The Need for Systematic Naming Software Tools for Exchange of Chemical Information

1
Advanced Chemistry Development Inc., 133 Richmond Street West, Suite 605, Toronto, Ontario, M5H 2L3 Canada
2
ACD, Inc., ul. Akademika Bakuleva, 6, str. 1, Moscow 117513 Russia
*
Author to whom correspondence should be addressed.
Molecules 1999, 4(9), 255-263; https://doi.org/10.3390/40900255
Submission received: 20 June 1999
/
Accepted: 31 July 1999
/
Published: 1 September 1999
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The availability of systematic names can enable the simple textual exchange of chemical structure information. The exchange of molecular structures in graphical format or connection tables has become well established in the field of cheminformatics and many structure drawing tools exist to enable this exchange. However, even with the availability of systematic naming rules, software tools to allow the generation of names from structures, and hopefully the reversal of these systematic names back to the original chemical structure, have been sorely lacking in capability and quality. Here we review the need for systematic naming as well as some of the tools and approaches being taken today in this area.
Introduction
“Well honey, I couldn’t convince the doctor that I needed 5-{2-ethoxy-5-[(4-methyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one to help my high (3S,9S,14S,17R)-17-(1,5-dimethylhexyl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15, 16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol levels. Sorry.”
Results and Discussion
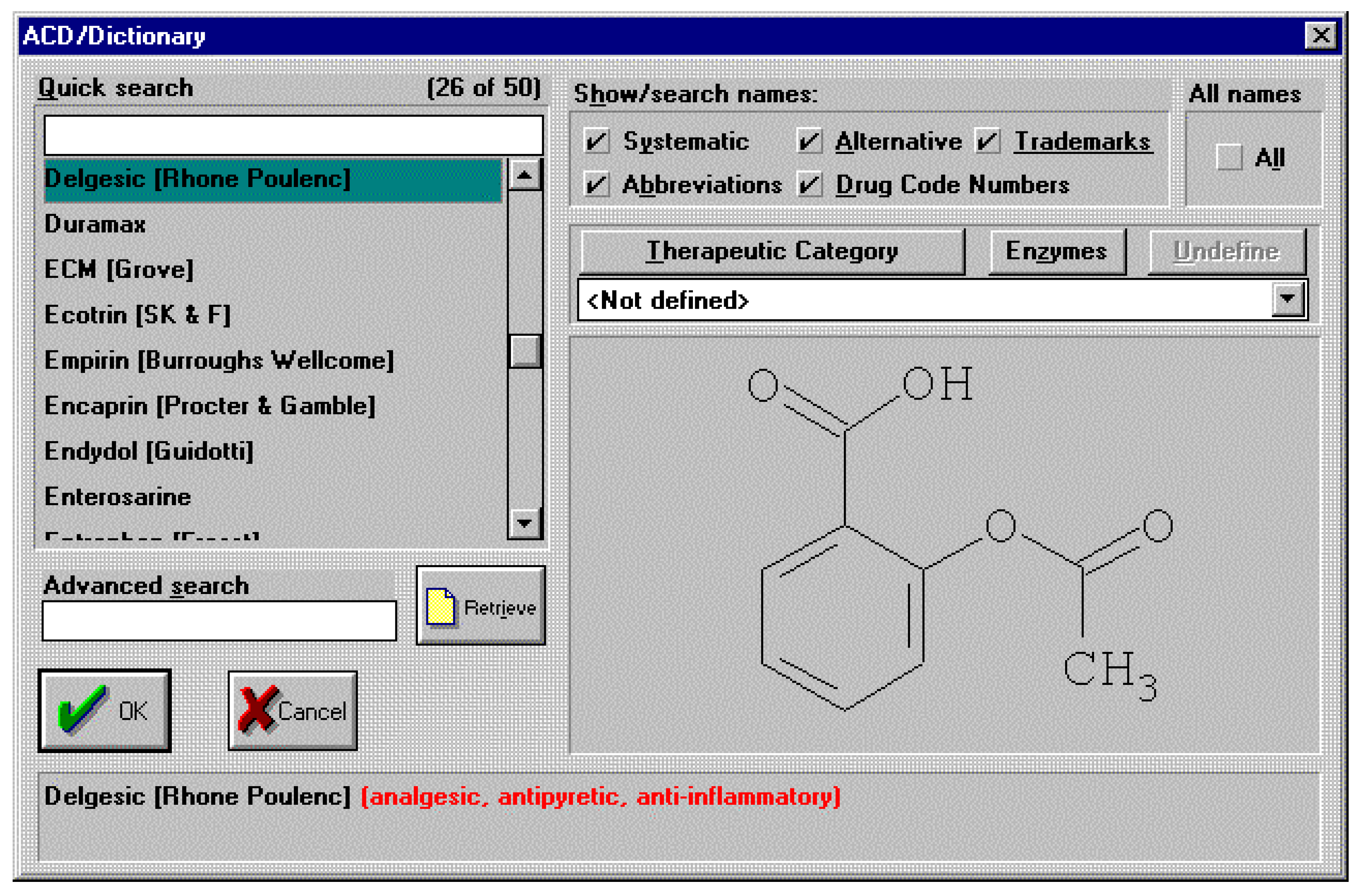
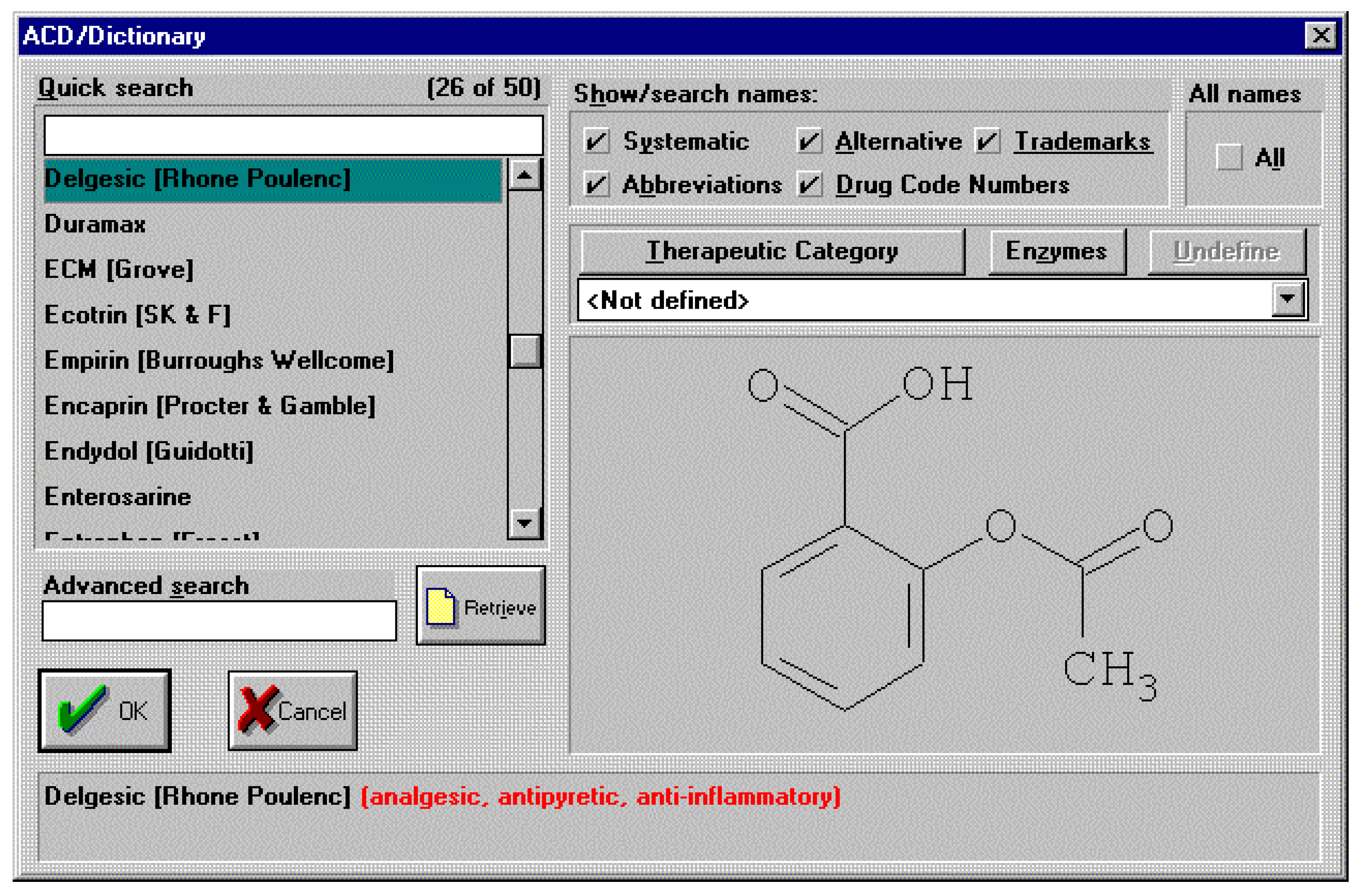
It’s truly unlikely that this type of conversation between a man and his wife, after his visit to the doctor, will ever happen in any household in the near future. As well as the sheer effort of remembering the details of a particular chemical name, the subtleties of pronunciation alone are more than enough to warrant the use of more general names when discussing pharmaceutical drugs for example. So, we can assume that the need for generic names will remain for the foreseeable future. From a skilled chemists point of view most of us will know that aspirin is in fact 2-(acetyloxy)benzoic acid. However, how many of us would know that 2-(acetyloxy)benzoic acid is also the chemical for the drugs with generic names of acenterine, aceticyl, acetophen, acetosal, acetosalin, acetylin, aspro, caprin, claradin, duromax, ecotrin, helicon, levius, rhodine, xaxa and over 35 other variants! An exhaustive list can be found inside the electronic chemical dictionaries available today. An example screen shot is shown below for the ACD/Dictionary. This dictionary, like many others, is integrated to a structure drawing package for defining the chemical structure of interest by which to search the dictionary.
Figure 1.
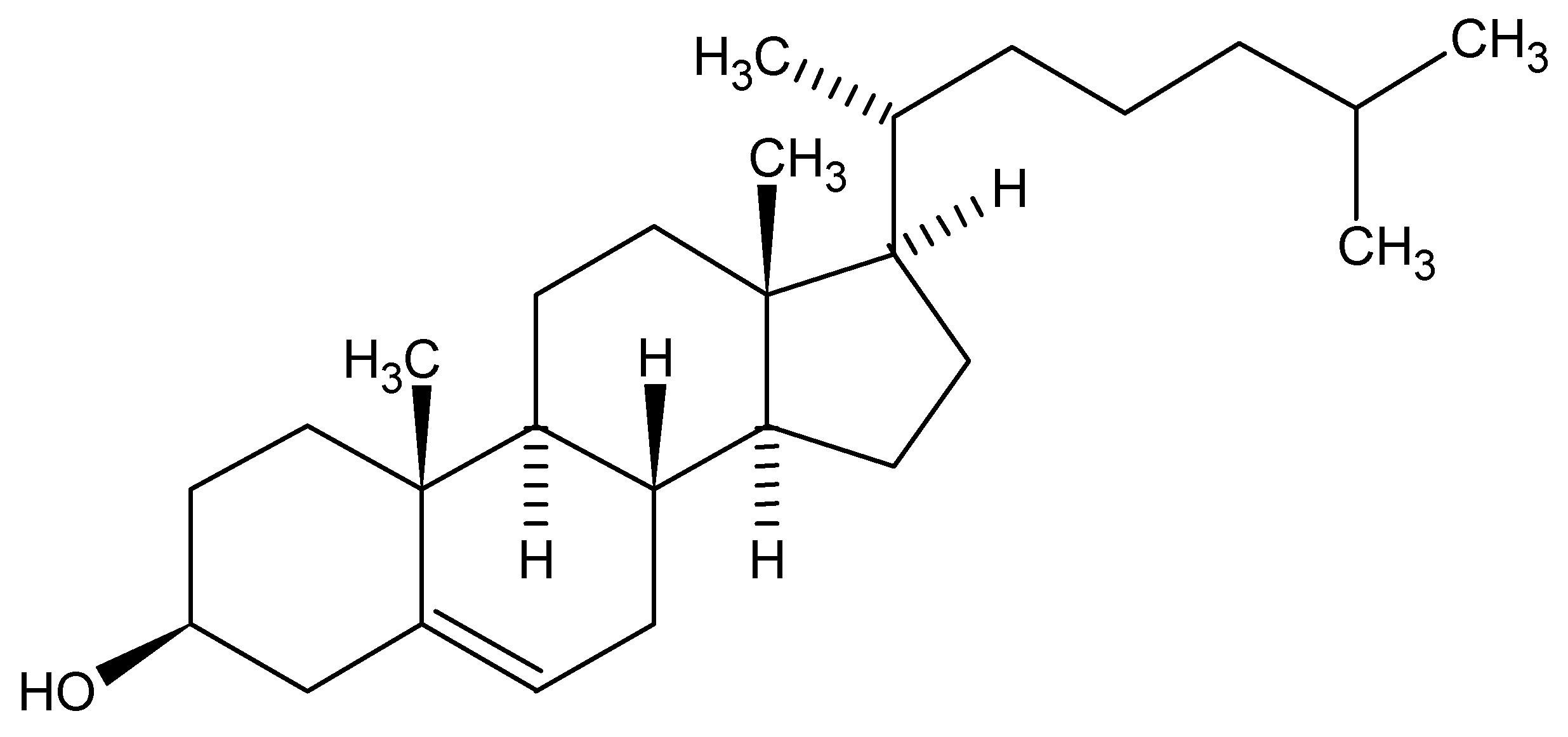
So, with such possible variations in generic names, but all applicable to a single chemical structure, we can acknowledge that the chosen manner for communication would be the molecular structure itself. Herein lies a problem. How can I orally or even in a written format communicate a structure when a molecular structure exists as a 2D or 3D rendering? As an example, cholesterol is a pretty demanding structure to draw, never mind communicate between colleagues. Surely we all acknowledge the medical effects of high levels of the bad type and low levels of the good type of cholesterol but only as chemists are we concerned with exchanging actual structural detail. The twodimensional structure of cholesterol is shown below:
Figure 2.
Cholesterol.
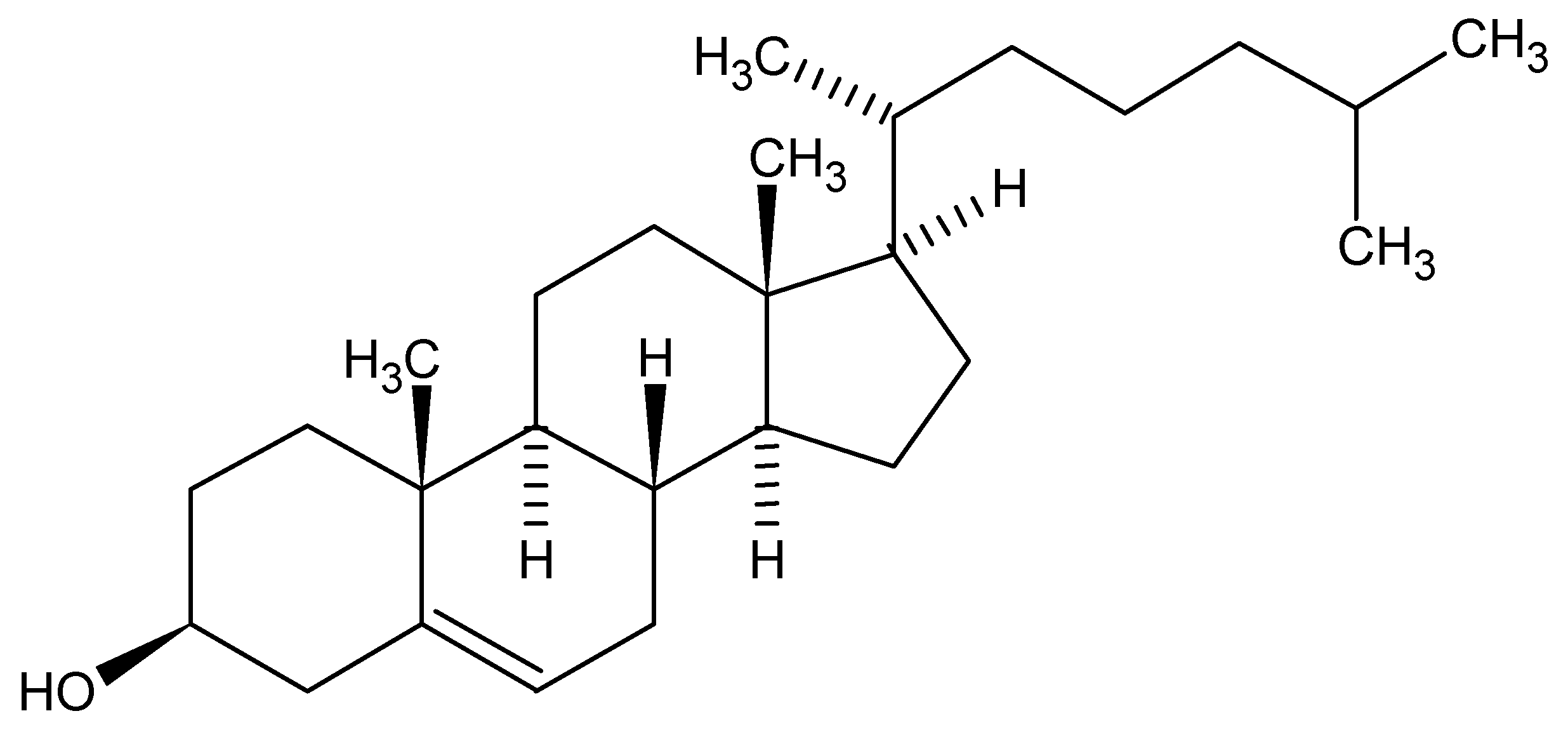
Note that the structure is carrying the stereochemical center descriptions in the bond orientations within the figure. Today, in the days of electronic exchange, via email attachments, FAX or simple global networks, there would have no problem in communicating issues around cholesterol since we would just send an image of the structure (as above captured as a word processor image), or an actual structure in some standard file format, such as Molfile, which all structure drawing packages should be able to exchange easily. Alternatively, for this structure I could just mention the word Cholesterol and most of us would immediately acknowledge the structure to be a steroid core and the detailed structure could easily be found in a high school chemistry text or at the library, however time demanding and frustrating that process would turn out to be.
Returning to the issue in question however is oral or written, not graphical communication of a chemical structure. Patent attorneys, pharmacists and benchtop chemists do not all work in a consistent exchange format of molecular structure information. Thus the need exists for simple exchange between the multiple formats of graphical structures and text. We have already explained lookup dictionaries to deal with generic names but the gap which remains is a systematic nomenclature system whereby a chemical structure can be named according to agreed upon systematic rules. Such systems do exist but herein lies yet another problem.
There are actually two widely adopted nomenclature rule bases. These are those of the International Union of Pure and Applied Chemistry (IUPAC) and the Chemical Abstracts Service (CAS). These different sets of rules are similar but different in some very distinct ways. The rules were developed over the years and are continually in some state of flux to deal with new components of molecular diversity and possible conflicts or confusions that have been identified with the rules that exist today.
The application of rules to deal with all of historical structural space has actually been well achieved. The results are rules which can be applied to complex structures in order to generate a text string which is a direct representation of these rules, and, if the rules are applied correctly by separate parties, the same systematic name will result. Rather than require human intervention at this stage however, it would only make sense to allow software to assume the generation of a systematic name directly from a chemical structure since any rules based system can be translated into an electronic conversion engine, in this case from structure to name. Such software must be correct to the largest extent possible and should encompass as many of the systematic rules available today to cover as much of electronic structure space as possible. There have been a number of attempts to deal with electronic generation of systematic names. These have included programs such as Beilstein Autonom, Cheminnovation Nomenclator and the most complete of these packages, Advanced Chemistry Development’s IUPAC Name and Index Name programs which cover both the IUPAC and CAS-rules based system.
In each case these software packages have delivered to the marketplace a structure drawing interface which is used as the input screen for the chemical structures [1]. Also, each of the applications has also been integrated with other well known structure drawing packages available in the marketplace such as MDL/Isis and Chemdraw [2]. As a result, there are no shortages of interfaces to input a molecular structure requiring systematic naming. The accuracy of the resulting names generated is by far the most exacting test of the software but the ease of use and the richness and flexibility of the interface are also crucial points. A comparison of the capabilities of each of the naming software packages mentioned above is available [3]. Here we will review the issues of accurate naming only using ACD/Name as the test module.
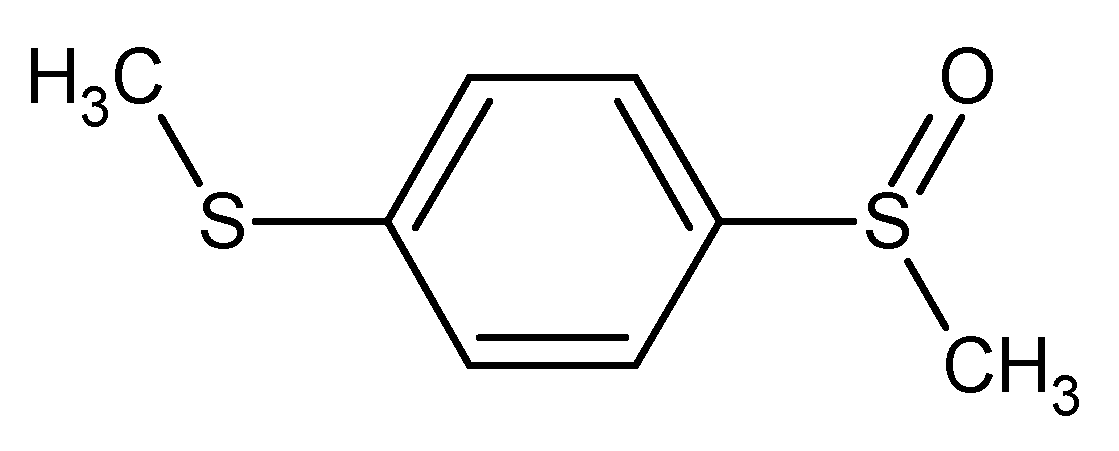
In an ideal world the systematic naming rules developed by either of the two bodies would result in a unique name. In this way, a single name is a unique descriptor which, like the molecular structure itself is a direct and non-confusing representation of the molecular detail. Unfortunately, this is not the case. A single molecular structure can result in a series of different names due to the application of the naming rules.
Figure 3.
1-(methylsulfanyl)-4-(methylsulfinyl)benzene* methyl 4-(methylsulfanyl)phenyl sulfoxide 1-(methylsulfinyl)-4-(methylthio)benzene** methyl 4-(methylthio)phenyl sulfoxide bis(methylthio)benzene monooxide
Figure 3.
1-(methylsulfanyl)-4-(methylsulfinyl)benzene* methyl 4-(methylsulfanyl)phenyl sulfoxide 1-(methylsulfinyl)-4-(methylthio)benzene** methyl 4-(methylthio)phenyl sulfoxide bis(methylthio)benzene monooxide
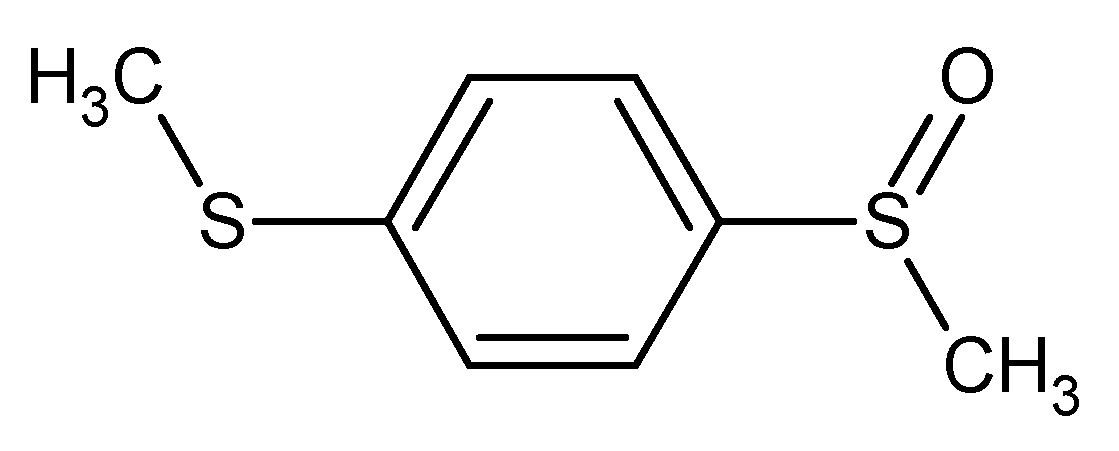
For example these five names could be considered as IUPAC names and the first (*) is the most preferred according to current recommendations, and the name labelled with ** is the CAS name.
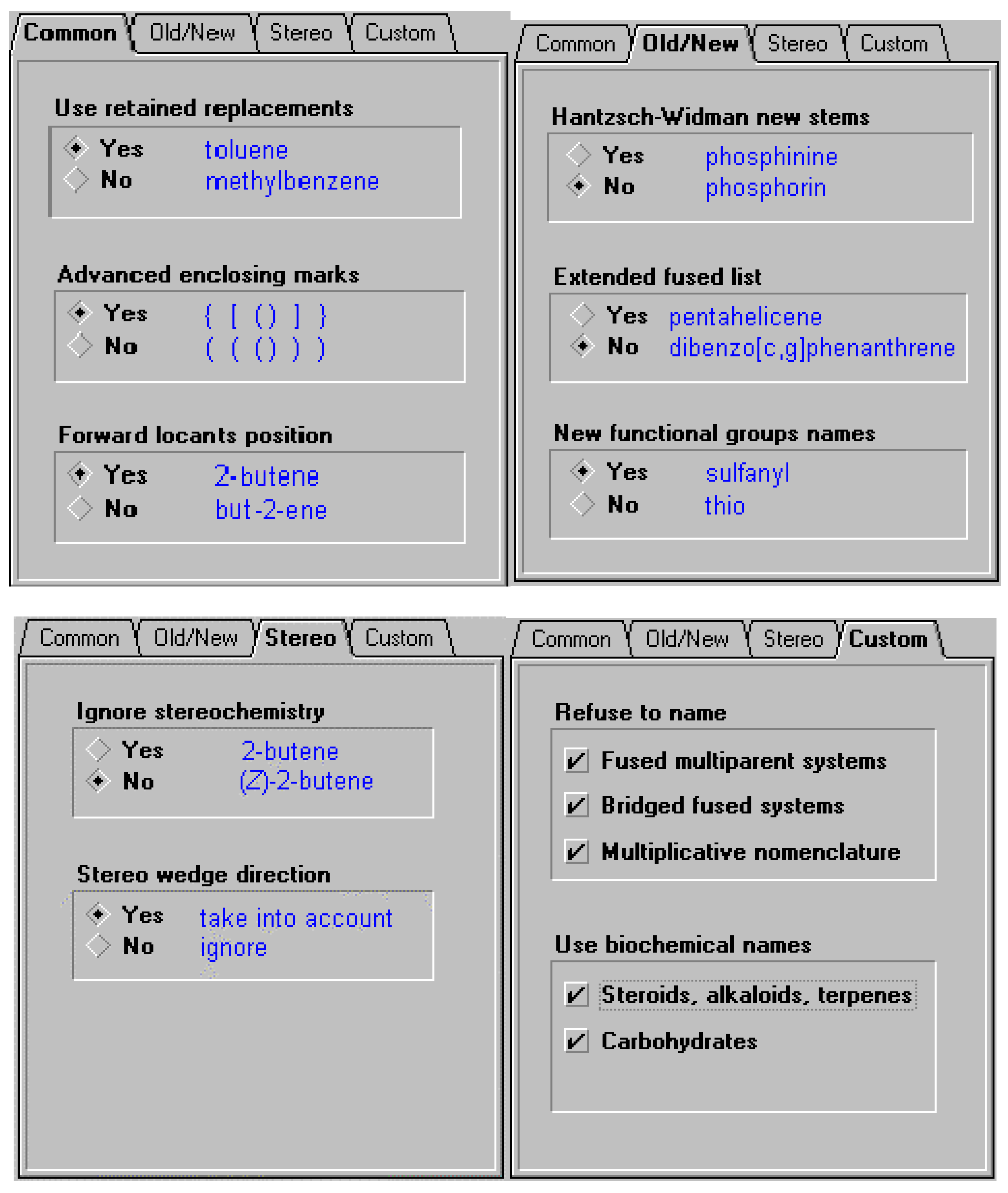
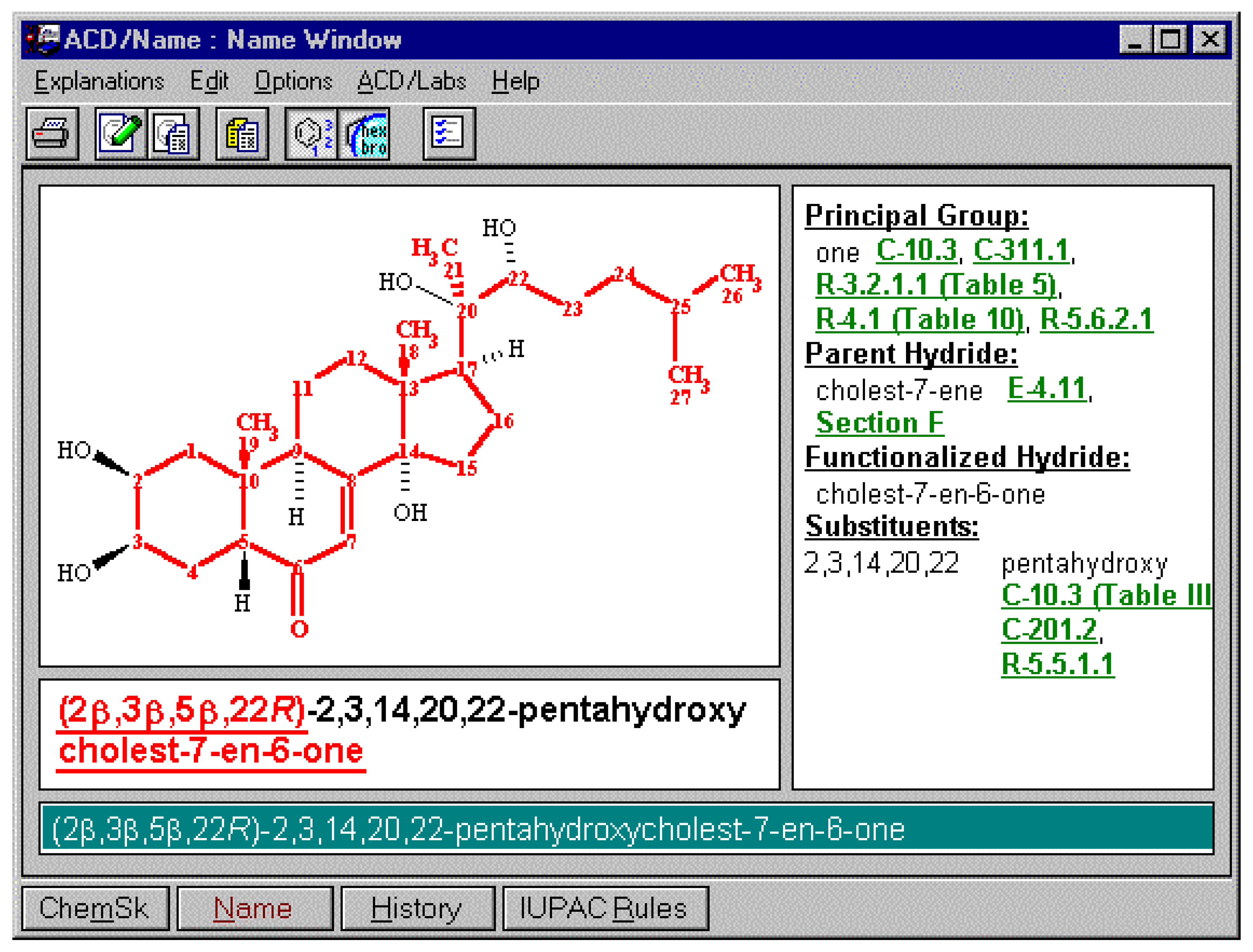
In order to offer the user the ability to use the different variations of naming options supported by the different bodies (or local conventions, for example, a corporate practice to use previously recommended thio and mercapto nomenclature instead of the current sulfanyl, or alternatively the forward locant position instead of internal locant ), certain options may need to be supported. These include some of the options listed below. So, in the case of cholesterol, there are a number of names that can be generated based on settings. In the latter case the option to support naming according to IUPAC and CAS Index rules for steroids, alkaloids and terpenes was selected. Some of the options available for naming are illustrated below.
Figure 4.
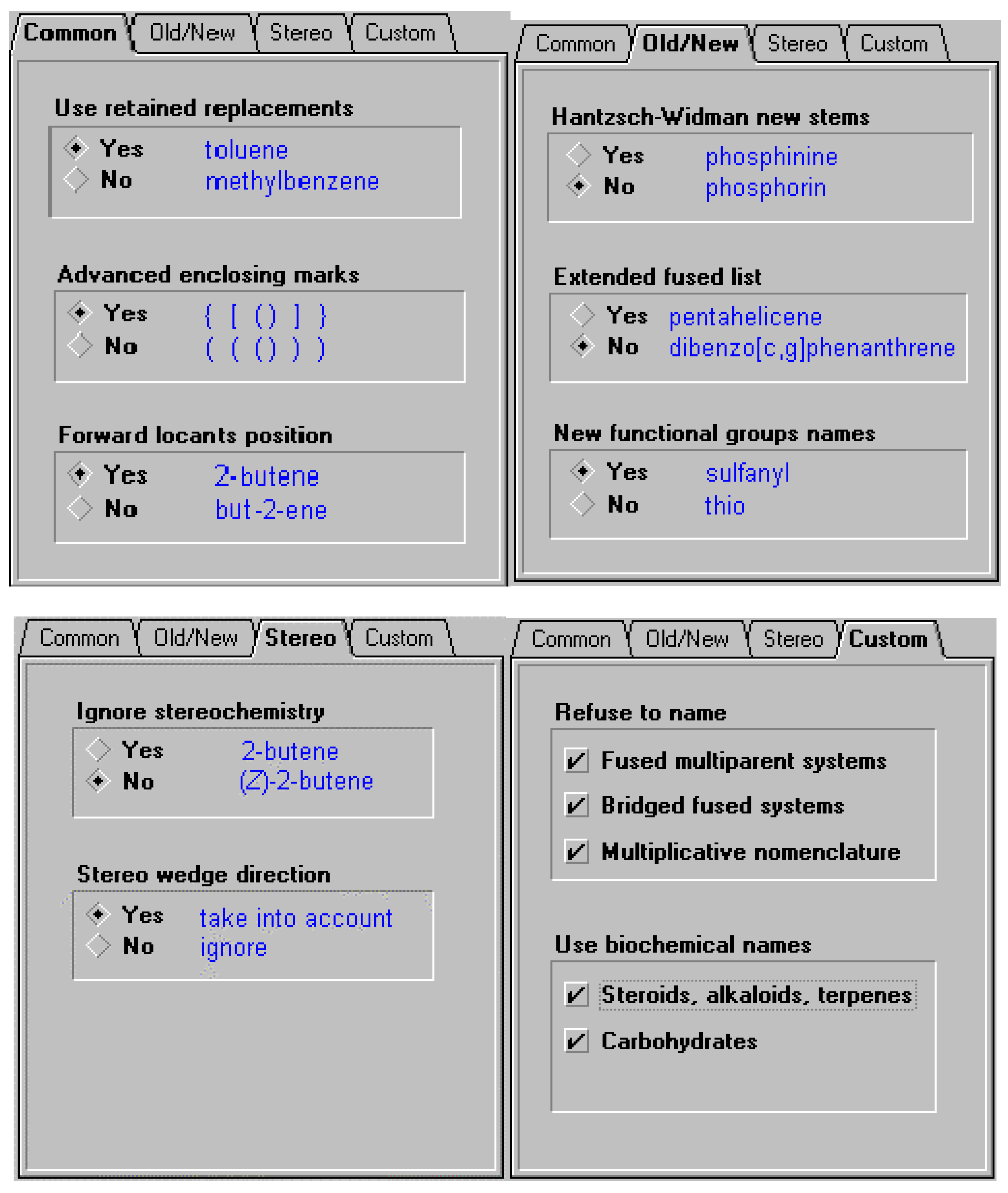
Figure 5.
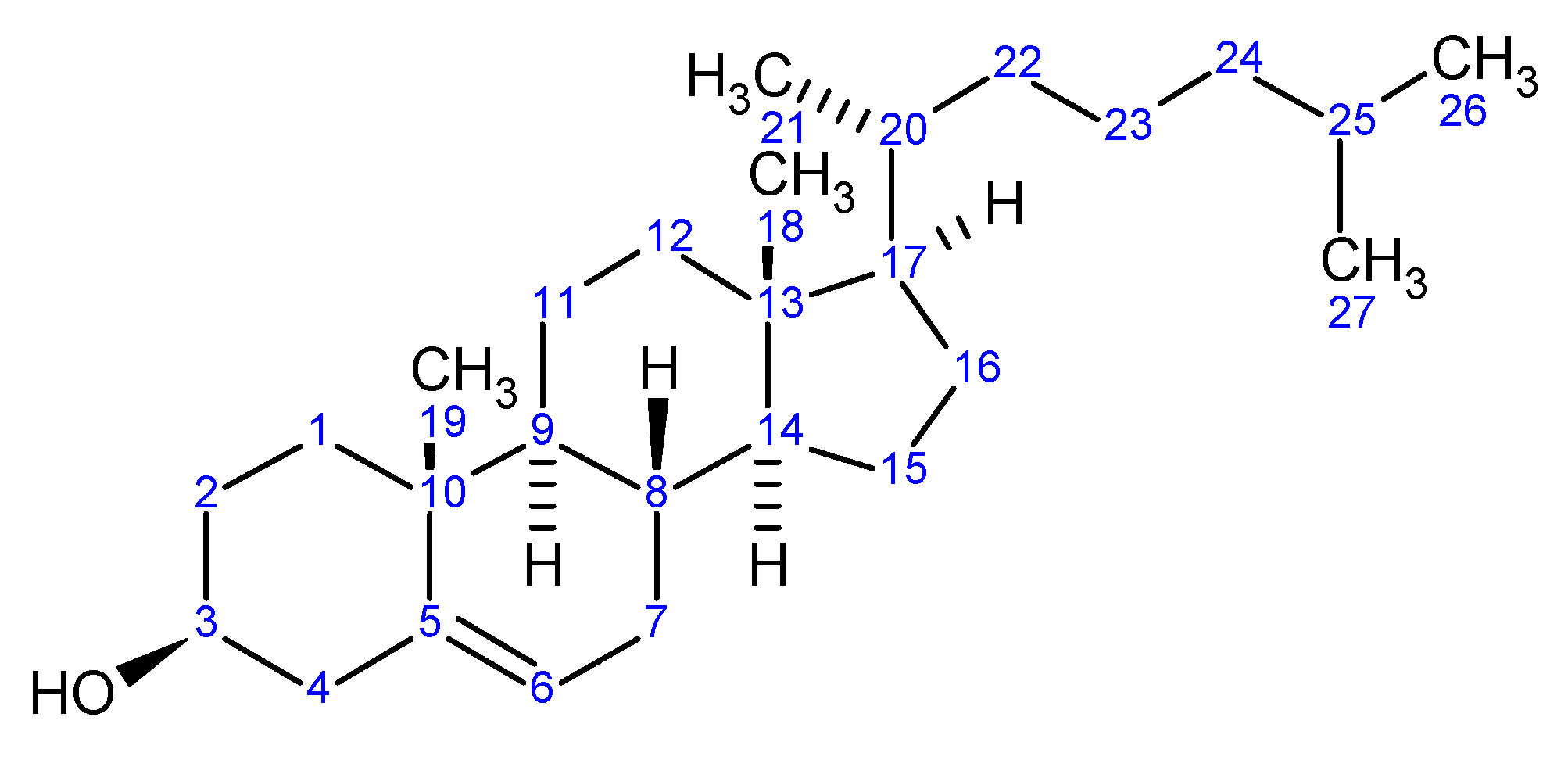
1. Using the IUPAC Naming system: (3S,9S,14S,17R)-17-(1,5-dimethylhexyl)-10,13-dimethyl - 2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol
2. Using IUPAC Rules system and option for steroids naming: (3β)-cholest-5-en-3-ol
3. [4] Using CAS Index Rules system: cholest-5-en-3-ol, (3β)-
Notice that in the example above the details regarding stereochemistry is available in the stereochemical designations for each of the centers. However, when the option to name according to approved steroidal cores is selected, the naming produces solely the unique aspects of the cholest-5-en-3-ol molecule which is the 3β-stereochemistry. Using the steroidal molecular core as the basis for naming, substitution of the molecule will result in a IUPAC name containing the (3β)-cholest-5-en-3-ol name component as well as all of the substitution detail (see below):
Figure 6.
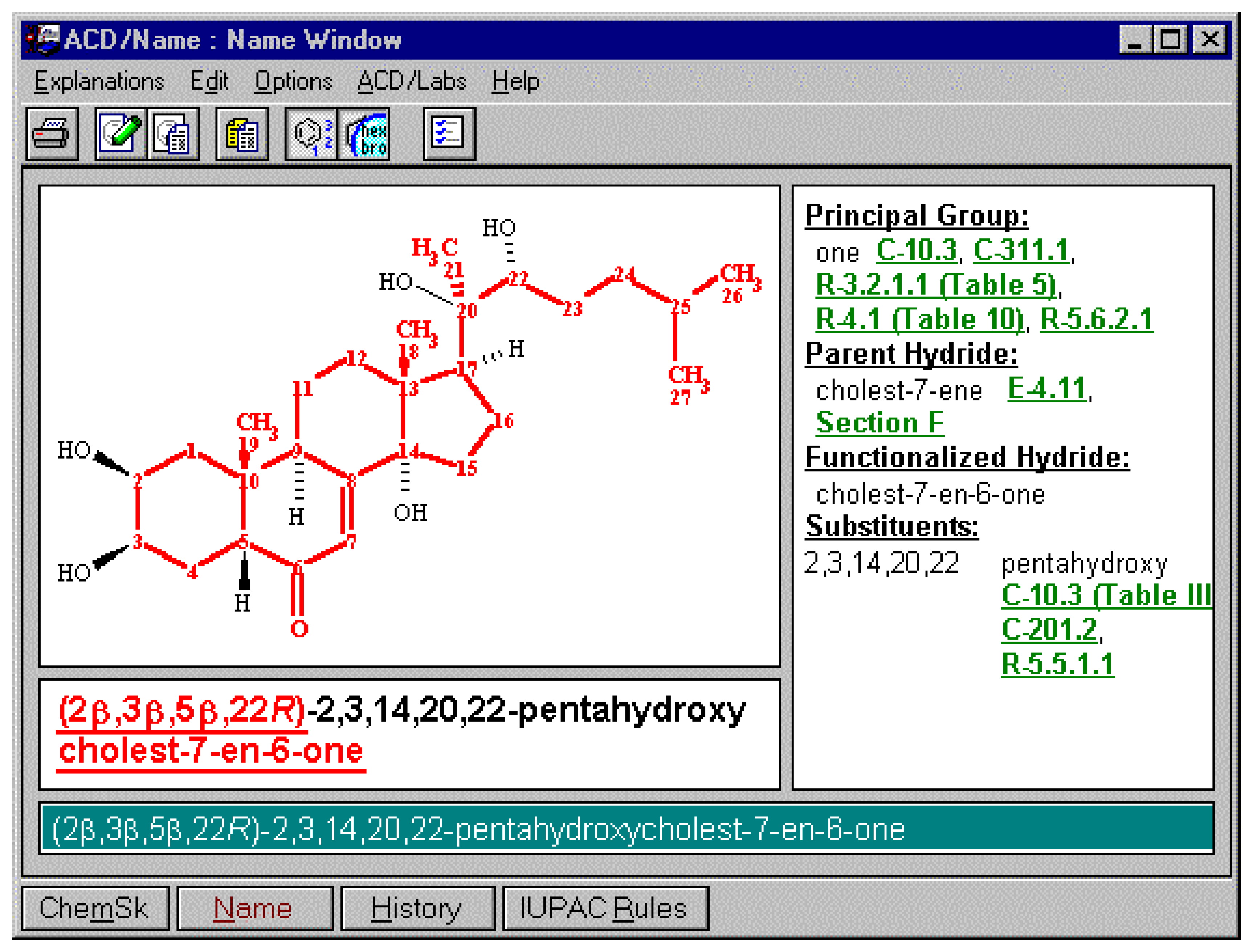
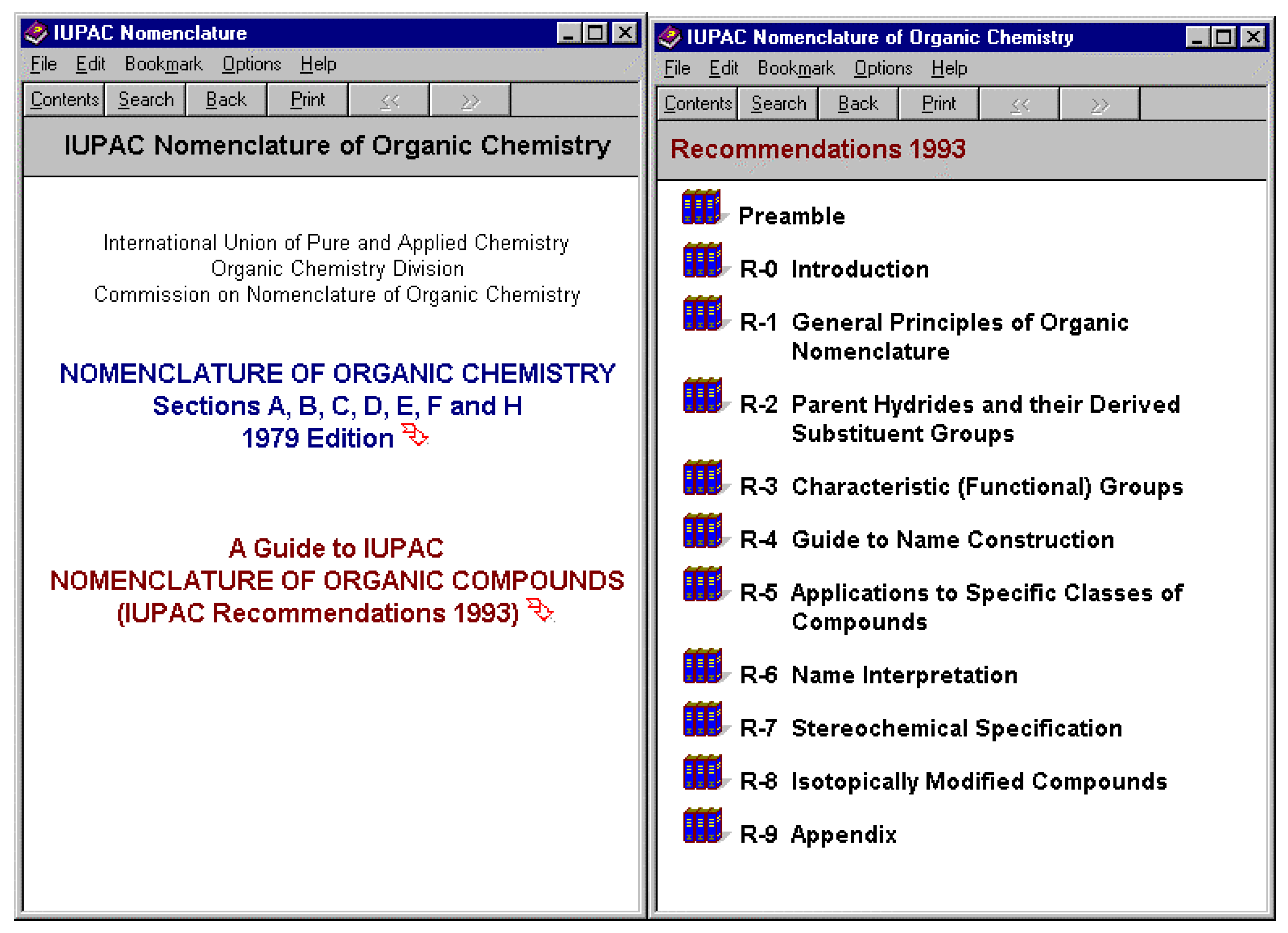
Systematic generation of names would be insufficient without allowing the user to access the details of the rules that have been applied. As a result of this need the IUPAC rules have been made available as a part of the suite and the appropriate selection can be made from the connecting links listed above to the appropriate IUPAC rule. The IUPAC rules in this searchable format are also available for download from www.acdlabs.com.
Figure 7.
It should be added that there is another form of nomenclature that can be applied to molecular structures and this is the SMILES string. A SMILES string is simply a string of characters that carries within its code the details of the elements and their connectivies. Once again, this can be converted from its string format back through a connectivity table into the form of a 2D molecular display using the appropriate SMILES to structure converter. For cholesterol a valid SMILES string is::
SMILES:
CC(C)CCC[C@@H](C)[C@H]1CC[C@H]2[C@@H]3CC=C4C[C@@H](O)CC[C@]4(C)[C@H]3CC[C@]12C
There are as many valid SMILES strings for cholesterol as there are ways to correctly order the atoms and this can be. There are numerous SMILES converters available but the general application is to use SMILES strings as the textual input describing a structure and convert the string to an actual connection table in order to provide the structure in a form that can be used as input to a prediction or search engine. Therefore the reverse process of SMILES to structure conversion is a necessary component of any SMILES parser. The SMILES definition has certain limitations in regard to completeness but for general organics is a simple and widely used “nomenclature” .
So far we have seen that the rigorous application of systematic naming rules according to specified conventions is necessary in order to generate an appropriate name for a chemical compound. These rules need to be applied faithfully and correctly with an ideal of zero errors in naming. However, flexibility in the different preferences available for naming can immediately lead to confusion.
The reverse process of taking a chemical name and generating the chemical structure, a process espoused to be of more general application to benchtop chemists, is also of general utility. There are three software packages available today that can perform this name to structure conversion. These are ChemInnovation’s NamExpert, a module within Cambridgesoft’s ChemDraw Ultra and also a module from Advanced Chemistry Development presently under beta-test. It is to be expected that the critical application of rules for reverse generation of a structure from a name will need to be as definitively stipulated as those required to generate a name from a structure. A recent presentation [5] emphasized that the actual generation of structures from names is a simpler task than the application of systematic rules to name a structure. However, we remain extremely cautious when it comes to accuracy, even in the reverse direction of name to structure, as some of the resulting errors generated by software are extremely noteworthy [3].
It is generally obvious that software for the generation of systematic names either in a structure to name or reverse mode can be an extremely useful tool for the benchtop synthetic chemist looking for a manner by which to transfer chemical information in a textual format. Care must be taken in the decisions used to generate such names including accuracy of the software tool applied, the preference settings during the naming process and appropriate representation of the structure prior to naming since stereochemical details are captured within the naming procedures. It is possible to test the naming process and accuracy online at no charge using the ACD/Interactive Lab at www.acdlabs.com/ilab using the integrated structure drawing and online prediction service should you wish.
While it is unlikely that the conversation “Well honey, I couldn’t convince the doctor that I needed 5-{2-ethoxy-5-[(4-methyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-propyl-1,6-dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-one to help my (3S,9S,14S,17R)-17-(1,5-dimethylhexyl)-10,13-dimethyl- 2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol levels. Sorry.” will ever occur. Who knows whether the converse, with appropriate trivial names in place, will also ever happen? “Well honey, I couldn’t convince the doctor that I needed Viagra to help my cholesterol levels”.
References and Notes
- Advanced Chemistry Development provides their structure drawing software, ACD/Chemsketch at no charge from their website at www.acdlabs.com/download. The package includes 2D drawing, 3D optimization, tautomer identifier, 3D viewer with different graphic modes. Over 20,000 copies have been downloaded at the time of writing (June 15th 1999).
- Free modules integrating to both Cambridgesoft ChemDraw and MDL/ISIS are available from www.acdlabs.com/download. For ChemDraw these are certain physical property prediction modules and for MDL/ISIS this is the integrated 3D structure display module.
- Review of different naming products including Beilstein Autonom, Cheminnovation Nomenclator, and Advanced Chemistry Development’s ACD/Name (IUPAC rules) and ACD/Index Name (CAS-based rules) - www.acdlabs.com/products/name
- The correct CAS Index name is generated if the preference for steroid naming is selected. The possibility to disable natural product names is included according to the CAS practice to add additional systematic names for some natural products. However, the main name is based on the steroid. It is possible to generate a more complete CAS Name by switching off the natural product preference.
- Jonathan Brecher, Cambridgesoft, ACS Spring Meeting 1999.
© 1999 by the authors. Reproduction of this article, by any means, is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Williams, A.; Yerin, A. The Need for Systematic Naming Software Tools for Exchange of Chemical Information. Molecules 1999, 4, 255-263. https://doi.org/10.3390/40900255
AMA Style
Williams A, Yerin A. The Need for Systematic Naming Software Tools for Exchange of Chemical Information. Molecules. 1999; 4(9):255-263. https://doi.org/10.3390/40900255
Chicago/Turabian StyleWilliams, Antony, and Andrey Yerin. 1999. "The Need for Systematic Naming Software Tools for Exchange of Chemical Information" Molecules 4, no. 9: 255-263. https://doi.org/10.3390/40900255