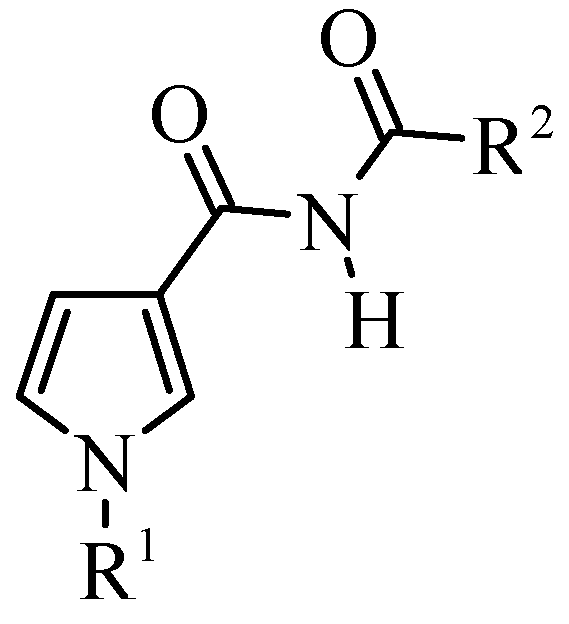
General
Chemicals were purchased from
Fluka AG, Aldrich Chemical Company, Inc.,
Merck GmbH, or
Lancaster Synthesis Ltd. Solvents used in reactions were distilled and dried or purchased in
absolute quality. Tetrahydrofuran was freshly distilled from Na/K. TLC:
Merck silica gel 60 F
254 precoated glass plates. Column chromatography: flash-chromatography procedure of
Still et al. [
4]; columns with water cooling;
Merck Kieselgel 60, 40-63 μm.
M.p.: Kofler hot stage, corrected. IR: Perkin-Elmer FT-IR 1600; KBr pellet or liquid film between NaCl plates. NMR: Varian Gemini 300 (1H: 300 MHz, 13C: 75 MHz); chemical shifts δ in ppm relative to TMS (0 ppm); coupling constants J in Hz; multiplicities of 13C resonances from APT and/or 1H,13C COSY experiments; * means that similar assignments may be interchanged within the same spectrum. MS: VG 70-250 (Dr. H. Nadig); FAB MS: VG ZAB HF (courtesy Ciba-Geigy AG, Basel).
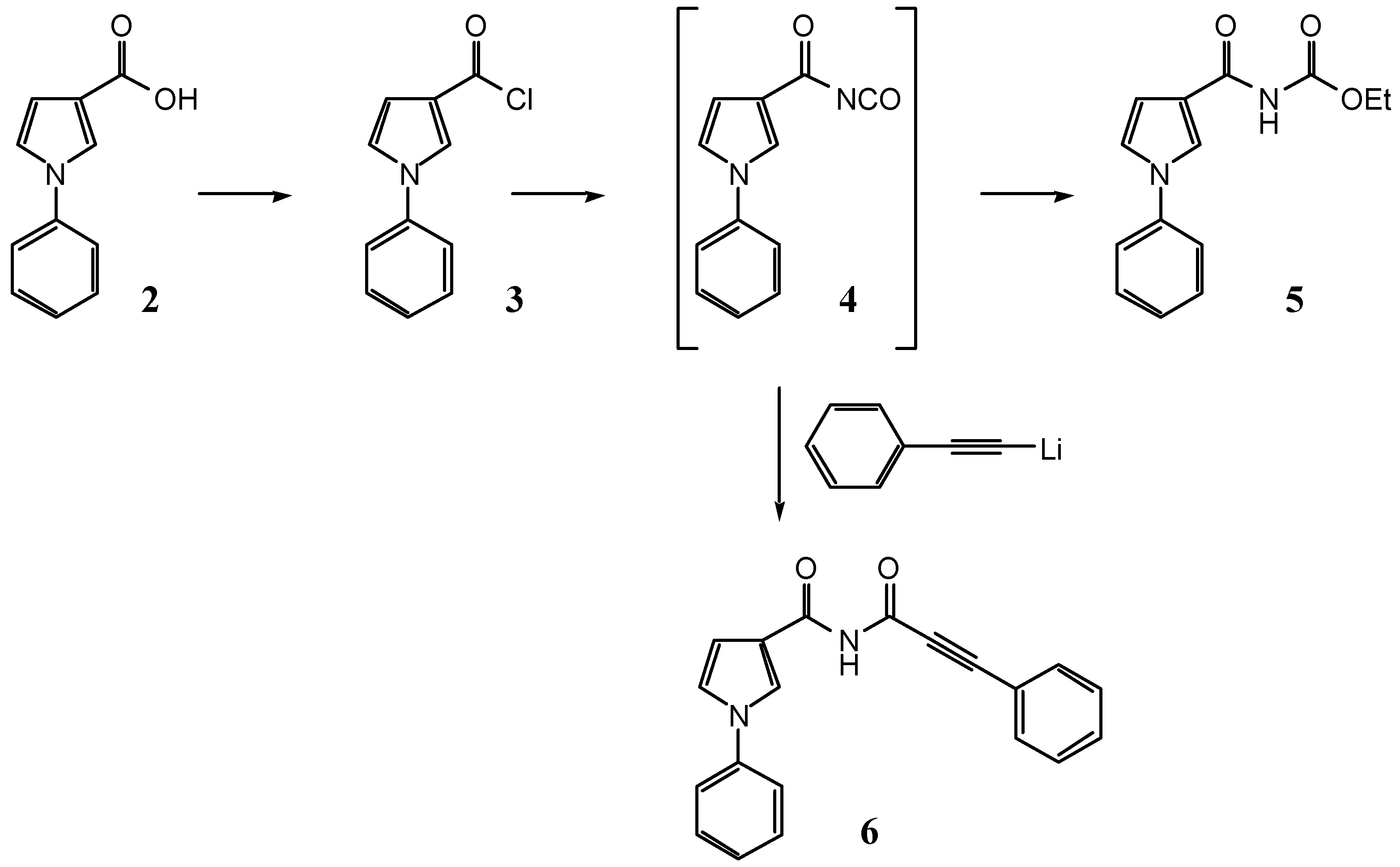
1-Phenyl-1H-pyrrole-3-carbonyl chloride (3)
A solution of 1-phenyl-1
H-pyrrole-3-carboxylic acid [
5] (
2, 104 mg, 556 μmol) and thionyl chloride (700 μL, 9.62 mmol) in hexane (2 mL) was stirred under Ar for 5 h. The solvent was removed to give 126 mg of
3 as a brownish oil (containing 10-20% of the carboxylic acid), which was used without further purification [
6].
Ethyl N-(1-phenyl-1H-pyrrole-3-carbonyl)carbamate (5)
Tetrabutylammonium isocyanate (279 mg, 981 μmol) was dried in vacuo at 65° for 1 h, dissolved under Ar in tetrahydrofuran (1.7 mL), and the solution cooled to –78°. Ethanol (abs, 50 μL), and then a precooled solution of 3 (110 mg, 535 μmol) were added under Ar. The mixture was stirred for 3 h and then the solvent removed in vacuo. The residue was chromatographed on SiO2 (27 g, dichloromethane/ethyl acetate 8:1) to give 26 mg (19%) of 5 as a colorless solid, 20 mg (20%) of 1-phenyl-1H-pyrrole-3-carboxamide, and 37 mg of unidentified by-products.
Colorless solid, m.p. 44-46°.
IR (KBr): 3279; 3132; 2980; 2930; 1750; 1676; 1599; 1511; 1269; 1201; 1057; 1033; 901; 758; 692.
1H NMR (300 MHz, CDCl3): 8.01 (s, 1H, NH); 7.78 (t, J = 2.0, 1H, H-C(2)); 7.5-7.3 (m, 5H, phenyl-H); 7.05 (dd, J = 2.3, 3.1, 1H, H-C(5)); 6.67 (dd, J = 3.0, 1.7, 1H, H-C(4)); 4.30 (q, J = 7.1, 2H, CH2); 1.33 (t, J = 6.9, 3H, CH3).
13C NMR (75 MHz, CDCl3): 161.1 (C=O); 151.4 (O-C=O); 139.5 (phenyl C(1)); 129.8 (phenyl C(3), C(5)); 127.2 (phenyl C(4)); 124.0 (C(2)); 121.1 (C(5)); 121.0 (phenyl C(2), C(6)); 120.2 (C(3)); 109.5 (C(4)); 62.1 (CH2); 14.3 (CH3).
EI MS: 259 (3); 258 (20, M+); 212 (13); 171 (13); 170 (100, [M–NH-COOC2H5]+); 115 (13); 77 (17); 51 (12).
Anal. Calcd for C14H14N2O3 (258.28): C, 65.11; H, 5.46; N, 10.85; O, 18.58. Found: C, 64.91; H, 5.67; N, 10.04; O, 17.11.
N-(3-Phenyl-2-propynoyl)-1-phenyl-1H-pyrrole-3-carboxamide (6) from 1-phenyl-1H-pyrrole-3-carbonyl chloride (3)
To a solution of tetrabutylammonium cyanate (909 mg, 3.20 mmol) in tetrahydrofuran (12 mL), 1-phenyl-1H-pyrrole-3-carbonyl chloride (3, 465 mg, 2.26 mmol) was added under Ar. After stirring the mixture for 90 min at 0°, lithium phenylacetylide (2.26 ml, 1.0 M in tetrahydrofuran, 2.26 mmol) was added and the mixture stirred for additional 3 h at 0°. The solvent was removed in vacuo and the residue chromatographed on SiO2 (100 g, dichloromethane) to give 104 mg (15%) of 6 as a colorless solid.
Colorless needles (dichloromethane/pentane), m.p. 149-151°.
IR (KBr): 3251; 3151; 2230; 2196; 1709, 1637; 1336; 1250; 754; 747.
1H NMR (300 MHz, CDCl3): 9.79 (s br, 1H, NH); 8.03 (t, J = 2.0, 1H, H-C(2)); 7.67 (dd, J = 6.8, 1.5, 2H, H-C(2), C≡C-phenyl H-C(6)); 7.45-7.35 (m, 8H, phenyl-H); 7.10 (t, J = 3.1, 1H, H-C(5)); 6.89 (dd, J = 3.1, 1.8, 1H, H-C(4)).
13C NMR (75 MHz, CDCl3): 161.2, 153.8 (2 × C=O); 139.5 (N-phenyl C(1)); 133.1 (C≡C-phenyl C(2), C(6)); 130.7 (C≡C-phenyl C(4)); 129.8, 128.5 (N-phenyl C(3), C(5), and C≡C-phenyl C(3), C(5)); 127.1 (N-phenyl C(4)); 124.4 (C(2)*); 121.3 (C(5)*); 120.8 (N-phenyl C(2), C(6)); 120.1 (C≡C-phenyl C(1)*); 119.6 (C(3)*); 110.5 (C(4)); 93.5 (C≡C-CO); 83.3 (C≡C-CO).
EI MS (70 eV): 314 (7, M+); 168 (100); 140 (5); 118 (24); 105 (10); 90 (7); 77 (15, [C6H5]+); 51 (7).
CI MS (NH3): 315 (100, [M+H]+); 186 (11, [C6H5-C4H3N-CONH2]+); 164 (5).
EI HRMS (70 eV): Calcd for C20H14N2O2: 314.1055. Found: 314.1049.
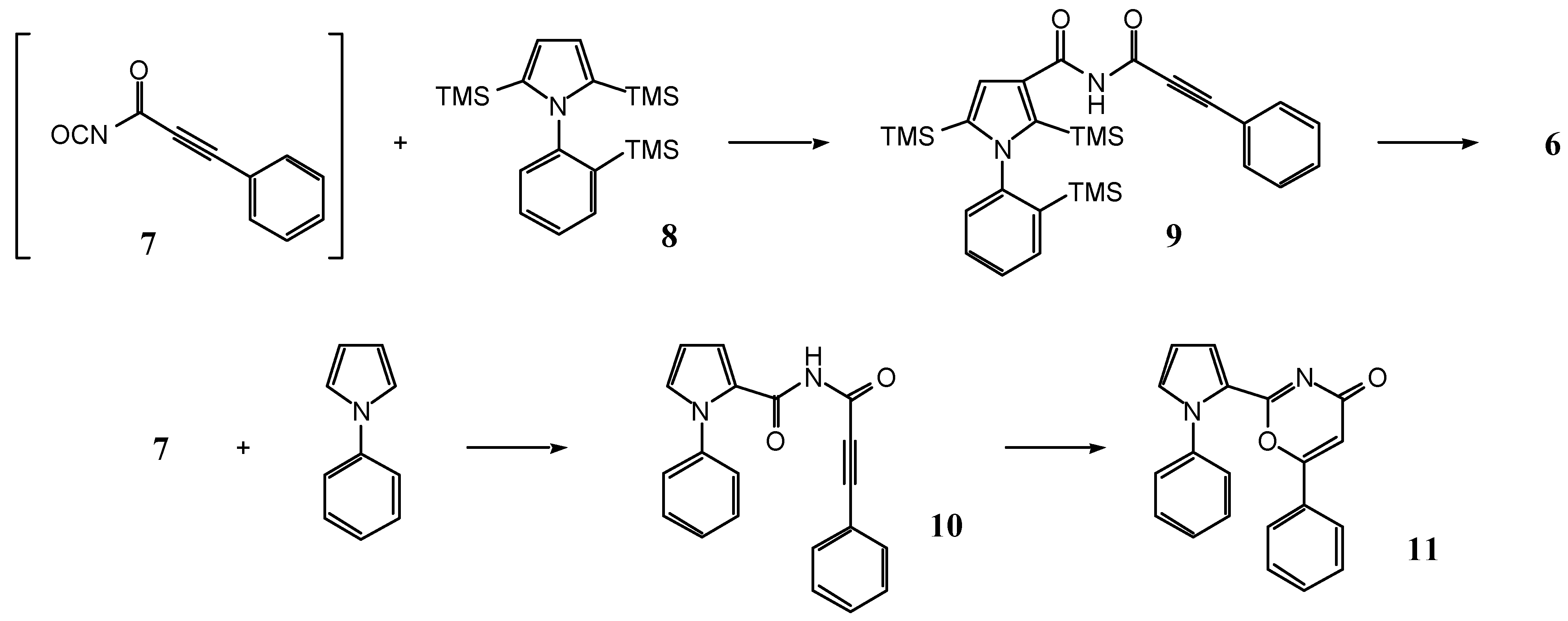
2,5-Bis(trimethylsilyl)-1-[2-(trimethylsilyl)phenyl]-1H-pyrrole (8)
Butyllithium (100 mL, 1.6 m in hexane, 160.0 mmol) was added under Ar to 1-phenyl-1H-pyrrole (5.09 g, 36.0 mmol) and N,N,N',N'-tetramethylethylenediamine (22.5 mL, 160.0 mmol). The mixture was refluxed for 23 h and then cooled to –78°. Trimethylchlorosilane (20.0 mL, 160.0 mmol) was added and the mixture stirred for 6 h at 0° and for 90 min at room temperature. After washing twice with sat. NH4Cl solution and then with water, the organic layer was dried (Na2SO4), filtered, and the solvent evaporated. The crude product (12.4 g of a yellowish oil) was purified in ten portions by chromatography on SiO2 (150 g, pentane/dichloromethane 12:1) to give 4.85 g (37%) of 8 as a yellowish oil. An analytically pure sample was obtained by kugelrohr distillation (175°/0.13 mbar).
IR (NaCl): 3060; 2956; 2897; 1479; 1247; 1166; 1120; 931; 837; 757.
1H NMR (300 MHz, CDCl3): 7.54 (dd, J = 7.3, 1.8, 1H, phenyl H-C(3)); 7.38 (td, J = 7.4, 1.5, 1H, phenyl H-C(4)*); 7.31 (td, J = 7.4, 1.7, 1H, phenyl H-C(5)*); 7.15 (dd, J = 7.7, 1.2, 1H, phenyl H-C(6)); 6.48 (s, 2H, H-C(3), H-C(4)); –0.01 (s, 9H, (CH3)3Si-phenyl); –0.09 (s, 18H, (CH3)3Si-pyrrole).
13C NMR (75 MHz, CDCl3): 147.9 (s, phenyl C(1)); 140.2 (s, phenyl C(2)); 140.0 (s, C(2), C(5)); 135.2 (d, phenyl C(3); 130.0, 128.4, 127.8 (3d, phenyl C(4), C(5), C(6)); 119.1 (d, C(3), C(4)); 0.3 (q, (CH3)3Si-pyrrole); –0.5 (q, (CH3)3Si-phenyl).
EI MS (70 eV): 359 (10, M+); 286 (24, [M–Si(CH3)3]+); 256 (23); 240 (5); 212 (9); 198 (14); 73 (100, [Si(CH3)3]+).
CI MS (NH3): 360 (100, [M+H]+); 288 (21, [M–Si(CH3)3+2H]+); 214 (7); 90 (30); 73 (6, [Si(CH3)3]+).
N-(3-Phenyl-2-propynoyl)-2,5-bis(trimethylsilyl)-1-[2-(trimethylsilyl)phenyl]-1H-pyrrole-3-carboxamide (9)
To a solution of 3-phenyl-2-propynamide [
7] (741 mg, 5.10 mmol, prepared from 3-phenyl-2-propynoyl chloride [
8]) in dichloromethane (12 mL), oxalyl chloride (482 μL, 5.62 mmol) was added under Ar at room temperature. The mixture was refluxed for 135 min and then cooled to 0°. 2,5-Bis(trimethylsilyl)-1-[2-(trimethylsilyl)phenyl]-1
H-pyrrole (
8, 820 μL, 2.10 mmol) in dichloromethane (5 mL) was added under Ar, followed by AlCl
3 (1.472 g, 11.04 mmol). The mixture was stirred for 22 h at 0° and then poured into ice/water (100 mL). After stirring for 2.5 h, the organic phase was separated, dried (Na
2SO
4), filtered, and the solvent evaporated
in vacuo. The residue was chromatographed on SiO
2 (95g, gradient dichloromethane → dichloromethane/methanol 99:1) to give 852 mg (76%) of
9 as a colorless solid.
Colorless prisms (tert-butyl methyl ether/pentane), m.p. 175-177°.
IR (KBr): 3272; 3059; 2954; 2896; 2234; 2199; 1703; 1634; 1336; 1215; 838; 760.
1H NMR (300 MHz, CDCl3): 8.87 (s br, 1H, NH); 7.66 (dt, J = 6.7, 1.6, 2H, C≡C-phenyl H-C(2), H-C(6)); 7.57 (dd, J = 7.4, 1.5, 1H, N-phenyl H-C(3)); 7.47-7.33 (m, 5H, phenyl-H); 7.12 (dd, J = 7.6, 1.5, 1H, N-phenyl H-C(6)); 6.78 (s, 1H, H-C(4)); 0.02, –0.01, –0.07 (3s, 3 × 9H, 3 × (CH3)3Si).
13C NMR (75 MHz, CDCl3): 161.7s, 152.8s (2 × C=O); 147.0s; 146.9s; 141.5s; 139.9s; 135.4d; 133.2d; 130.6d; 129.7d; 128.8d; 128.6d; 128.5d; 126.3s; 120.4s; 118.8d; 91.8s (C≡C-CO); 83.2s (C≡C-CO); 0.1q, –0.1q –0.7q (3 × (CH3)3Si).
EI MS (70 eV): 530 (6, M+); 515 (9, [M–CH3]+); 457 (7, [M–Si(CH3)3]+); 384 (28, [M–2×Si(CH3)]+); 311 (23, [M–3×Si(CH3)3]+); 129 (17); 118 (9); 89 (5); 77 (5, [C6H5]+); 73 (100, [Si(CH3)3]+ ); 45 (15).
Anal. Calcd for C29H38N2O2Si3 (530.89): C, 65.61; H, 7.22; N, 5.28. Found: C, 65.72; H, 7.30; N, 5.10.
N-(3-Phenyl-2-propynoyl)-1-phenyl-1H-pyrrole-3-carboxamide (6) from (9)
N-(3-Phenyl-2-propynoyl)-2,5-bis(trimethylsilyl)-1-[2-(trimethylsilyl)phenyl]-1H-pyrrole-3-carboxamide (9, 87 mg, 164 μmol) and tetrabutylammonium fluoride trihydrate (218 mg, 691 μmol) were stirred in tetrahydrofuran for 50 min at 60°. The mixture was then taken up with dichloromethane and washed with water. The organic phase was separated, dried over Na2SO4, filtered, and the solvent removed in vacuo. The residue was chromatographed on SiO2 (15 g, dichloromethane/methanol 99:1) to give 12 mg (23%) of 6 as a colorless solid.
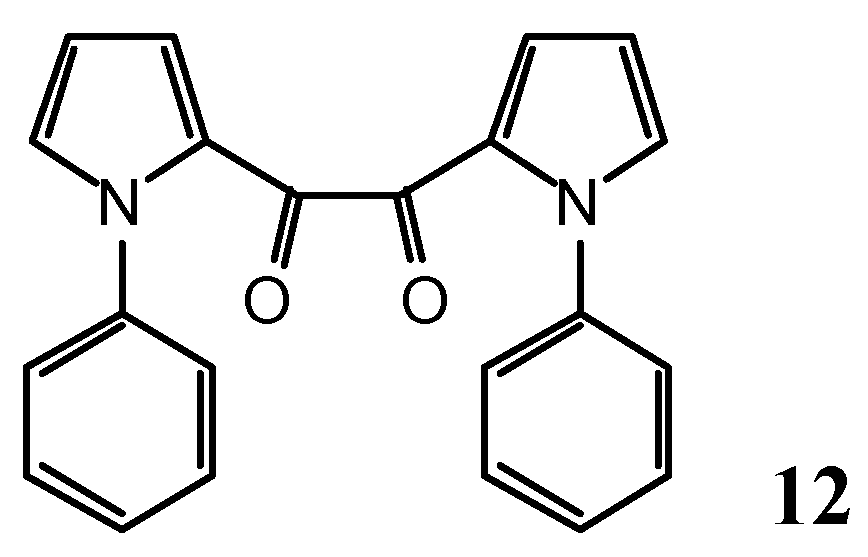
N-(3-Phenyl-2-propynoyl)-1-phenyl-1H-pyrrole-2-carboxamide (10) and 1,2-bis-(1-phenyl-1H-pyrrol-2-yl)-ethane-1,2-dione (12)
To a solution of 3-phenyl-2-propynamide [
7] (812 mg, 5.59 mmol) in dichloromethane (12 mL), oxalyl chloride (527 μL, 6.15 mmol) was added under Ar at room temperature. The mixture was refluxed for 150 min. 1-Phenyl-1
H-pyrrole (820 mg, 5.73 mmol) in dichloromethane (3 mL) was added and the mixture refluxed for 45 h. The solvent was removed and the residue chromatographed on SiO
2 (100 g, gradient dichloromethane → dichloromethane/methanol 99:1 → dichloromethane/methanol 98:2) to give 813 mg (46%) of
10.
Colorless needles (dichloromethane/pentane), mp. 125-127°.
IR (KBr): 3215; 3125; 2211; 1702; 1654; 1598; 1498; 1261; 1187; 1170; 754; 744; 694.
1H NMR (300 MHz, CDCl3): 8.74 (s br, 1H, NH); 7.55 (dd, J = 6.9, 1.5, 2H, C≡C-phenyl H-C(2), H-C(6)); 7.47-7.30 (m, 8H, phenyl-H); 7.06 (dd, J = 4.0, 1.7, 1H, H-C(3)); 7.04 (dd, J = 2.7, 1.7, 1H, H-C(5)); 6.35 (dd, J = 4.0, 2.7, 1H, H-C(4)).
13C NMR (75 MHz, CDCl3): 157.0s, 152.9s (2 × C=O); 139.8s (N-phenyl C(1)); 132.9d (C≡C-phenyl C(2), C(6)); 131.4d (C(5)); 130.5d (C≡C-phenyl C(4)); 128.8d (C≡C-phenyl C(3), C(5)); 128.3d (N-phenyl C(3), C(5)); 127.8d (N-phenyl C(4)); 125.8d (N-phenyl C(2), C(6)); 124.4s ( C(2)); 119.8s (C≡C-phenyl C(1)); 118.3d (C(3)); 109.7d (C(4)); 92.4s (C≡C-CO); 82.9s (C≡C-CO); assignments from 1H,13C COSY.
EI MS (70 eV): 314 (25, M+); 291 (13); 168 (100); 140 (12); 118 (55); 105 (28); 90 (16); 77 (36, [C6H5]+); 51 (22).
Anal. Calcd for C20H14N2O2 (314.34): C, 76.42; H, 4.49; N, 8.91; O, 10.18. Found: C, 76.33; H, 4.48; N, 8.85; O, 10.23.
As a by-product, 1,2-bis-(1-phenyl-1H-pyrrol-2-yl)-ethane-1,2-dione (12, 168 mg, 9%) was isolated.
Yellow leaflets (dichloromethane/pentane), m.p. 127.5-130.5°.
IR (KBr): 3127, 3064; 1644; 1629; 1495; 1407; 1351; 777; 753; 733; 698.
1H NMR (300 MHz, CDCl3): 7.41-7.27 (m, 10H, phenyl-H); 7.10 (dd, J = 4.1, 1.7, 2H, pyrrole H-C(3)); 7.03 (dd, J = 2.5, 1.7, 2H, pyrrole H-C(5)); 6.28 (dd, J = 4.1, 2.5, 2H, pyrrole H-C(4)).
13C NMR (75 MHz, CDCl3): 181.2 (C=O); 139.8 (phenyl C(1)); 132.9 (pyrrole C(3)*); 128.6 (phenyl C(3'), C(5)); 128.2 (pyrrole C(2)); 127.8 (phenyl C(4')); 125.7 (phenyl C(2), C(6)); 124.9 (pyrrole C(5)*); 110.4 (pyrrole C(4')).
EI MS (70 eV): 340 (7, M+); 170 (100, [C6H5C4H3NCO]+); 115 (31); 77 (5, C6H5]+).
Anal. Calcd for C20H16N2O2 (340.38): C, 77.63; H, 4.74; N, 8.23. Found: C, 77.15; H, 4.73; N, 8.19.
6-Phenyl-2-(1-phenyl-1H-pyrrol-2-yl)-[1,3]oxazin-4-one (11)
N-(3-phenyl-2-propynoyl)-1-phenyl-1H-pyrrole-2-carboxamide (10, 33 mg, 105 mmol) was heated under Ar to 160° for 25 min. After cooling, the brownish oil was chromatographed on SiO2 (24 g, dichloromethane/methanol 99:1) to give 22 mg (67%) of 11 as a brownish solid.
Yellowish prisms (hexane/dichloromethane), m.p. 174-176°.
IR (KBr): 3075; 2922; 1671, 1640; 1553; 1495; 1448; 1348; 948; 767; 734; 698.
1H NMR (300 MHz, CDCl3): 7.56 (dd, J = 4.0, 1.8, 1H, pyrrole H-C(3)); 7.49-7.39 (m, 6H, N-phenyl-H and C=C-phenyl H-C(4)); 7.26 (t, J = 8, 2H, C=C-phenyl H-C(3), H-C(5)); 7.09 (dd, J = 2.6, 1.8, 1H, pyrrole H-C(5)); 6.94-6.90 (m, 2H, C=C-phenyl H-C(2), H-C(6)); 6.48 (s, 1H, H-C(5)); 6.45 (dd, J = 4.0, 2.6, 1H, pyrrole H-C(4)).
13C NMR (75 MHz, CDCl3): 168.1s (C(4)); 162.2s, 158.8s (C(2), C(6)); 140.7s (N-phenyl C(1)); 132.2d (pyrrole C(5)); 131.7d (C=C-phenyl C(4)); 129.2s (C=C-phenyl C(1)); 128.2d (N-phenyl C(4)); 129.5d, 128.8d, 126.0d, 125.3d (C=C-phenyl and N-phenyl C(3), C(5), C(2), C(6)); 123.1s (pyrrole C(2)); 122.3d (pyrrole C(3)); 111.0d (pyrrole C(4)); 103.8d (C(5)); assignments from 1H,13C COSY.
EI MS (70 eV): 314 (22, M+); 168 (100); 140 (7); 115 (10); 77 (19, [C6H5]+).
Anal. Calcd for C20H14N2O2 (314.34): C, 76.42; H, 4.49; N, 8.91; O, 10.18. Found: C, 76.42; H, 4.62; N, 8.64; O, 10.22.
Crystals obtained from ethanol/water were subjected to X-ray structure determination [
3].

{kind=link}
{kind=link}
{kind=link}
{kind=link}