Synthesis of (3Z)-Dodecenyl-(E)-2-butenoate, the Pheromone of Sweet Potato Weevil

{kind=link}
Abstract
:Introduction
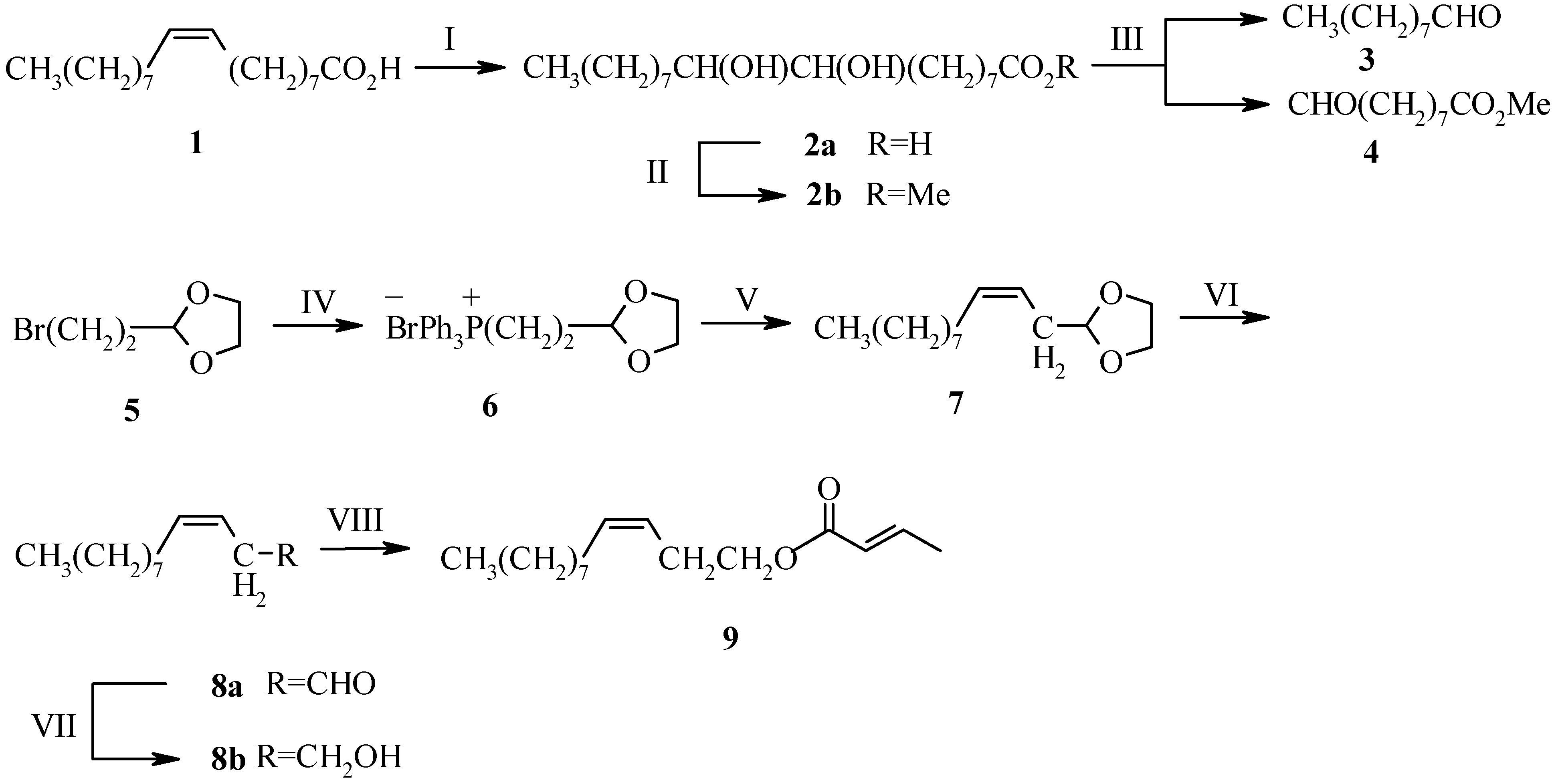
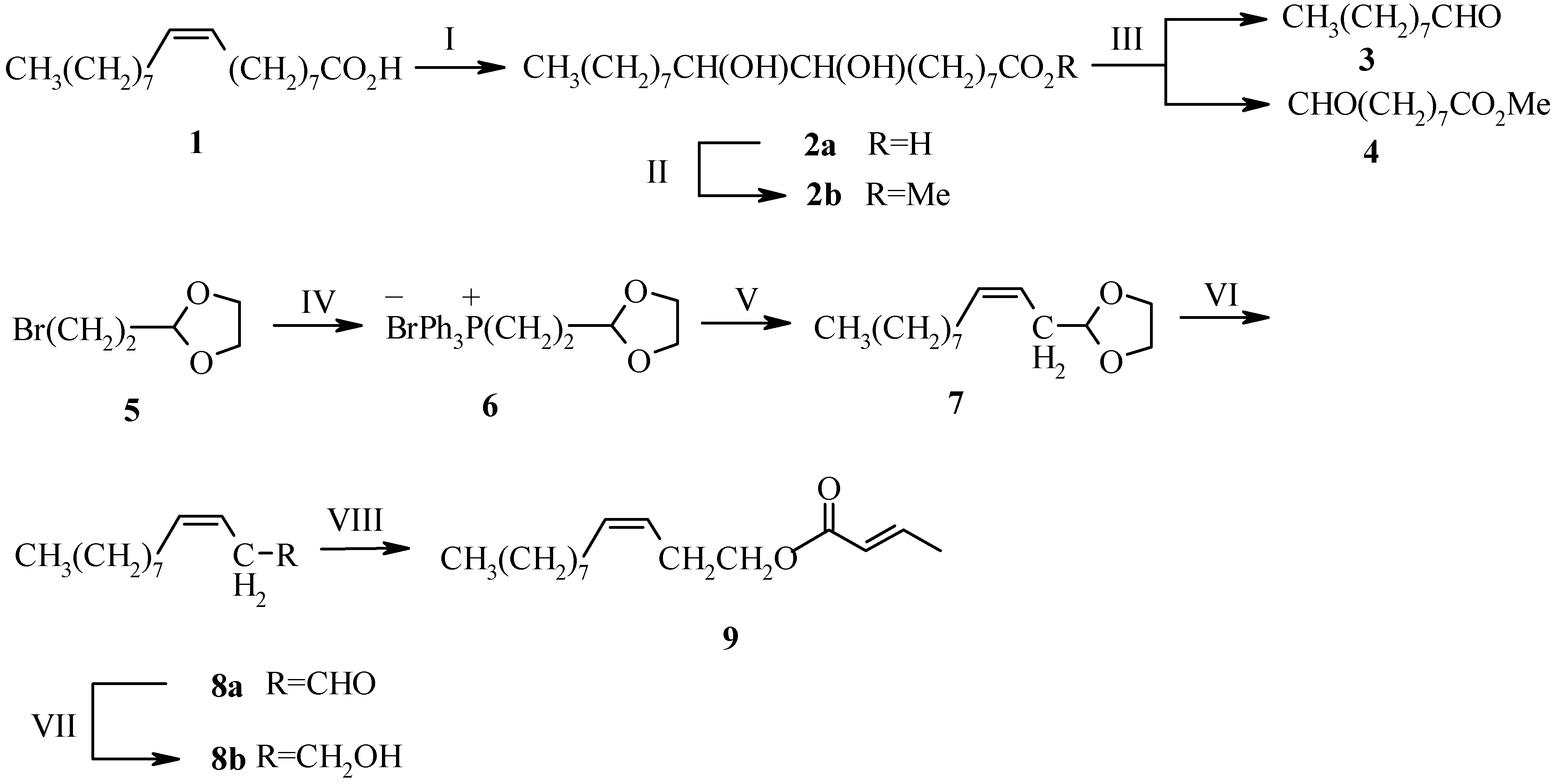
Experimental
Methyl 9,10-Dihydroxyoctadecanoate (2b)
Nonanal (3)
3-Bromopropanal Ethyleneglycol Acetal (5)
1,3-Dioxolan-2-yl-methyltriphenylphosphonium Bromide (6)
(3Z)-Dodecenal (8a)
(3Z)-Dodecen-1-ol (8b)
(3Z)-Dodec-3-enyl-(E)-2-butenoate (9)
References and Notes
- Martin, F. W. Proc. Am. Soc. Horti. Sci. Trop. Sect. 1983, 27B, 61.
- Heath, R. R.; Coffelt, J. A.; Sonnet, P. E.; Proshold, F. I.; Dueben, B.; Tumlinson, J. H. J. Chem. Ecol. 1986, 12, 1489. [PubMed]
- Nesbitt, B. F.; Beevor, P. S.; Cork, A.; Hall, D. R.; Murillo, R. M.; Leal, H. R. Entomol. Exp. Appl. 1985, 38, 81.
- Wilemse, L. P. M.; Booji, C. J. H.; Voerman, S. J. Appl. Entomol. 1987, 103, 508.
- Carvalho, J. F.; Prestwich, G. D. J. Org. Chem. 1984, 49, 1251.
- Handong, L.; Yanneng, D.; Hansheng, X.; Li, T.; Huisheng, Q. Youji Huaxue 1988, 8, 167, Chem. Abstr. 1989, 110, 23584x..
- Pawar, A. S.; Chattopadhyay, S.; Mamdapur, V. R. Indian J. Chem. 1993, 32B, 463.
- Mani, N. S.; Nair, M. S. Indian J. Chem. 1993, 32B, 1151.
- Singh, A. N.; Mhaskar, V. V.; Sukhdev. Tetrahedron 1978, 34, 595.
- Buchi, G.; Wuest, H. J. Org. Chem. 1969, 34, 1122.
- Heath, R. R.; Coffelt, J. A.; Sonnet, P. E.; Proshold, F. I.; Dueben, B.; Tumlinson, J. H. J. Chem. Ecol. 1986, 12, 489.
- Doree, C.; Pepper, A. C. J. Chem. Soc. 1942, 477.
- Samples Availability: Available from the authors.
© 1999 by the authors. Reproduction of this article, by any means, is permitted for noncommercial purposes.
Share and Cite
Mithran, S.; Subbaraman, A.S. Synthesis of (3Z)-Dodecenyl-(E)-2-butenoate, the Pheromone of Sweet Potato Weevil. Molecules 1999, 4, 159-164. https://doi.org/10.3390/40600159
Mithran S, Subbaraman AS. Synthesis of (3Z)-Dodecenyl-(E)-2-butenoate, the Pheromone of Sweet Potato Weevil. Molecules. 1999; 4(6):159-164. https://doi.org/10.3390/40600159
Chicago/Turabian StyleMithran, S., and A. S. Subbaraman. 1999. "Synthesis of (3Z)-Dodecenyl-(E)-2-butenoate, the Pheromone of Sweet Potato Weevil" Molecules 4, no. 6: 159-164. https://doi.org/10.3390/40600159
APA StyleMithran, S., & Subbaraman, A. S. (1999). Synthesis of (3Z)-Dodecenyl-(E)-2-butenoate, the Pheromone of Sweet Potato Weevil. Molecules, 4(6), 159-164. https://doi.org/10.3390/40600159