56 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.65, 150.96, 140.34, 129.28, 128.71, 128.34, 125.99, 116.40, 114.01, 113.08, 58.26, 55.12, 50.67, 49.39, 44.88, 40.54, 37.51, 36.08, 34.14, 29.14, 21.39, 19.08. HRMS-ESI (m/z): [M+H]+ calc for C24H33N2O: 365.2593; found, 365.2594. [α]20D −30.3 (c 1.0, CHCl3). mp: 134.2–135.3 °C. CHN calc for C24H32N2O • 0.03 H2O • 0.24 CHCl3: C, 73.95; H, 8.27; N, 7.12. Found: C, 74.09; H, 8.36; N, 6.95.
(1R,5R)-9-(Methoxymethylene)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonane (
48) [
5]. 1
R,5
R-
4 (2.20 g, 1 equiv, 2.86 mmol) was transferred to an oven-dried flask charged with (methoxymethyl)triphenylphosphonium chloride (6.47 g, 3 equiv, 8.58 mmol). The solution was cooled down to 0 °C, to which was added lithium bis(trimethylsilyl)amide (4.21 g, 25.2 mL, 1 molar, 4 equiv, 11.4 mmol) dropwise. The reaction was gradually allowed to warm to room temperature and stirred overnight. The reaction was then cooled down to 0 °C, quenched with MeOH, and stirred for 10 min. The reaction was concentrated under vacuum conditions, and the residue was taken up in water and chloroform. The pH of the aqueous layer was adjusted to 9 with a saturated aqueous NH
4OH solution. The aqueous phase was extracted with 9:1 CHCl
3/MeOH (3 × 25 mL), and the combined organic layer was dried over anhydrous Na
2SO
4, filtered, and concentrated under vacuum conditions. The residue was purified using flash chromatography (gradient 0–100% EtOAc in hexanes) to give rise to
48 as a mixture of E/Z isomers (1.70 g, 71%) that were immediately used in the next step.
1-(1R,5S,9R)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)-N-methylmethanamine (50) and 1-((1R,5S,9S)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-9-yl)-N-methylmethanamine (51). The enol ether 48 (0.700 g, 1 equiv, 1.85 mmol) was dissolved in tetrahydrofuran (5 mL), and 6 N HCl (15 mL) was added to it. The reaction was stirred under nitrogen conditions at room temperature overnight. The reaction was cooled down to 0 °C and basified with aqueous NH4OH. The aqueous solution was then extracted with CHCl3/MeOH (9:1) (5 × 10 mL). The organic layers were combined and washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum conditions to give rise to a mixture of aldehydes as a bluish green oil. The aldehydes 49 were immediately used in the next step without purification. The mixture of aldehydes, methylamine (2.31 mL, 33% in EtOH, 10 equiv, 18.5 mmol), and titanium(IV) isopropoxide (1.05 g, 1.12 mL, 2 equiv, 3.71 mmol) was added to a clean, dry-pressure vial. The reaction was stirred under argon at 60 °C overnight. The reaction was cooled to 0 °C, and sodium borohydride (140 mg, 2 equiv, 3.71 mmol) was added. The reaction was stirred for 2 h, quenched with water, and basified to pH 9 with an NH4OH solution. The solids were filtered off over celite, and the filtrate was extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with brine, dried over anhydrous MgSO4, and filtered, and the solvent was removed in vacuo to give rise to a yellow residue, a mixture of diastereomers that were separated using flash chromatography (0–5% CHCl3/MeOH/NH4OH) to give 50 (1R,5S,9R) and 51 (1R,5S,9S) as pale-yellow oils (0.301 g, 43%). 1H NMR (400 MHz; CDCl3): δ 7.28–7.25 (m, 3H), 7.22 (d, J = 8 Hz, 2H), 7.17 (t, J1 = 4 Hz, J2 = 8 Hz, 1H), 6.93 (d, J = 8Hz, 1H), 6.87 (s, 1H), 6.71 (d, J = 8 Hz, 1H), 3.80 (s, 3H), 3.10–3.05 (m, 2H), 2.99 (s, 1H), 2.85–2.68 (m, 5H), 2.31–2.27 (m, 2H), 2.12 (s, 3H), 2.11–2.08 (m, 1H), 1.98–1.94 (m, 2H), 1.92–1.79 (m, 2H), 1.82–1.64 (m, 3H), 1.49–1.41 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 159.46, 151.78, 140.97, 129.02, 128.67, 128.10, 125.76, 117.87, 112.03, 109.93, 56.69, 55.11, 52.95, 49.38, 48.82, 45.87, 42.87, 38.21, 36.90, 34.05, 30.15, 25.82, 23.32. HRMS-ESI (m/z): [M+H]+ calc for C25H35N2O: 379.2749; found, 379.2756.
For 1R,5S,9S-51, the product was obtained as a pale-yellow oil. 1H NMR (400 MHz; CDCl3) δ: 7.30–7.17 (m, 6H), 7.01 (d, J = 8.0 Hz, 1H), 6.95 (s, 1H), 6.72 (dd, J = 8.1, 2.2 Hz, 1H), 3.79 (s, 3H), 3.20 (bs, 1H), 3.03–2.99 (m, 2H), 2.88–2.77 (m, 4H), 2.49 (t, J = 11.1 Hz, 1H), 2.39–2.35 (m, 1H), 2.27 (s, 3H), 2.24–2.18 (m, 2H), 2.02–1.87 (m, 4H), 1.81–1.71 (m, 3H), 1.55–1.48 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 159.50, 151.61, 140.73, 129.08, 128.73, 128.30, 125.90, 117.99, 112.07, 110.29, 58.29, 55.15, 53.54, 50.03, 49.78, 45.45, 41.62, 37.86, 36.76, 34.70, 29.33, 21.74, 18.59.
3-((1R,5S,9R)-9-((Methylamino)methyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (52). The ether 50 (0.130 g, 1 equiv, 343 µmol) was dissolved in anhydrous dichloromethane (5 mL), and the solution was cooled to −78 °C. To this cooled solution was added boron tribromide (430 mg, 162 µL, 5 equiv, 1.72 mmol) dropwise. The reaction was stirred at −78 °C for 15 min and then warmed up to room temperature for 2 h. The reaction was cooled down to 0 °C, quenched with MeOH, and stirred for 30 min. Then, 1 N HCl (10 mL) was added and distilled at 100 °C (to remove the volatile solvents) under nitrogen conditions for 1 h. The reaction was allowed to cool down and basified to pH 8.5. The aqueous phase was extracted with CHCl3 (3 × 10 mL). The organic layers were combined and washed with brine (20 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was removed in vacuo to give rise to 52 as a pale-yellow oil (120 mg, 96%). 1H NMR (400 MHz; CDCl3): δ 7.28–7.10 (m, 6H), 6.77–6.76 (m, 2H), 6.58 (d, J = 8 Hz, 1H), 4.62 (bs, 1H), 3.07–3.02 (m, 2H), 2.93 (s, 1H), 2.84–2.73 (m, 4H), 2.69–2.64 (m, 1H), 2.31–2.18 (m, 3H), 2.14 (dd, J = 8 Hz, 1H), 2.06 (s, 3H), 1.96–1.79 (m, 3H), 1.72–1.68 (m, 1H), 1.64–1.60 (m, 1H), 1.43–1.39 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.48, 151.03, 140.82, 129.26, 128.63, 128.18, 125.86, 116.20, 113.58, 112.85, 56.58, 53.59, 49.64, 48.73, 44.85, 42.85, 38.04, 36.19, 33.89, 29.95, 25.68, 23.26. HRMS-ESI (m/z): [M+H]+ calc for C24H33N2O: 365.2593; found, 365.2591. [α]20D −7.7° (c 1.1, CHCl3). Dioxalate salt: mp 150–152 °C. CHN calcd for C28H36N2O9: C, 61.75; H, 6.66; N, 5.14. Found: C, 61.98; H, 6.93; N, 5.29.
3-((1R,5S,9S)-9-((Methylamino)methyl)-2-phenethyl-2-azabicyclo[3.3.1]nonan-5-yl)phenol (53). The ether 51 (630 mg, 1 equiv, 1.66 mmol) was dissolved in anhydrous dichloromethane (25 mL), and the solution was cooled to −78 °C. To this cooled solution was added boron tribromide (2.08 g, 787 µL, 5 equiv, 8.32 mmol) dropwise. The reaction was stirred at −78 °C for 15 min and then warmed up to room temperature for 2 h. The reaction was cooled down to 0 °C, quenched with MeOH, and stirred for 30 min. Then, 1 N HCl (20 mL) was added and distilled at 100 °C (to remove the volatile solvents) under nitrogen conditions for 1 h. The reaction was allowed to cool down and basified to pH 8.5. The aqueous phase was extracted with CHCl3 (3 × 20 mL). The organic layers were combined and washed with brine (20 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was removed in vacuo to give rise to 53 as a white foam (400 mg, 66%). 1H NMR (400 MHz; CD3OD): δ 7.28–7.22 (m, 4H), 7.18–7.14 (m, 1H), 7.12 (t, J = 8.0 Hz, 1H), 6.88 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 1.9 Hz, 1H), 6.59 (dd, J = 7.9, 1.9 Hz, 1H), 3.20 (s, 1H), 3.05 (td, J = 12.1, 5.0 Hz, 1H), 2.99–2.94 (m, 1H), 2.89–2.77 (m, 4H), 2.56 (t, J = 11.3 Hz, 1H), 2.43 (d, J = 10.4 Hz, 1H), 2.24–2.16 (m, 5H), 2.03–1.95 (m, 4H), 1.83–1.79 (m, 1H), 1.71 (dt, J = 11.7, 2.4 Hz, 1H), 1.62–1.55 (m, 1H). 13C NMR (101 MHz; CDCl3): δ 157.57, 150.99, 140.36, 129.28, 128.71, 128.34, 125.99, 116.46, 113.99, 113.04, 58.27, 55.15, 50.71, 49.39, 44.92, 40.54, 37.52, 36.12, 34.17, 29.14, 21.39, 19.10. HRMS-ESI (m/z): [M+H]+ calc for C24H33N2O: 365.2593; found, 365.2595. [α]20D +31.9° (c 1.0, CHCl3). Mp: 138.1–139.0 °C. CHN calcd for C24H32N2O • 0.05 H2O • 0.35 CHCl3: C, 71.82; H, 8.03; N, 6.88. Found: C, 71.71; H, 7.93; N, 7.06.


 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















