
Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship
by
 and
and
Anna Laura Sanna
1,
Tatiana Pachova
2,
Alessandra Catellani
3,
Arrigo Calzolari
3,* and
Giuseppe Sforazzini
1,2,*
1
Department of Chemical and Geological Sciences, Università degli Studi di Cagliari, SS 554, Bivio per Sestu, 09042 Cagliari, Italy
2
Laboratory of Macromolecular and Organic Materials, Institute of Material Science and Engineering, Ecole Polytechnique Federale de Lausanne (EPFL), 1015 Lausanne, Switzerland
3
CNR-NANO, Istituto Nanoscienze, Via Giuseppe Campi, 213, 41125 Modena, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2024, 29(9), 1929; https://doi.org/10.3390/molecules29091929
Submission received: 3 April 2024
/
Revised: 18 April 2024
/
Accepted: 19 April 2024
/
Published: 23 April 2024
(This article belongs to the Special Issue Advanced Materials for Energy and Sustainability)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:This article presents a comprehensive investigation into the functionalization of methoxyphenylazobenzene using electron-directing groups located at the meta position relative to the azo group. Spectroscopic analysis of meta-functionalized azobenzenes reveals that the incorporation of electron-withdrawing units significantly influences the absorption spectra of both E and Z isomers, while electron-donating functionalities lead to more subtle changes. The thermal relaxation process from Z to E result in almost twice as prolonged for electron-withdrawing functionalized azobenzenes compared to their electron-rich counterparts. Computational analysis contributes a theoretical understanding of the electronic structure and properties of meta-substituted azobenzenes. This combined approach, integrating experimental and computational techniques, yields significant insights into the structure–property relationship of meta-substituted asymmetrical phenolazobenzenes.
1. Introduction
Azobenzene is a widely studied molecule that has garnered significant attention in diverse fields including chemistry, physics, materials science, biology, and pharmacology [1,2,3]. Its unique photoresponsive properties, arising from its ability to undergo E/Z isomerization upon absorption of light, have made it a popular choice for designing light-responsive molecular switches [4,5,6]. The facile and reversible nature of the photoisomerization process, which can be triggered by different wavelengths of light, has enabled a plethora of applications for azobenzene-containing systems, including photo-controlled cargo delivery [7,8,9,10], surface patterning [11,12,13,14], mechanical actuators [15,16,17,18,19], and biological process regulators [20,21,22]. The E/Z photoisomerization of azobenzenes occurs by electron transition from an occupied π molecular orbital (MO) to an unoccupied π* MO localized on the N=N bond. At the same time, the empty π* MO of the double bond can be accessed from the non-bonding orbital (n) of the N lone electron pairs. These π–π* and n–π* transitions give rise to distinct and well-separated absorption bands in the spectra of azobenzenes. Due to the higher energy levels of n electrons compared to π electrons, the excitation energy required for n–π* transitions is generally lower. Consequently, the absorption band corresponding to the n–π* excitation is commonly red-shifted relative to the π–π* band. Typically, the π–π* transition is observed as an intense band around 320 nm, while the n–π* transition is observed as a weak broad band centered around 450 nm. The conversion of azobenzenes from the stable isomer E to the metastable isomer Z is exclusively initiated by light, while the reverse interconversion can take places either through photochemical or thermal processes. The spectroscopic characteristics and the isomerization behavior of azobenzenes are primarily governed by the azo group, along with the nature and the position of substituents on the two phenyl rings. Within this context, considerable effort has been put into reducing the spectral overlap between π–π* and n–π* transition bands, and in tailoring the Z→E thermal relaxation time for numerous uses. Over the years, extensive and comprehensive studies have been undertaken to achieve these objectives by functionalizing azobenzenes at both their ortho and para positions relative to the N=N group [1,4,5,6]. Substituting azobenzene’s para position (4-position) with electron-donating groups (e.g., hydroxy, amino, etc.) induces a red-shift in the π–π* band (400–450 nm), while the n–π* band remains unaffected, causing convergence and partial overlap of the two bands.
This functionalization is frequently employed to introduce a reactive site into azobenzenes for attaching solubilizing chains and linker, and it has been successfully used to develop photoswitchable surfactants [23,24] and a photo-responsive assembly monolayer on surfaces and on nanoparticles [25,26]. Introduction of strong electron-withdrawing groups (e.g., cyano, nitro) at the second azobenzene para position (4′-postion) forms pseudo-stilbene-type azobenzenes (Figure 1). These molecules exhibit overlapping π–π* and n–π* absorption bands within the visible spectrum, reducing E/Z isomerization yield due to concurrent reverse Z/E isomerization triggered by the same wavelength. Pseudo-stilbene azobenzenes find commonly applications in nonlinear optics [27] and light-responsive polymeric materials [28]. Functionalization at the para position also plays a role in reducing the half-life of Z isomers. This reduction is notable, typically decreasing on the order of seconds for 4-substituted azobenzenes and milliseconds for 4,4′-azobenzenes, in contrast to the hours required for the original azobenzene configuration.
The concept of ortho substitution, introduced by Hecht and Woolley (Figure 1), strategically places electron-directing groups at all four ortho positions of azobenzene to form tetra-ortho-substituted azobenzenes. Substituting electron-withdrawing (e.g., fluorine, chlorine, bromine) [29] or electron-donating groups (e.g., methoxy, S-alkyl) [30] separates n–π* bands of isomers, enhancing Z isomer stability from hours to years. Given their distinct band separation, red-shifted spectrum, and the slow thermal relaxation of the Z isomers, tetra-ortho azobenzenes have emerged as promising candidates for effectively controlling the structure and functionality of biomolecules through photoactivation [31,32].
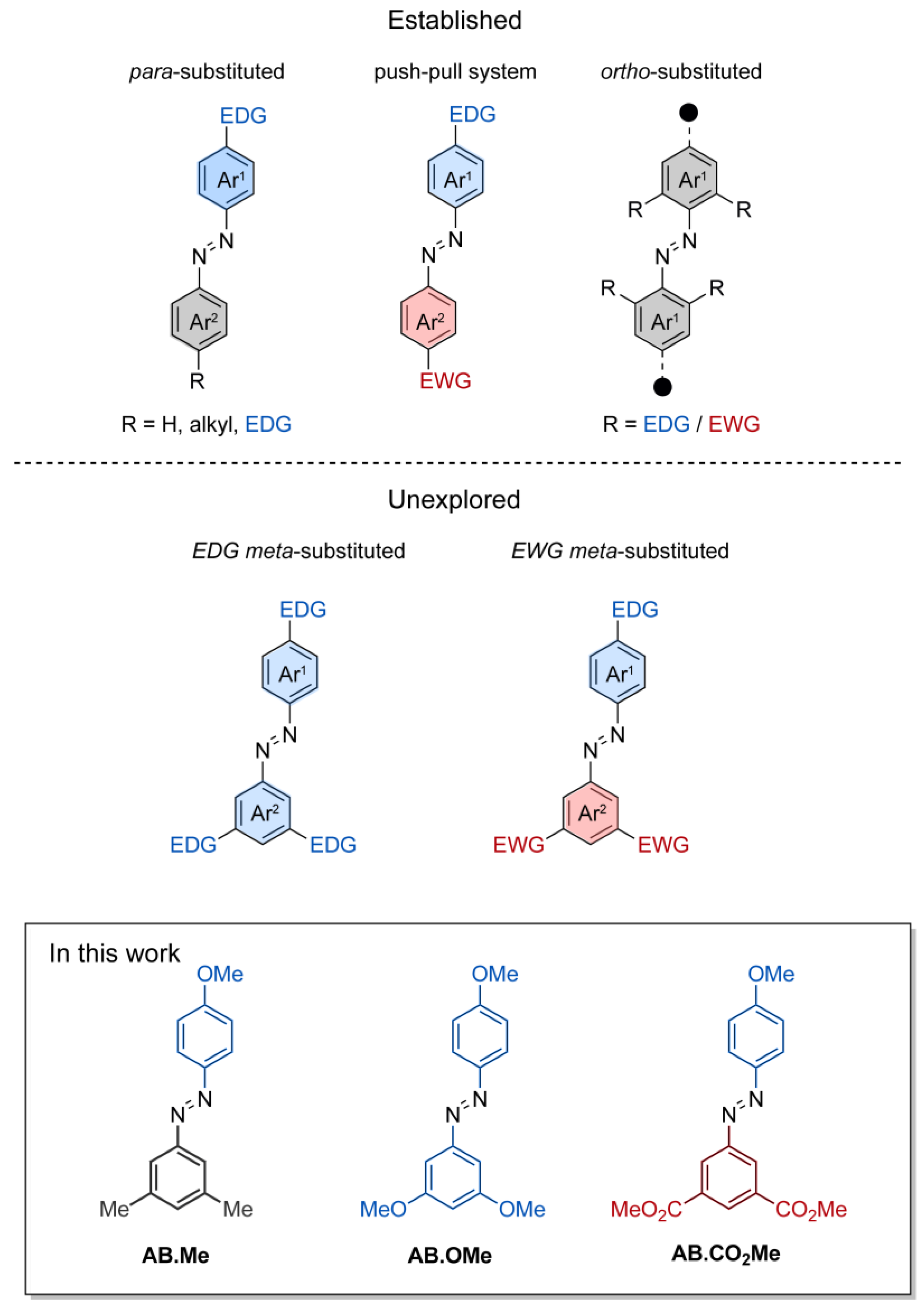
Substitutions at the meta positions of azobenzenes are a common occurrence. Meta-substitutions are frequently employed to serve structural purposes, enabling the creation of intricate molecular architectures. Substituents at the meta positions of azobenzene have been utilized in the development of light-responsive elastomers [33], molecular cages [34], and organic semiconductors [16]. However, there exists a notable gap in the understanding regarding the profound influence that electron-directing groups impart on the photoswitching and thermal relaxation dynamics of meta-substituted azobenzenes. While these functionalities are anticipated to not directly influence the electronic delocalization along the whole molecular π system by mesomeric effects, their spatial arrangement and electron localization on each individual phenyl ring may play a crucial role in the physical properties of azobenzenes. To date, the majority of studies focusing on understanding the effects of functionalization in the meta position have been primarily dedicated to symmetric azobenzenes containing alkyl functionalities or halogens [35,36]. With the aim of expanding existing knowledge of the structure–activity relationship of azobenzene, we undertook a comprehensive investigation involving three distinct types of substituents located at the meta positions of alkoxyl-azobenzenes, Figure 1. These compounds where investigated spectroscopically to provide information on the photoswitching activity and the thermal relaxation, as well as by computational approach to pinpoint the electronic characteristics of their E and Z isomers.
2. Results and Discussion
2.1. Design and Synthesis
The design of the azobenzene was envisioned to incorporate two methoxy groups as electron-donating functionalities, two carbonyl groups as electron-withdrawing functionalities, and two methyl groups as a non-electron-directing reference compound. The inclusion of the hydroxyl functionality on a phenyl ring was intended to complement the investigation of 4-substituted electron-directing azobenzenes. The compounds under investigation were synthesized by reaction of phenol with the azonium salts of meta di-substituted amino benzenes, resulting in the production of the corresponding meta-di-substituted hydroxyl azobenzenes. Subsequently, these were subjected to alkylation through iodomethane, leading to the desired compounds AB.Me, AB.OMe, and AB.CO2Me (Figure 1; for more details, refer to Section 3). The alkylation of the hydroxyl group was conducted in anticipation of potential applications, requiring the insertion of solubilizing groups, linker to connect polymers, and biomolecules.
2.2. UV–vis Characterization
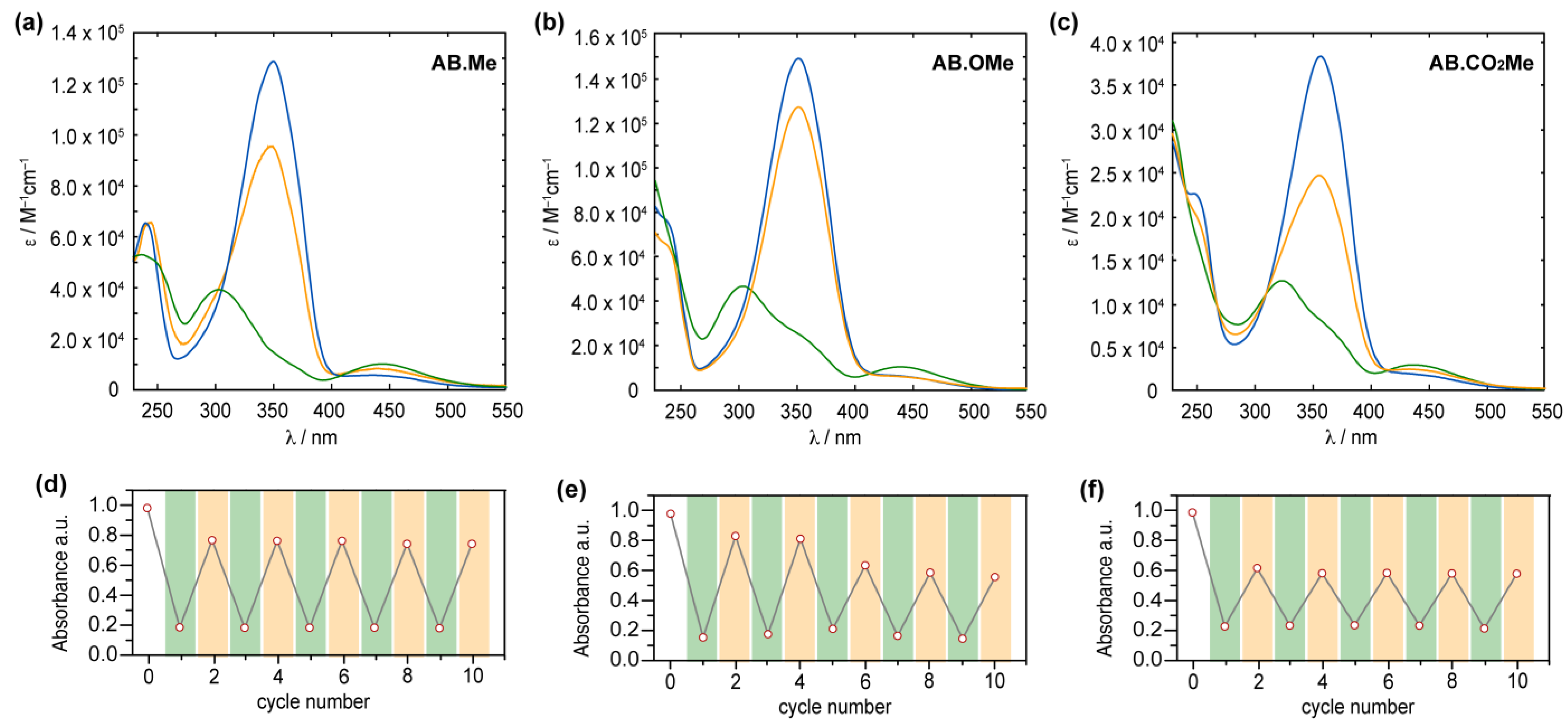
The obtained compounds were initially investigated by UV–vis absorption spectroscopy. The absorption spectrum of meta-methylated azobenzene in its E-form displays a characteristic pattern with two separate bands. The first, a prominent peak around 350 nm, derives from π–π* excitation, while the second, a weaker band centered at 444 nm, corresponds to the n–π* transition (Figure 2a). Additionally, a further band at higher energy is observed around 237 nm, which is associated with the π–π* transition resulting from molecular orbitals primarily localized on one of the two aromatic rings [15,35]. The value measured for the first two bands are in good agreement with those reported in literature for alkoxylazobenzens [23,25,26], so highlighting that the alkylation on meta position has a negligible effect on the absorption spectra features. Replacement of the two methyl units with two electron donating methoxy groups induced modest changes to all the three bands.
Compound AB.OMe exhibits a λmax at 353 nm deriving from the π–π* transition, and a signal at 440 nm, for the n–π* counterpart. In this case, however, the signal correlated with the high-energy π–π* is blue-shifted to overlap the solvent cut off area, resulting in undefined band. The limited spectral variation deriving from the introduction of electron donating functionalities at the meta position of azobenzenes suggests that the electronic delocalization over the molecules remains largely unaffected (see also molecular orbital analysis below). Differently, introduction of two carbonyl electron-withdrawing groups at the meta positions influence more effectively the spectra signature of the azobenzene. The resulting functionalized AB.CO2Me exhibit a bathochromic shift of approximately 8 nm in the low-energy π–π* transition band, with a λmax at 358 nm, and a red-shift of approximately 14 nm in the high-energy π–π* transition, centered at 251 nm. Interestingly, the n–π* transition signal undergoes a hypsochromic shift of around 7 nm compared to AB.Me. In a less pronounced manner, this scenario is reminiscent of what is observed for azobenzene within a push–pull system, where the primary band arising from π–π* transition shifts towards longer wavelengths, while the n–π* band experiences minimal changes. The spectral shift observed for the high excitation band is of particular interest as this can be exploited to promote the Z→E isomerization when the n–π* transition band is not easily accessible. This situation has been observed in various scenarios, including constrained azobenzenes macrocycle [15], intricate structures with overlapping absorption spectra [16], and E-4-aminobenzenes [36]. The redshift of the high-energy π–π* band can offer a distinct advantage by reducing its overlap with the absorption spectra of the solvents used in the sample preparation process, thus facilitating easier access to this band. Upon irradiation at 365 nm, the E isomers of azobenzenes are converted to their corresponding Z counterparts (Figure 2). The resulting absorption spectra exhibit the characteristic signature of azobenzene in its Z conformation, featuring an approximate 87% reduction in intensity for the π–π* band in the case of AB.Me, an 83% reduction for AB.OMe, and an 79% reduction for AB.CO2Me, accompanied by an enhancement in the n–π* band. Significantly, the π–π* band in the Z isomer of AB.CO2Me exhibits a distinct bathochromic shift of approximately 21 nm compared to AB.Me and AB.OMe.
It is noteworthy that the introduction of electron-withdrawing groups in the meta position predominantly affects the electronic transitions associated with the Z isomer, while exerting a negligible impact on those of the E form. All three compounds can undergo back-isomerization to the E isomer through light-assisted processes. When irradiated at 450 nm, meta-substituted azobenzenes, AB.Me, AB.OMe, and AB.CO2Me, exhibit a recovery of approximately ~75%, ~85%, and ~60%, respectively, in the initial absorption intensity of the E isomer. This process reaches a photostationary state (PSS), as illustrated in Figure 2. The photoisomerization between the two isomers during alternating irradiation cycles at λ = 365 nm and λ = 450 nm, respectively, did not lead to any noticeable degradation for the compounds AB.Me and AB.CO2Me. This highlights that the introduction of electron withdrawing groups at the meta position does not compromise the photochemical stability of the azobenzene structure. However, when subjecting AB.OMe to alternating cycles of irradiation, a gradual decrease in both the E and Z absorption intensities is observed. This phenomenon suggests the presence of a competitive process alongside light-induced isomerization, possibly indicating a gradual photodegradation.
2.3. Thermal Relaxation
In all three compounds, the metastable Z isomer can spontaneously revert to the E form through thermal relaxation. The Z-to-E isomerization process was observed to proceed at a slower rate in electron-withdrawing functionalized azobenzenes compared to the alkylated and alkoxylated counterpart. At a temperature of 25 °C, the half-life (τ1/2) for the Z-to-E isomerization was approximately 8.5 h for AB.Me and AB.OMe, in contrast to approximately 9.5 h for AB.CO2Me. Complete back isomerization of m-substituted azobenzenes is achieved within approximately 1.8 days for AB.Me and AB.OMe, whereas an additional day is required for AB.CO2Me, totaling 2.7 days (see Figure 3). The analysis of the kinetics of the back isomerization from Z to E underscores the varying rates at which each compound undergoes thermal relaxation, with a trend following the order of AB.Me > AB.OMe > AB.CO2Me, with a constant rate (kR) of 7.0 × 10−2 h−1, 5.9 × 10−2 h−1, and 4.3 × 10−2 h−1, respectively.
A plausible explanation for this phenomenon can be attributed to the enhanced stability of the Z isomer conferred by the electron-directing groups. This increased stability is primarily ascribed to the electronic effects of the electron-directing functionality, as opposed to the bulkiness of the group, which would otherwise promote the destabilization of the Z isomers. This scenario is especially pronounced with the carbonyl group, being the largest functionality among the three, yet exhibiting a slower thermal relaxation rate. The recent literature on azobenzenes suggests that the thermal isomerization mechanism involves both singlet and triplet electronic states, engaging in inversion and rotation processes [37,38]. Notably, the rotation mechanism through triplet states has been recognized as the predominant.
The rotational process encompasses out-of-plane CNNC-torsional motion, facilitated by the -N=N- π-bond rupture, ultimately reaching the CNNC-torsional angle characteristic of a nearly perpendicularly twisted conformation [37]. In such a scenario, the isomerization of azo aromatic groups through rotation at its transition state can be conceptualized as a phenylnitrenium ion. Investigation on the impact of substitution with electron-directing groups in the meta position of phenylnitronium ions has demonstrated a substantial influence on the stability of the triplet state [39]. The process is attributed to the transfer of an electron from a nonbonding orbital on the electron-directing substituent(s) to the out-of-plane nonbonding orbital on the nitrenium ion center. In essence, this mechanism could theoretically be applied to any species featuring a strong electron acceptor and a strong donor conjugated with non-disjoint π orbitals [39]. As a result, we hypothesize that while meta-functionalization modestly affects the π conjugation delocalization of azobenzenes, it nonetheless contributes to the stabilization of the triplet states that govern the rotation mechanism during the thermal isomerization.
2.4. Computational Studies
To gain insight into the stability of the E and Z isomers, we analyzed their frontier HOMO and LUMO orbitals resulting from first-principles calculations based on density functional theory (DFT). The results for the E and Z conformations are summarized in Figure 4 (see Section S5 of Supplementary Materials for further details).
All E isomers have a p-character spread over the phenyl groups, with minor or no contributions on the attached groups, regardless their electron attractive/withdrawing character. The structural folding in Z configurations put the aromatic rings closer, promoting an enhanced p-overlap between them. The LUMO states are hardly affected by the different functionalization for all the geometries. On the other hand, HOMO states are more sensitive to the position of the attached group and isomerization. In particular, the HOMO state for the E configuration of AB.OMe differs from the other orbitals shown in Figure 4a for both symmetry and for the contribution of the attached methoxy groups. The methyl and the carbonyl terminations do not contribute to the HOMO states of the corresponding molecules. These characteristics of the AB.OMe HOMO states change in the Z configurations, where both the symmetry and the methoxy contribution results more similar to other azobenzenes. The overall similarities of the frontier orbitals concur with the orbital transition analysis deduced from the absorption spectra. The different electron donor/acceptor character of the attached groups changes the overall polarization of the molecules. The specific dipolar interaction affects the energy position of the molecular orbitals and, therefore, the different energy transitions (i.e., peak energy) detected in the optical spectra. As shown in Figure 4c, AB.Me and AB.OMe have very similar orbital energy distributions. The electron withdrawing character of AB.CO2Me accumulates charge in one part of the molecule increasing the internal dipole. The effect is a downshift of frontier orbitals (for both E and Z configuration) of ~30 meV (see Section S5 of Supplementary Materials for details). We further remark an overall redistributions of the entire energy spectrum (especially HOMO-3, HOMO-4, LUMO+1, and LUMO+2 states) of AB.CO2Me and increased difference between the E and Z isomers, not observed for the other two azobenzene moieties. This is agrees with the stability analysis described above.
2.5. 1H-NMR Analysis
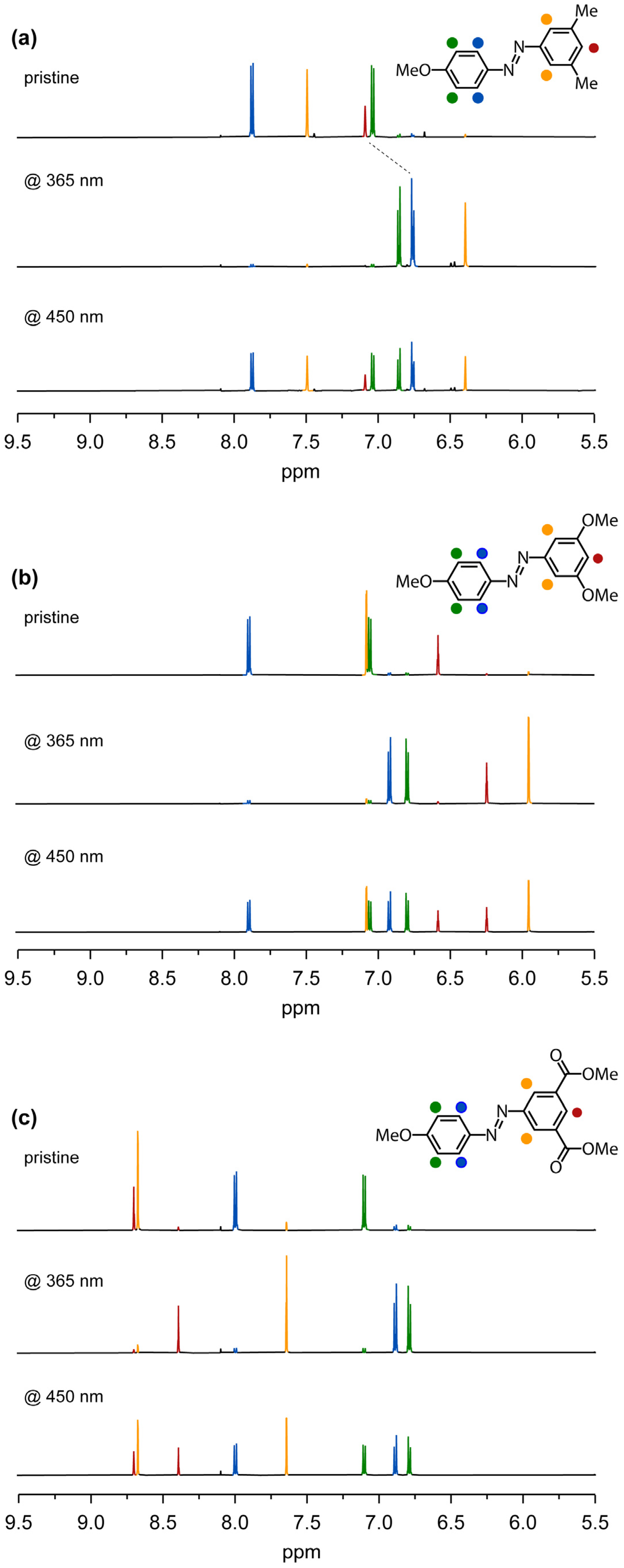
Finally, to assess the amount of E and Z isomers generated after photoexcitation at both 365 nm and 450 nm, we utilized nuclear magnetic resonance spectroscopy. Pristine E isomers display distinct spectral signatures. A comparison of the 1H-NMR spectral profiles of AB.OMe and AB.CO2Me with that of the reference compound AB.Me in Figure 5 reveals a significant displacement of the aromatic proton peaks within the meta-substituted benzene ring.
Specifically, in the reference compound AB.Me, the protons of the dimethylphenyl moiety are positioned at 7.50 and 7.09 ppm, respectively. Upon substituting the methyl group with the electron-donating group, the protons experience a shielding effect, resulting in a downfield shift to 7.07 and 6.57 ppm. Conversely, the introduction of carbonyl functionalities in place of the methyl group induces a deshielding effect, shifting the proton peaks to a lower magnetic field at 8.69 and 8.67 ppm. Upon irradiation at 365 nm, all the compounds adopt their respective Z forms. The 1H-NMR spectral profiles reveal the positions of protons in AB.Me at 6.75 and 6.40 ppm, in AB.OMe at 6.23 and 5.94 ppm, and in AB.CO2Me at 8.38 and 7.64 ppm. Upon comparing the 1H-NMR signal intensities of the E and Z isomers, we determined that photoisomerization of AB.Me occurs with a yield of 93%, while AB.OMe and AB.CO2Me exhibit yields of 90% and 89%, respectively. Irradiation at 450 nm facilitates the generation of the initial E isomer, ultimately attaining a PSS with yields of 51%, 49%, and 51% for AB.Me, AB.OMe, and AB.CO2Me, respectively.
3. Materials and Methods
3.1. General Experimental Details
All solvents and reagents were used as commercially supplied, without further purification. All the solvents (dichloromethane, ethyl acetate, and petroleum ether 40–60 °C) were purchased from Carlo Erba Reagents. Iodomethane was purchased from Sigma Aldrich, St. Louis, MO, USA; dimethylformamide (DMF, 99.0%) and 3,5-Dimethylaniline were purchased from Fluorchem, Glossop, UK; concentrated hydrochloric acid 37%, phenol, NaNO2, NaOH, and K2CO3 were purchased from Carlo Erba Reagents, Cornaredo, Italy; 3,5-Dimethoxylaniline was purchased from ABCR and dimethyl 5-aminoisophthalate was purchased from TCI. Reactions were monitored by thin-layer chromatography (TLC) performed on alugram precoated silica sheets, 0.2 mm plates (MN), and compounds were visualized under UV light (254 nm or 365 nm), depending on the substrate. Column chromatography was performed on silica gel 60A (particle size 40–63 μm, Carlo Erba) using positive air pressure. 1H and 13C liquid NMR spectra were recorded on a Bruker Advance III HD 600 MHz NMR spectrometer at 298 K. Deuterated chloroform was used for preparing the NMR sample and purchased from Carlo Erba Reagents. Chemical shifts (δH and δC) are expressed in parts per million (ppm) relative to the residual solvent peak and, for proton NMR, shown as follows: chemical shift, multiplicity (s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet), coupling constant (J, Hz), and integration. Mass analyses were performed using a Exploris 240 LC/GC-Orbitrap FTMS mass spectrometer.
3.2. Synthetic Procedures and Characterization
- Compound AB-OH.Me, (E)-4-((3,5-dimethylphenyl)diazenyl)phenol. This compound was synthesized as reported in the literature [15]. Concentrated hydrochloric acid (2 mL) was added to a suspension of 3,5-dimethylaniline Ph-NH2.Me (0.606 g, 5.00 mmol, 1 eq) in water (20 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.380 g, 5.50 mmol, 1.1 eq) in water (3 mL) was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.999 g, 25.0 mmol, 5 eq) was added to a solution of phenol (0.565 g, 6.00 mmol, 1.2 eq) in water (2 mL), and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The resultant precipitate was filtered with vacuo and purified by silica chromatography (dichloromethane/ethyl acetate 9.5:0.5) to give an orange powder (0.404 g, 35.7%). 1H NMR (600 MHz, CDCl3): δ = 7.86 (d, J = 8.8 Hz, 2H), 7.49 (s, 2H), 7.09 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 5.08 (s, 1H, OH), 2.41 (s, 6H). 13C NMR (151 MHz, CDCl3): δ = 158.15, 153.06, 147.47, 138.85, 132.32, 125.00, 120.51, 115.90, 21.47. HRMS (ESI, positive mode) m/z calculated for C14H15N2O [M + H]+: 227.1174; found: 227.1179 [40].
- Compound AB-OH.OMe, (E)-4-((3,5-dimethoxyphenyl)diazenyl)phenol. This compound was synthesized adapting a procedure from the literature [40]. Concentrated hydrochloric acid (1.3 mL) was added to a suspension of 3,5-dimethoxyanline Ph-NH2.OMe (0.500 g, 3.26 mmol, 1 eq) in tetrahydrofuran (5 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.247 g, 3.59 mmol, 1.1 eq) in 1.95 mL of water was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.652 g, 16.3 mmol, 5 eq) was added to a solution of phenol (0.368 g, 3.91 mmol, 1.2 eq) in 2.6 mL of water, and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The reaction mixture was extracted with dichloromethane, the organic layers collected, dried over Na2SO4, filtered, and solvent removed with vacuo. The crude product was purified by silica chromatography using a gradient eluent (dichloromethane 100% → ethyl acetate 100%) to obtain the desired product as a red dark powder (0.377 g, 44.8%). 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 8.8 Hz, 2H), 7.08 (s, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.57 (s, 1H), 5.17 (s, 1H, OH), 3.87 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 161.22, 158.49, 154.68, 147.16, 125.22, 115.96, 103.55, 100.81, 55.75. HRMS (ESI, positive mode) m/z calculated for C14H15N2O3 [M + H]+: 259.1083; found: 259.1082.
- Compound AB-OH.CO2Me, (E)-dimethyl 5-((4-hydroxyphenyl)diazenyl)isophthalate. This compound was synthesized adapting a procedure from the literature [40]. Concentrated hydrochloric acid (0.95 mL) was added to a suspension of dimethyl 5-aminoisophthalate Ph-NH2.CO2Me (0.500 g, 2.39 mmol, 1 eq) in tetrahydrofuran (5 mL). The mixture was cooled to 0 °C in an ice bath and stirred vigorously. A solution of NaNO2 (0.182 g, 2.63 mmol, 1.1 eq) in 1.42 mL of water was then added dropwise to the cooled suspension, and the mixture was stirred for 1 h. In a separate flask, NaOH 5 M (0.476 g, 11.9 mmol, 5 eq) was added to a solution of phenol (0.270 g, 2.87 mmol, 1.2 eq) in 1.92 mL of water, and cooled to 0 °C. The former mixture was then slowly transferred to the solution of phenol and vigorously stirred for 1 h at 0 °C. The reaction mixture was extracted with ethyl acetate, the organic layers collected, dried over Na2SO4, filtered, and solvent removed with vacuo. The crude product was purified by silica chromatography using dichloromethane/ethyl acetate (9.5:0.5) as an eluent, to obtain the desired product as bright orange powder (0.515 g, 68.6%). 1H NMR (600 MHz, CDCl3) δ = 8.75 (s, 1H), 8.70 (s, 2H), 7.94 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 5.32 (s, 1 H, OH), 4.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ = 166.06, 159.13, 153.04, 147.11, 131.87, 131.82, 127.74, 125.66, 116.10, 52.72. HRMS (ESI, positive mode) m/z calculated for C16H15N2O5 [M + H]+: 315.0981; found: 315.0983.
- Compound AB.Me, (E)-1-(3,5-dimethylphenyl)-2-(4-methoxyphenyl)diazene. A solution of compound AB-OH.Me (0.100 g, 0.442 mmol, 1 eq), iodomethane (0.033 mL, 0.530 mmol, 1.2 eq), K2CO3 (0.091 g, 0.663 mmol, 1.5 eq), and dimethylformamide (2 mL) was stirred at 7 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica chromatography (petroleum ether/ethyl acetate = 8:2) to obtain the desired product as a bright orange oil (0.088 g, 83.0%). 1H NMR (600 MHz, CDCl3) δ 7.91 (d, J = 9.0 Hz, 2H), 7.50 (s, 2H), 7.09 (s, 1H), 7.02 (d, J = 9.0 Hz, 2H), 3.89 (s, 3H), 2.41 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 162.06, 153.11, 147.25, 138.81, 132.22, 124.75, 120.48, 114.33, 55.70, 21.40. HRMS (ESI, positive mode) m/z calculated for C15H16N2O [M + H]+: 241.1341; found: 241.1341.
- Compound AB.OMe, (E)-1-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)diazene. A solution of compound AB-OH.OMe (0.146 g, 0.566 mmol, 1 eq), iodomethane (0.042 mL, 0.680 mmol, 1.2 eq), K2CO3 (0.117 g, 0.849 mmol, 1.5 eq), and dimethylformamide (2.5 mL) was stirred at 70 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by silica chromatography (petroleum ether/ethyl acetate = 8:2) to obtain the desired product as a bright orange powder (0.084 g, 54.5%). 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 9.4 Hz, 2H), 7.09 (d, 2H), 7.03 (d, J = 9.4 Hz, 2H), 6.58 (s, 1H), 3.90 (s, 3H), 3.88 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 162.32, 161.22, 154.76, 146.99, 124.98, 114.38, 103.49, 100.77, 76.95, 55.74. HRMS (ESI, positive mode) m/z calculated for C15H16N2O3 [M + H]+: 273.1239; found: 273.1238.
- Compound AB.CO2Me, (E)-dimethyl 5-((4-methoxyphenyl)diazenyl)isophthalate. A solution of compound AB-OH.CO2Me (0.100 g, 0.318 mmol, 1 eq), iodomethane (0.024 mL, 0.382 mmol, 1.2 eq), K2CO3 (0.066 g, 0.477 mmol, 1.5 eq) and dimethylformamide (1.5 mL) was stirred at 70 °C for 24 h. The mixture was allowed to reach room temperature, diluted with ethyl acetate, transferred in a separatory funnel, and washed with water. The organic layers were collected, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by silica chromatography (dichloromethane 100%) to obtain the desired product as a bright yellow powder (0.033 g, 31.7%). 1H NMR (600 MHz, CDCl3) δ = 8.75 (s, 1H), 8.70 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 3.99 (s, 6H), 3.91 (s, 3H). 13C NMR (151 MHz, CDCl3) δ = 166.00, 162.90, 153.04, 146.86, 131.75, 127.67, 125.39, 114.48, 55.76, 52.67. HRMS (ESI, positive mode) m/z calculated for C17H16N2O5 [M + H]+: 329.1137; found: 329.1130.
4. Conclusions
We investigated azobenzene derivatives functionalized at their meta position with electron-directing substituents. The spectral and photoswitching changes induced by the presence of electron-donating and electron-withdrawing groups were compared to a reference compound—an azobenzene inclusive of two methyl units at its meta positions. The azobenzene functionalized with electron-donating groups (OMe) exhibits modest changes in the absorption spectrum in both E and Z isomers, closely resembling the spectral signatures of the reference methylated counterpart. In contrast, functionalization with electron-withdrawing groups (CO2Me) induced an 11 nm bathochromic shift in the π–π* high-energy band of the E isomer and a 21 nm shift in the main π–π* band of its Z counterpart. The time of thermal relaxation Z→E is limitedly affected by the presence of methoxyl functionality, whereas longer time (over a day more) is needed for the carbonyl groups. Regardless, the τ1/2 of the thermal relaxation of three azobenzenes take place within similar time range with ~2 h of difference (6–8 h). Our computational studies confirmed that introducing electron-directing substituents at the meta position affects the energy levels of the frontier orbitals of azobenzene and enhances the stability of the Z and E isomers for the electron-withdrawing carbonyl groups. Our findings enhance our understanding of the structure–property relationship of azobenzene, enabling us to selectively tune its optical properties and thermal relaxation behavior. This newfound knowledge represents a valuable addition to the toolkit for designing functional molecular switches.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29091929/s1. All of the 1H and 13C NMR spectra are available in the Supplementary Materials. Absorption measurements and cyclability test, E/Z isomerization experiments, thermal relaxation mathematical models, and DFT calculations are explored in more detail in the Supplementary Materials. Scheme S1: Synthetic scheme for the preparation of the meta-substituted azobenzenes. Table S1: Irradiation time and sample concentration for AB.Me, AB.OMe, and AB.CO2Me. Table S2: Cyclability: irradiation time for AB.Me, AB.OMe, and AB.CO2Me. Tables S3 and S4: Thermal relaxation parameters and fitting results (Figure S8, logistic modeland logarithmic model). Table S5: Total energy of azobenzene derivatives in E and Z configurations. Table S6: Bandgap (Eg) and energy level of HOMO and LUMO states of azobenzene derivatives in E (a) and Z (b) configuration. Zero energy reference is set to the vacuum level. Figure S1 aromatic region of the 1H-NMR spectrum of compound AB.Me (a), AB.OMe (b), AB.CO2Me (c) in chloroform-d. Figures S2, S4, S6, panoramic 1H-NMR spectra of E form (a), Z form (b), and PSS state (c) for AB.Me, AB.OMe, and AB.CO2Me in chloroform-d. Figures S3, S5, S7, panoramic 1H-NMR spectra of E form (a), Z form (b), and PSS state (c) for AB.Me, AB.OMe, and AB.CO2Me in THF-d8. Figure S8: Experimental data points of thermal relaxation for AB.Me, AB.OMe, and AB.CO2Me. Figure S9: Density of states (DOS) of azobenzene derivatives in E (a) and Z (b) configuration. Zero energy reference is set to HOMO state of each system. References [41,42,43,44,45,46] are citation in the Supplementary Materials.
Author Contributions
Conceptualization, A.C. (Arrigo Calzolari) and G.S.; investigation, A.L.S., T.P., A.C. (Arrigo Calzolari), A.C. (Alessandra Catalani) and G.S.; resources, data curation, A.L.S., T.P., A.C. (Arrigo Calzolari) and G.S.; writing—original draft preparation, A.C. (Arrigo Calzolari) and G.S. All authors have read and agreed to the published version of the manuscript.
Funding
G.S. would like to thank the Swiss National Science Foundation (SNSF) Ambizione program (PZ00P2_148050), PON Ricerca e Innovazione (annualità 2014-2020, grant no. DOT1304455-2), and Fondazione di Sardegna (annualità 2020, FdS grant no. F75F21001250007) for financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to specific ethical and privacy considerations.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jerca, F.A.; Jerca, V.V.; Hoogenboom, R. Advances and opportunities in the exciting world of azobenzenes. Nat. Rev. Chem. 2022, 6, 51–69. [Google Scholar] [CrossRef]
- Mukherjee, A.; Seyfried, M.D.; Ravoon, B.J. Azoheteroarene and Diazocine Molecular Photoswitches: Self-Assembly, Responsive Materials and Photopharmacology. Angew. Chem. Int. Ed. 2023, 62, e202304437. [Google Scholar] [CrossRef]
- Fedele, C.; Ruoko, T.; Kuntze, K.; Virkki, M.; Priimagi, A. New tricks and emerging applications from contemporary azobenzene research. Photochem. Photobiol. Sci. 2022, 21, 1719–1734. [Google Scholar] [CrossRef]
- Crespi, S.; Simeth, N.A.; König, B. Heteroaryl azo dyes as molecular photoswitches. Nat. Rev. Chem. 2019, 3, 33–146. [Google Scholar] [CrossRef]
- García-Amorós, J.; Velasco, D. Recent advances towards azobenzene-based light-driven real-time information-transmitting materials. Beilstein J. Org. Chem. 2012, 8, 1003–1017. [Google Scholar] [CrossRef]
- Chen, H.; Chen, W.; Lin, Y.; Xie, Y.; Liu, S.H.; Yin, J. Visible and near-infrared light activated azo dyes. Chin. Chem. Lett. 2021, 32, 2359–2368. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, T.; Wang, Z.; Zhao, Y. Triggered azobenzene-based prodrugs and drug delivery systems. J. Control. Release 2022, 325, 475–493. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, Y.; Chen, T.; Lu, D.; Zhao, Z.; Zhang, X.; Li, Z.; Yan, C.; Tan, W. Photon-Manipulated Drug Release from a Mesoporous Nanocontainer Controlled by Azobenzene-Modified Nucleic Acid. ACS Nano 2012, 6, 6337–6344. [Google Scholar] [CrossRef]
- Yadav, S.; Deka, S.R.; Verma, G.; Sharma, A.K.; Kumar, P. Photoresponsive amphiphilic azobenzene–PEG self-assembles to form supramolecular nanostructures for drug delivery applications. RSC Adv. 2016, 6, 8103. [Google Scholar] [CrossRef]
- Meng, X.; Gui, B.; Yuan, D.; Zeller, M.; Wang, C. Mechanized azobenzene-functionalized zirconium metal-organic framework for on-command cargo release. Sci. Adv. 2016, 2, e1600480. [Google Scholar] [CrossRef]
- Kryshenik, V.M.; Azhniuk, Y.M.; Kovtunenko, V.S. All-optical patterning in azobenzene polymers and amorphous chalcogenides. J. Non Cryst. Solids 2019, 512, 112–131. [Google Scholar] [CrossRef]
- Bakker, S.; Korver, E.; Fransen, M.; Kamer, E.; Metselaar, G.; Catarina, A.; Esteves, C.; Schenning, A.P.H.J. Optical Patterning in Photoresponsive Azobenzene-Based Waterborne Coatings. ACS Appl. Opt. Mater. 2023, 1, 403–411. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Kulikovsky, L.; Kulikovska, O.; Stumpe, J. Extremely high patterning efficiency in easily made azobenzene-containing polymer films. J. Mater. Chem. 2009, 19, 6103–6105. [Google Scholar] [CrossRef]
- Carroll, G.T.; Lee, K.M.; Mc Conney, M.E.; Hall, H.J. Optical control of alignment and patterning in an azobenzene liquid crystal photoresist. J. Mater. Chem. C 2023, 11, 2177–2185. [Google Scholar] [CrossRef]
- Maciejewski, J.; Sobczuk, A.; Claveau, A.; Nicolai, A.; Petraglia, R.; Cervini, L.; Baudat, E.; Miéville, P.; Fazzi, D.; Corminboeuf, C.; et al. Photochromic Torsional Switch (PTS): A Light-Driven Actuator for the Dynamic Tuning of π-Conjugation Extension. Chem. Sci. 2017, 8, 361–365. [Google Scholar] [CrossRef]
- Jozeliūnaitė, A.; Rahmanudin, A.; Gražulis, S.; Baudat, E.; Sivula, K.; Fazzi, D.; Orentas, E.; Sforazzini, G. Light-Responsive Oligothiophenes Incorporating Photochromic Torsional Switches (PTS). Chem. Eur. J. 2022, 28, e202202698. [Google Scholar] [CrossRef]
- Jousselme, B.; Blanchard, P.; Gallego-Planas, N.; Delaunay, J.; Allain, M.; Richomme, P.; Levillain, E.; Roncali, J. Photomechanical Actuation and Manipulation of the Electronic Properties of Linear π-Conjugated Systems. J. Am. Chem. Soc. 2003, 125, 2888–2889. [Google Scholar] [CrossRef]
- Jousselme, B.; Blanchard, P.; Allain, M.; Levillain, E.; Dias, M.; Roncali, J. Structural Control of the Electronic Properties of Photodynamic Azobenzene-Derivatized π-Conjugated Oligothiophenes. J. Phys. Chem. 2006, 110, 3488–3494. [Google Scholar] [CrossRef]
- Azov, V.A.; Cordes, J.; Schlüter, D.; Dülcks, T.; Böckmann, M.; Doltsinis, N.L. Light-Controlled Macrocyclization of Tetrathiafulvalene with Azobenzene: Designing an Optoelectronic Molecular Switch. J. Org. Chem. 2014, 79, 11714–11721. [Google Scholar] [CrossRef]
- Mart, R.J.; Allemann, R.K. Azobenzene photocontrol of peptides and proteins. Chem. Commun. 2016, 52, 12262. [Google Scholar] [CrossRef]
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Barrett, C.J. Photo-Control of Biological Systems with Azobenzene Polymers. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3058–3070. [Google Scholar] [CrossRef]
- Butler, C.S.G.; King, J.P.; Giles, L.W.; Marlow, J.B.; Vidallon, M.L.P.; Sokolova, A.; de Campo, L.; Tuck, K.L.; Tabor, R.F. Design and synthesis of an azobenzene–betaine surfactant for photo-rheological fluids. J. Colloid Interface Sci. 2021, 594, 669–680. [Google Scholar] [CrossRef]
- Wu, Z.; Li, X.; Jiang, X.; Xie, T.; Li, H.; Zhang, G.; Jiang, J. Photoswitchable de/adsorption of an azobenzene-derived surfactant on a silica surface. Phys. Chem. Chem. Phys. 2019, 21, 21030–21037. [Google Scholar] [CrossRef]
- Chu, Z.; Han, Y.; Bian, T.; De, S.; Král, P.; Klajn, R. Supramolecular Control of Azobenzene Switching on Nanoparticles. J. Am. Chem. Soc. 2019, 141, 1949–1960. [Google Scholar] [CrossRef]
- Moldt, T.; Przyrembel, D.; Schulze, M.; Bronsch, W.; Boie, L.; Brete, D.; Gahl, C.; Klajn, R.; Tegeder, P.; Weinelt, M. Differing Isomerization Kinetics of Azobenzene-Functionalized Self-Assembled Monolayers in Ambient Air and in Vacuum. Langmuir 2016, 32, 10795–10801. [Google Scholar] [CrossRef]
- Cuétara-Guadarrama, F.; Vonlanthen, V.; Sorroza-Martínez, K.; González-Méndez, I.; Rivera, E. Photoisomerizable azobenzene dyes incorporated into polymers and dendrimers. Influence of the molecular aggregation on the nonlinear optical properties. Dyes Pigm. 2021, 194, 109551. [Google Scholar] [CrossRef]
- Cunha, M.P.; Foelen, Y.; Engels, T.A.P.; Papamichou, K.; Hagenbeek, M.; Debije, M.G.; Schenning, P.H.J. On Untethered, Dual Magneto- and Photoresponsive Liquid Crystal Bilayer Actuators Showing Bending and Rotating Motion. Adv. Opt. Mater. 2019, 7, 1801604. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef]
- Beharry, A.A.; Sadovski, O.; Woolley, G.A. Azobenzene Photoswitching without Ultraviolet Light. J. Am. Chem. Soc. 2011, 133, 19684–19687. [Google Scholar] [CrossRef]
- Kerckhoffs, A.; Bo, Z.; Penty, S.E.; Duarte, F.; Langton, M.J. Red-shifted tetra-ortho-halo-azobenzenes for photo-regulated transmembrane anion transport. Org. Biomol. Chem. 2021, 19, 9058–9067. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Beharry, A.A.; OSadovski, O.; McCormick, T.M.; Babalhavaeji, A.; Tropepe, V.; Woolley, G.A. Photoswitching Azo Compounds in Vivo with Red Light. J. Am. Chem. Soc. 2013, 135, 9777–9784. [Google Scholar] [CrossRef] [PubMed]
- Ohzono, T.; Koyama, E. Effects of photo-isomerizable side groups on the phase and mechanical properties of main-chain nematic elastomers. Polym. Chem. 2022, 13, 2694–2704. [Google Scholar] [CrossRef]
- Togari, Y.; Hirota, S.; Kitagawa, H.; Tsukamoto, Y.; Kobayashi, K. Hydrogen-bonded six-component assembly for capsule formation based on tetra(4-pyridyl) cavitand and isophthalic acid linker and its application to photoresponsive capsule. Org. Biomol. Chem. 2018, 16, 7626–7635. [Google Scholar] [CrossRef] [PubMed]
- Di Berardin, C.; Strauss, M.A.; Schatz, D.; Wegner, H.A. An Incremental System To Predict the Effect of Different London Dispersion Donors in All-meta-Substituted Azobenzenes. Chem. Eur. J. 2022, 28, e202104284. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Yadav, M.K.; Singla, L.; Kumar, A.; Karanam, M.; Dev, S.; Choudhury, A.R. Understanding of the Kinetic Stability of cis- Isomer of Azobenzenes through Kinetic and Computational Studies. ChemistrySelect 2020, 5, 13957–13962. [Google Scholar] [CrossRef]
- Singer, N.K.; Schlögl, K.; Zobel, J.P.; Mihovilovic, M.D.; González, L. Singlet and Triplet Pathways Determine the Thermal Z/E Isomerization of an Arylazopyrazole-Based Photoswitch. J. Phys. Chem. Lett. 2023, 14, 8956–8961. [Google Scholar] [CrossRef]
- Reimann, M.; Teichmann, E.; Hecht, S.; Kaupp, M. Solving the Azobenzene Entropy Puzzle: Direct Evidence for Multi-State Reactivity. J. Phys. Chem. Lett. 2022, 13, 10882–10888. [Google Scholar] [CrossRef]
- Winter, A.H.; Falvey, D.E.; Cramer, C.J. Effect of meta Electron-Donating Groups on the Electronic Structure of Substituted Phenyl Nitrenium Ions. J. Am. Chem. Soc. 2004, 126, 9661–9668. [Google Scholar] [CrossRef]
- Song, Y.M.; Ha, Y.M.; Kim, J.A.; Chung, K.W.; Uehara, Y.; Lee, K.J.; Chun, P.; Byun, Y.; Chung, H.Y.; Moon, H.R. Synthesis of novel azo-resveratrol, azo-oxyresveratrol and their derivatives as potent tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7451–7455. [Google Scholar] [CrossRef]
- Ślusarek, J.; Nowoświat, A.; Olechowska, M. Logistic Model of Phase Transformation of Hardening Concrete. Materials 2022, 15, 4403. [Google Scholar] [CrossRef] [PubMed]
- Jerca, V.V.; Jerca, F.A.; Rau, I.; Manea, A.M.; Vuluga, D.M.; Kajzar, F. Advances in understanding the photoresponsive behavior of azobenzenes substituted with strong electron withdrawing groups. Opt. Mater. 2015, 48, 160–164. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Berland, K.; Cooper, V.R.; Lee, K.; Schröder, E.; Thonhauser, T.; Hyldgaard, P.; Lundqvist, B.I. van der Waals forces in density functional theory: A review of the vdW-DF method. Rep. Prog. Phys. 2015, 78, 066501. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of functionalized azobenzenes. Functionalizations in either one or two para positions, functionalization in push–pull system, and functionalization in ortho positions (top half); functionalization in meta (bottom half); and the compound investigated in this work (framed).
Figure 1.
Schematic representation of functionalized azobenzenes. Functionalizations in either one or two para positions, functionalization in push–pull system, and functionalization in ortho positions (top half); functionalization in meta (bottom half); and the compound investigated in this work (framed).
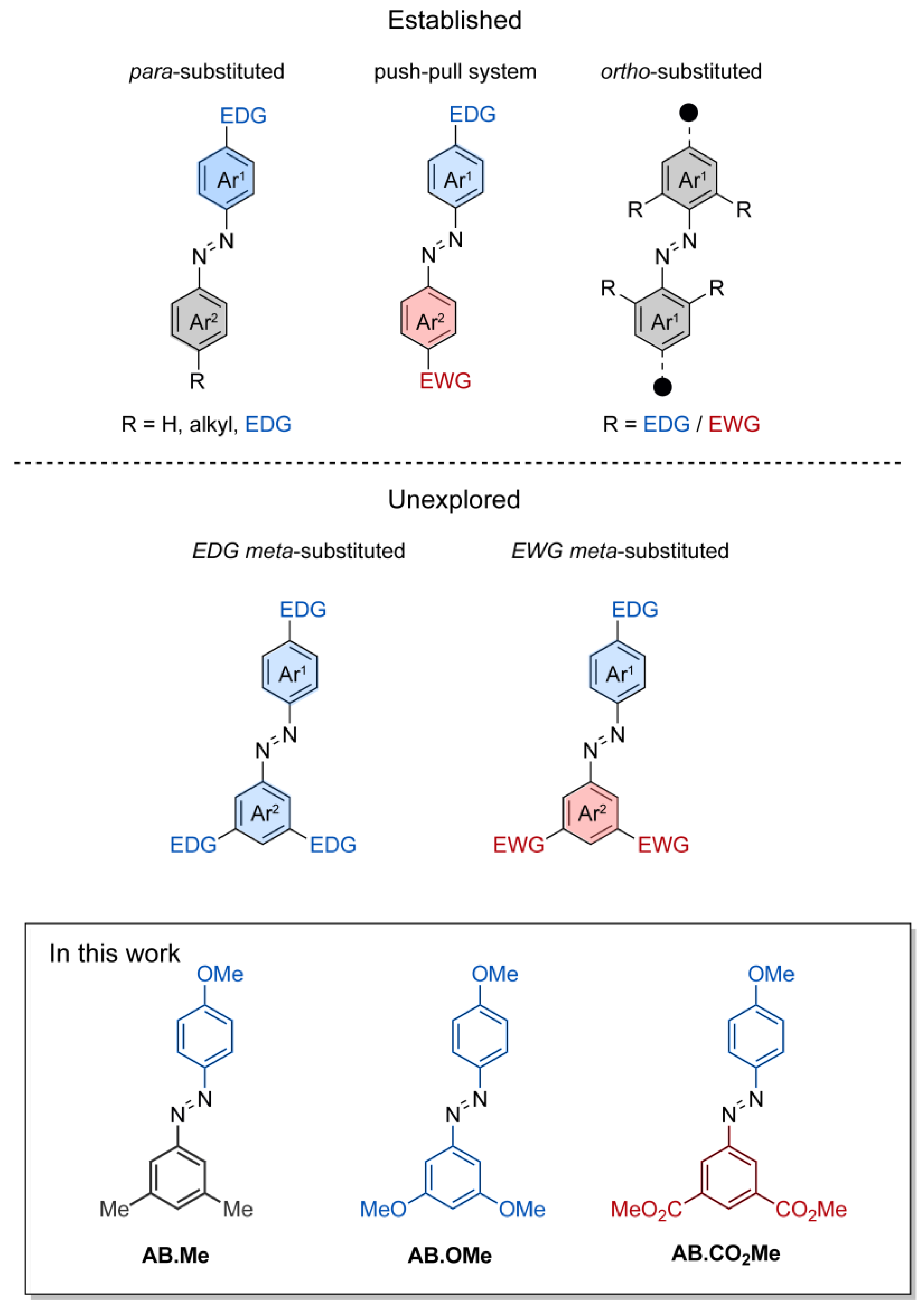
Figure 2.
Absorption spectra of AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in tetrahydrofuran (THF) solution; E isomer (blue), Z isomer (green), and photostationary state PSS450 nm (orange). Measured absorbance at λmax for AB.Me (d), AB.OMe (e), and AB.CO2Me (f) in THF solutions by alternately irradiating the sample at λ = 365 nm (green) and λ = 450 nm (orange) in repeated switching cycles.
Figure 2.
Absorption spectra of AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in tetrahydrofuran (THF) solution; E isomer (blue), Z isomer (green), and photostationary state PSS450 nm (orange). Measured absorbance at λmax for AB.Me (d), AB.OMe (e), and AB.CO2Me (f) in THF solutions by alternately irradiating the sample at λ = 365 nm (green) and λ = 450 nm (orange) in repeated switching cycles.
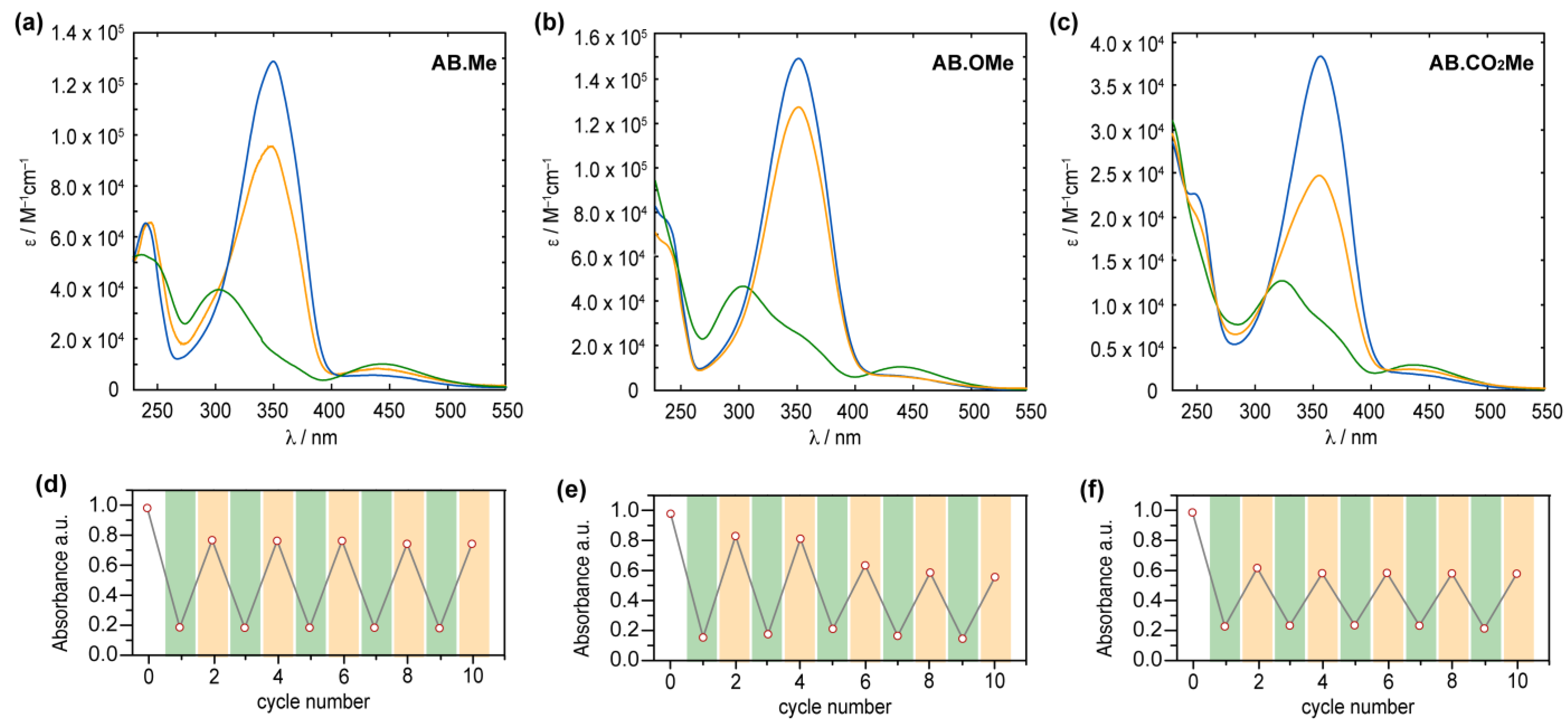
Figure 3.
Measured absorption spectra for the Z (blue line) to E (green line) thermal relaxation of AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in diluted THF solutions at 25 °C. Measured absorbance at λmax for AB.Me (yellow triangle), AB.OMe (blue circle), and AB.CO2Me (red square) in THF solutions over time; a logarithmic fit (d) and a linear fit were used to estimate relaxation rate constants (e).
Figure 3.
Measured absorption spectra for the Z (blue line) to E (green line) thermal relaxation of AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in diluted THF solutions at 25 °C. Measured absorbance at λmax for AB.Me (yellow triangle), AB.OMe (blue circle), and AB.CO2Me (red square) in THF solutions over time; a logarithmic fit (d) and a linear fit were used to estimate relaxation rate constants (e).

Figure 4.
Calculated molecular orbitals HOMO and LUMO for all the E (a) and Z (b) isomers studied in this work, and corresponding energy levels (c).
Figure 4.
Calculated molecular orbitals HOMO and LUMO for all the E (a) and Z (b) isomers studied in this work, and corresponding energy levels (c).

Figure 5.
Aromatic region of the 1H-NMR spectrum of compounds AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in THF-d8. Pristine (top spectrum), after irradiation at 365 nm (middle spectrum), and after irradiation at 450 nm (bottom spectrum). 1H-NMR spectra in chloroform-d are reported in the Supplementary Materials.
Figure 5.
Aromatic region of the 1H-NMR spectrum of compounds AB.Me (a), AB.OMe (b), and AB.CO2Me (c) in THF-d8. Pristine (top spectrum), after irradiation at 365 nm (middle spectrum), and after irradiation at 450 nm (bottom spectrum). 1H-NMR spectra in chloroform-d are reported in the Supplementary Materials.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sanna, A.L.; Pachova, T.; Catellani, A.; Calzolari, A.; Sforazzini, G. Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship. Molecules 2024, 29, 1929. https://doi.org/10.3390/molecules29091929
AMA Style
Sanna AL, Pachova T, Catellani A, Calzolari A, Sforazzini G. Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship. Molecules. 2024; 29(9):1929. https://doi.org/10.3390/molecules29091929
Chicago/Turabian StyleSanna, Anna Laura, Tatiana Pachova, Alessandra Catellani, Arrigo Calzolari, and Giuseppe Sforazzini. 2024. "Meta-Substituted Asymmetric Azobenzenes: Insights into Structure–Property Relationship" Molecules 29, no. 9: 1929. https://doi.org/10.3390/molecules29091929