Synthesis of the Formal Diels-Alder Adducts of N-substituted Dehydromaleimides and Anthracene

Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200F, 3001 Heverlee, Belgium
*
Author to whom correspondence should be addressed.
Molecules 2000, 5(2), 179-188; https://doi.org/10.3390/50200179
Submission received: 12 January 2000
/
Accepted: 14 February 2000
/
Published: 21 February 2000
{kind=link}
{kind=link}
{kind=link}
Abstract
:A new class of roof-shaped dibenzobarrelenemaleimide derivatives was prepared by the condensation of a bridged maleic anhydride with amines. A para-substituted bifunctional derivative was proven to be a 1:2 complex with acetone by an X-ray crystallographic study.
Introduction
Anthracene can behave as a diene, affording bridged adducts which are derivatives of dibenzobarrelene, in combination with a number of dienophiles. Anthracene adducts have been used by Dougherty et al. to prepare cyclophanes which can act as receptors for ammonium ions [1]. Roof-shaped derivatives of dibenzobarrelene have been used by Weber and coworkers as hosts for inclusion compounds [2]. Hart et al. have prepared a family of macromolecules, the iptycenes, which are based on the triptycene (tribenzobarrelene) skeleton, by a strategy of multiple Diels-Alder cycloadditions [3]. Some of the ipticenes were shown to form inclusion complexes. Porous polymer films, containing anthracene adducts, have been found to behave as sensors for dinitrotoluene, possibly allowing the application of this system as a land mine detector [4].
Results and Discussion
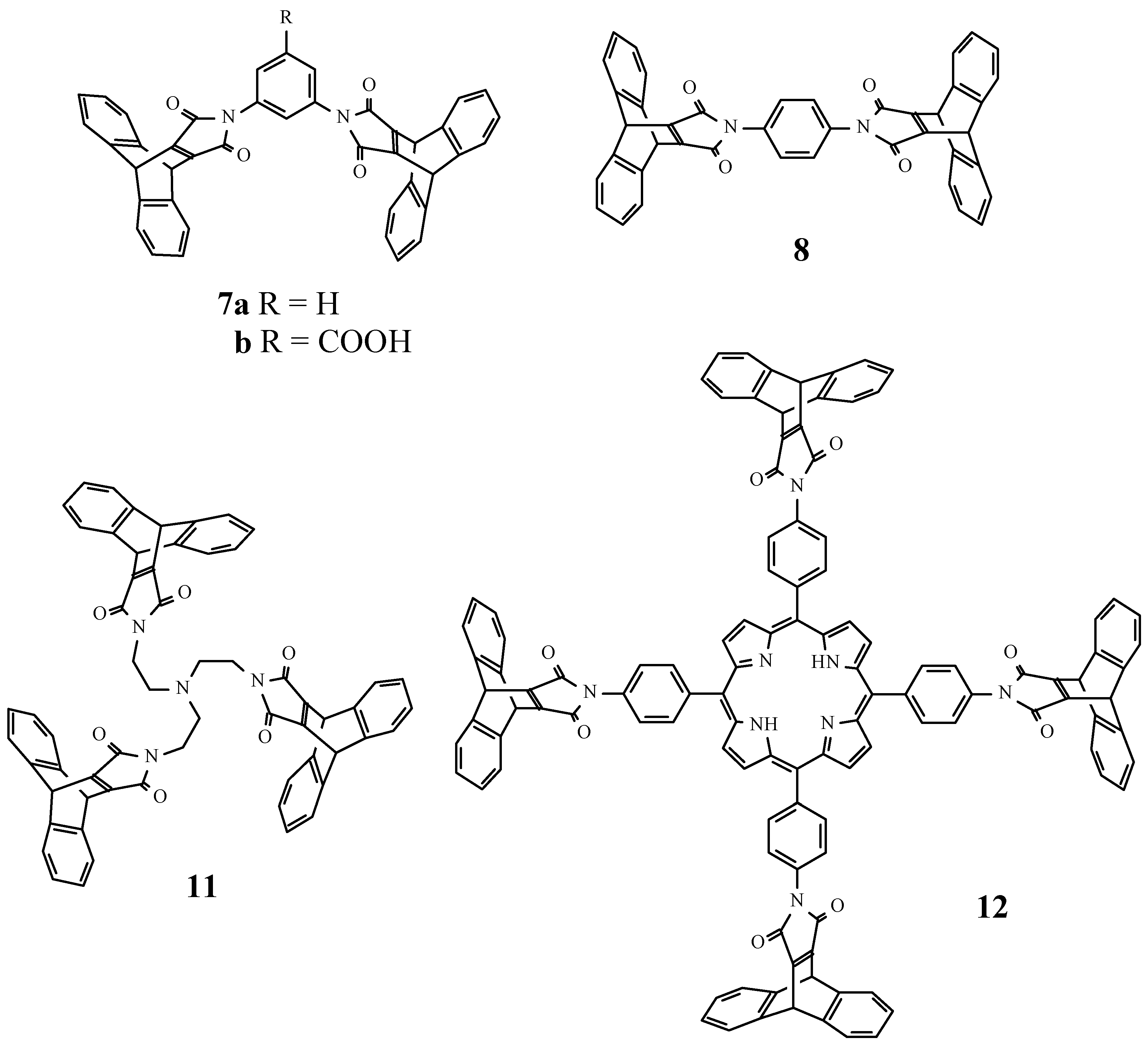
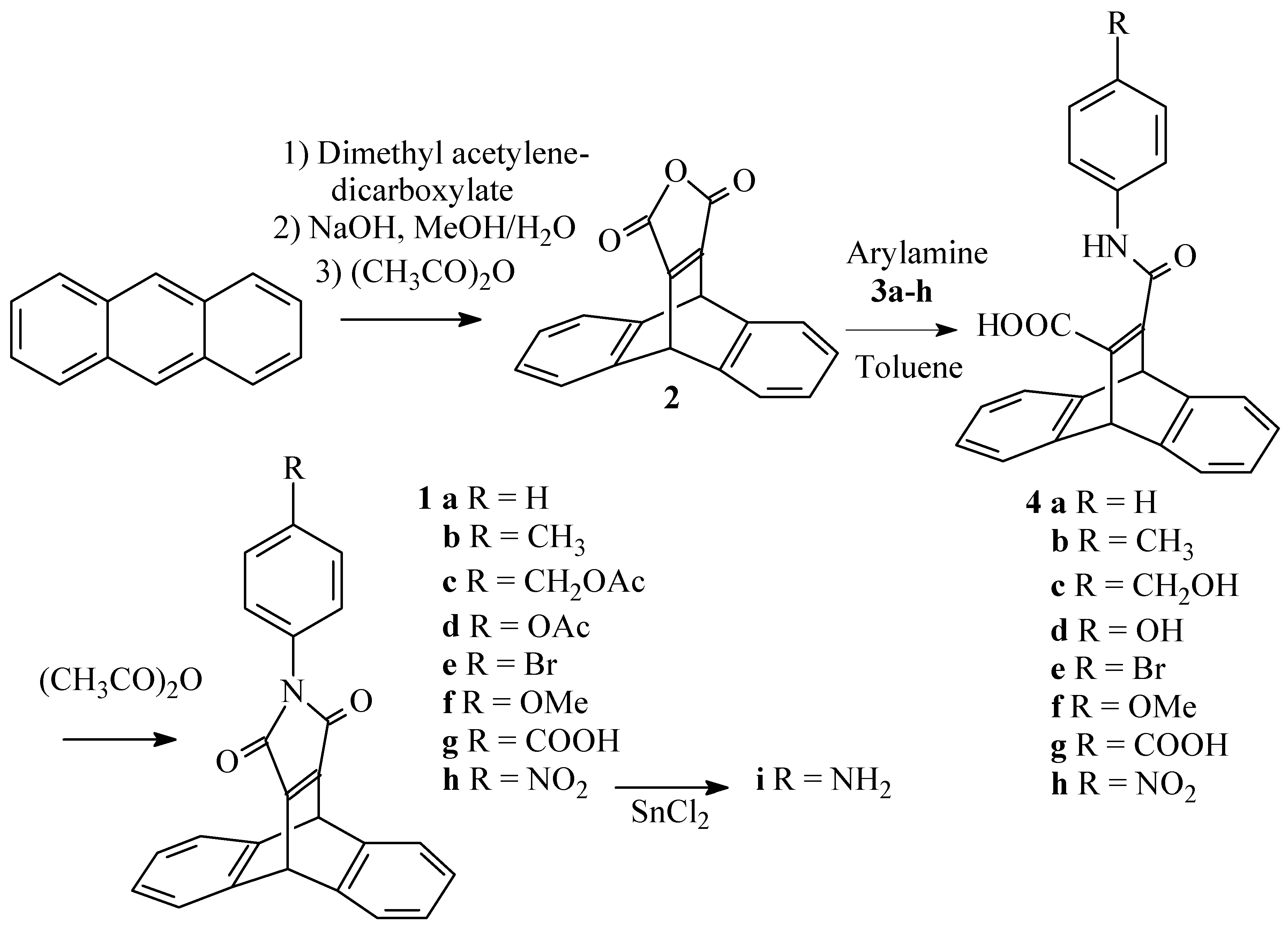
We were interested to introduce several dibenzobarrelene units within a single molecule. In order to have a maximum of symmetry, the connection could be made through the nitrogen of a pyrrole ring fused to the dibenzobarrelene, as in the maleimides 1 (Scheme 1). To the best of our knowledge, these products have not been described before. The dehydromaleimides required to prepare 1 in a direct Diels-Alder cycloaddition reaction obviously are not available, therefore we had to devise an alternative procedure.
The anhydride 2 (Scheme 1) was prepared in good overall yield starting from anthracene and dimethyl acetylenedicarboxylate by a slight modification of the original procedure of Diels and Alder [5]. Treating 2 with amines 3a-h gave the ring opened amides 4a-h which could be isolated, but more conveniently were cyclized immediately with acetic anhydride to yield the maleimides 1a-f. In the case of 1c,d, the hydroxy functions of 4c,d were acetylated at the same time as the cyclization. The nitro function of 1h could be reduced to afford amine 1i. This transformation is compatible with the reactive maleimide part of the molecule. This variation in substituents will allow a choice of methods for connecting dibenzobarrelenemaleimide units to given molecules.
We have investigated if direct introduction of several maleimides into one molecule was possible, starting from oligoamines and anhydride 2. The meta- and para-diaminobenzenes 5a,b and 6 were transformed into the bismaleimides 7a,b and 8, respectively. Ortho-diaminobenzene gave no maleimide product under the same circumstances. Tris- and even tetrakis-substituted derivatives 11 and 12 were obtained in good yield starting from the corresponding tris- and tetrakisamines 9 and 10. The derivatives 7a, 8, 11, and even the porphyrin 12 have high solubilities in organic solvents such as toluene, chloroform and dichloromethane, which allowed their characterisation by 1H and 13C NMR spectroscopy. The tetrakismaleimide 12 was significantly better soluble in organic solvents than the corresponding tetrakisamine 10 [6], which demonstrates the ability of the dibenzobarrelene moieties to prevent aggregation (Figure 1).
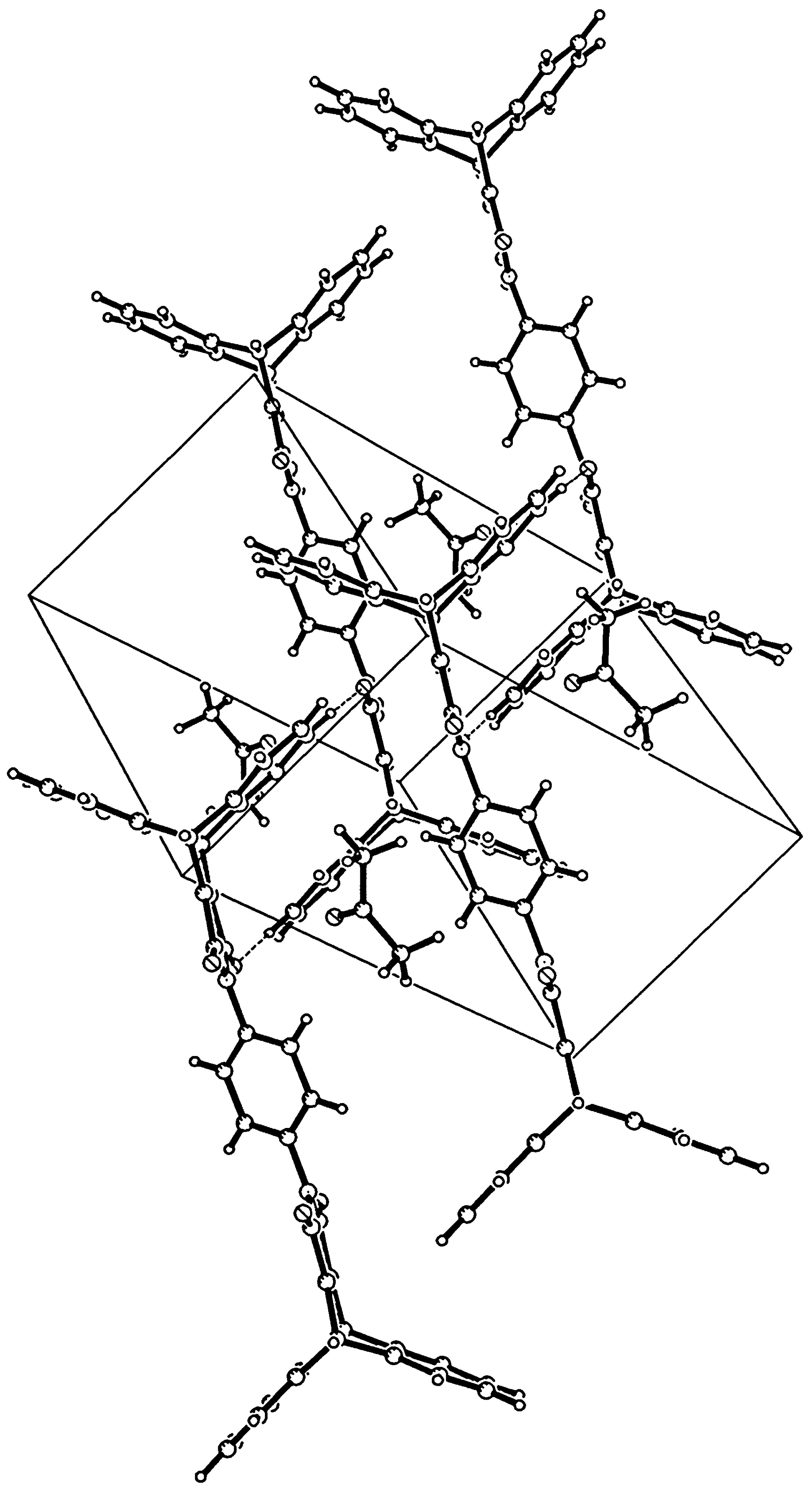
Crystals from 8 were grown by slow evaporation from an acetone solution, extensively dried and an X-ray crystallographic study was undertaken. In the solid state the molecule possesses a crystallographic inversion centre at the midpoint of the central phenyl ring. The maleimide ring is rotated by 52.4° with respect to the central phenyl ring. The holes in the crystal packing (Figure 2) are in an ordered manner filled with acetone, of which the oxygen is oriented towards the maleimide nitrogen atom (O…N 3.301 Å). Taking the contents of the unit cell into account, a 1:2 inclusion complex with acetone is formed. The inclusion of acetone in the crystals of 8 bodes well for the application of these novel 9,10-pyrroloanthracenes, and further study is underway to evaluate their use as host compounds.
Experimental
General
All reagents and solvents were purchased from Acros Organics and used without further purification. Each new compound was fully characterized by IR (Perkin Elmer 1600 FTIR), mass spectrometry (Perkin Elmer, EI 70 eV) in addition to 1H-NMR and 13C-NMR (Bruker AMX 400 MHz or Brucker WM 250 MHz). Chemical shifts are relative to TMS as an internal reference.
Synthetic procedures and spectral data
9,10-Dihydro-9,10-ethenoanthracene-11,12-dicarboxylic anhydride (2) [5]
A suspension of anthracene (15 g; 0.084 mol) and dimethyl acetylenedicarboxylate (15 ml) was heated for 45 minutes at 170°C, after which heating is continued for another 5 minutes at 180°C. The reaction mixture is cooled and the adduct crystallized from methanol. This compound was taken up in a solution of 5g of NaOH in a mixture of 75 ml of methanol and 25 ml of water, and the resulting suspension was refluxed for 1 hour. The reaction mixture was cooled down and left overnight at -20°C. The crystals formed were collected by filtration and concentrated hydrochloric acid was added to this salt to liberate the free acid. This was collected by filtration and taken up in warm acetic anhydride (50 ml) in the presence of sodium acetate (0.5 g). The resulting suspension was heated at 80°C for 30 minutes. The reaction mixture as cooled down and the solvent was evaporated in vacuo. The resulting solid was crystallized from ether/hexane (1/2) (60 ml) and obtained in 71% yield (overall): mp : 256°C (lit.4 247°C) ; IR (KBr, cm-1) : 1767 (s br, CO), 1635 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.54 (s, 2H, 9,10-H), 7.00-7.10 (m, 4H, 2,3,6,7-H), 7.36-7.48 (m, 4H, 1,4,5,8-H); 13C-NMR (62.5 MHz/CDCl3) δ (ppm) :47.8,124.9,126.2,142.9,160.4; MS (EI, m/z): 274 M+.
General procedure for the preparation of the maleimides (1a-h)
A suspension of the anhydride (2) (3.62 mmol) and the amine (3a-h) (3.80 mmol) in toluene (10 ml) was refluxed for 30 minutes. The resulting suspension was evaporated in vacuo and acetic anhydride (5 ml) and sodium acetate (0.1 g) were added. The mixture was heated for another 30 minutes at 80°C. The resulting clear solution was again evaporated in vacuo and the compound crystallized from methanol. For compound 1g, the thick slurry, obtained after evaporation of the acetic anhydride is first treated with a few drops of water and stirred for 5h, to destroy the formed mixed anhydride).
N-Phenyl-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1a)
It was obtained following the general procedure in 87% yield: mp: 245°C; IR (KBr, cm-1) : 1768 (s,CO), 1705 (s br, CO), 1630 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) :5.61 (s, 2H, 9,10-H), 7.01-7.10 (m, 4H,2,3,6,7-H), 7.19-7.25 (m,2H, m-H), 7.28-7.33 (m, 1H, p-H), 7.36-7.42 (m, 2H, o-H), 7.42-7.49(m, 4H, 1,4,5,8-H); ); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.2, 124.6, 125.6, 126.5, 127.6, 128.9, 131.7, 144.0, 156.2, 165.3; MS (EI, m/z): 349 M+.
N-(4-Methylphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1b)
It was obtained following the general procedure in 94% yield: mp: 250°C; IR (KBr, cm-1) :1766 (s,C=O), 1699 (s br, C=O); 1H-NMR (400 MHz/CDCl3) δ (ppm) :2.34 (s, 3H, CH3), 5.61 (s, 2H, 9,10- H) 7.05-7.07 (m, 4H, 2,3,6,7-H), 7.09 (d, J=8.5Hz, 2H, CHCH3), 7.20 (2H, J=8.5Hz, 2H, CHCHCH3), 7.43-7.45 (m, 4H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 21.1, 47.23, 124.6, 125.6, 126.5, 129.0, 129.7, 137.8, 144.0, 156.17, 165.5; MS (EI, m/z): 363 M+. EA(%C,%H,%N): Calcd.: 82.63, 4.71, 3.85, Found: 82.9, 5.0, 3.5.
N-(4-(Acetoxymethyl)phenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1c)
It was obtained following the general procedure in 92% yield: mp: 68°C; IR (KBr, cm-1) : 1714 (s, C=O), 1638 (m,C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : 2.09 (s, 3H, CH3), 5.09 (s, 2H, CH2), 5.61, (s, 2H, 9,10-H), 7.05-7.10 (m, 4H, 2,3,6,7-H), 7.24 (d, J=8.5Hz, 2H, CHCCH2), 7.40 (d, 2H, J=8.5Hz, CHN), 7.42-7.48 (m, 4H, 1,4,5,8-H), 13C-NMR (100 MHz/CDCl3) δ (ppm): 20.9, 47.6, 65.6, 124.6, 125.7, 126.5, 128.9, 131.6, 135.3, 143.9, 156.3, 165.2, 170.8; MS (EI, m/z): 421 M+, EA(%C,%H,%N): Calcd.: 76.95, 4.54, 3.32, Found: 77.2, 4.8, 3.1.
N-(4-Acetoxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1d)
It was obtained following the general procedure in 83% yield: mp: 296°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) :2.29 (s, 3H CH3) 5.60 (s, 2H, 9,10-H), 7.02-7.09 (m 4H, 2,3,6,7-H), 7.12 (d, J=8Hz, 2H, CHCO), 7.26 (d, J= 8Hz, CHCN), 7.41-7.48 (m, 4H, 1,4,5,8-H); MS (EI, m/z): 407 M+ EA(%C,%H,%N): Calcd.: 76.65, 4.21, 3.44, Found: 76.5, 4.3, 3.5.
N-(4-Bromophenyl)-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboximide (1e)
It was obtained following the general procedure in 90% yield: mp: 276°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) : 5.59 (s, 2H, 9,10-H), 7.04-7.06 (m, 4H, 2,3,6,7-H), 7.12 (d, J=8Hz, 2H, CHCBr), 7.42-7.45 (m, 4H, 1,4,5,8-H), 7.51 (d, J=8Hz, 2H CHCN); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.3, 121.3, 124.6, 125.7, 127.8, 130.8, 132.1, 143.9, 156.4, 164.9; MS (EI, m/z): 427/429 M+ EA(%C,%H,%N, %Br): Calcd.: 67.31, 3.29, 3.27, 18.66, Found: 67.3, 3.3, 3.0, 18.4.
N-(4-Methoxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1f)
It was obtained following the general procedure in 90% yield: mp: 301°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) :3.79 (s, 3H, OCH3), 5.60 (s, 2H, 9,10-H), 6.92 (d, J= 8.5Hz, 2H, CHCOCH3), 7.04-7.08 (m, 4H, 2,3,6,7-H), 7.12 (d, J=8.5Hz, 2H, CHCN), 7.42-7.46 (m, 4H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.3, 55.4, 114.4, 124.3, 125.7, 128.1, 144.1, 156.2, 159.0, 165.7; MS (EI, m/z): 379 M+ EA(%C,%H,%N): Calcd.: 79.14, 4.52, 3.69, Found: 79.1, 4.9, 3.2.
N-(4-Carboxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1g)
It was obtained following the general procedure in 90% yield: mp: 307°C; 1H-NMR (400 MHz/DMSO-d6) δ (ppm) :5.84 (s, 2H, 9,10-H), 7.05-7.07 (m, 4H, 2,3,6,7-H), 7.45 (d, J=7.5Hz, 2H, CHCN), 7.56-7.58 (m, 4H, 1,4,5,8-H), 7.98 (d, J=7.5Hz, 2H, CHCCOOH), 12-13 (s br, 1H COOH); 13C-NMR (100 MHz/DMSO-d6) δ (ppm): 46.1, 124.6, 125.3, 126.5, 129.4, 129.7, 135.8, 144.3, 155.8, 164.5, 166.7; MS (EI, m/z): 393 M+.
N-(4-Nitrophenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1h)
It was obtained following the general procedure in 95% yield: mp: 311°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) : 5.62 (s, 2H, 9,10-H), 7.07-7.09 (m, 4H, 2,3,6,7-H), 7.45-7.48 (m, 4H, 1,4,5,8- H), 7.55 (d, J = 7.5 Hz, 2H, CHCN), 8.27 (d, J = 7.5 Hz, 2H, CHCNO2); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.33, 124.4, 124.7, 125.7, 125.9, 137.8, 143.6, 145.9, 156.8, 164.4; MS (EI, m/z): 394 M+.
N-(4-Aminophenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1i)
A solution of 1h (0.3g; 0.77mmol) and anhydrous SnCl2 (4g) in THF (8ml) was stirred for 20 h. The mixture was poured on crushed ice (20g) and the resulting slurry was extracted with dichloromethane (3x30ml). The combined organic layers were dried over MgSO4 and evaporated in vacuo. The compound was obtained as a thick oil, after column chromatography (silica) with dichloromethane as the eluent, in 86% yield. 1H-NMR (250 MHz/CDCl3) δ (ppm) : 3.3-3.8 (sbr, 2H, NH2), 5.59 (s, 2H, 9,10-H), 6.63 (d, J=8Hz, 2H, CHCNH2), 6.92 (d, J=8Hz, 2H, CHCN), 7.02-7.08 (m, 4H, 2,3,6,7-H), 7.48-7.55 (m, 4H, 1,4,5,8-H); MS (EI, m/z): 364 M+.
General procedure for the preparation of the maleimides 7a,b and 8
A supension of the anhydride (2) (3.62 mmol) and the appropriate diamine (17mmol) in toluene (10 ml) was refluxed for 30 minutes. The resulting suspension was evaporated in vacuo and acetic anhydride (5 ml) and sodium acetate (0.1 g) were added. The mixture was heated for another 30 minutes at 80°C. The resulting clear solution was again evaporated in vacuo and the compound crystallized from methanol. For compound 7b, the thick slurry, obtained after evaporation of the acetic anhydride is first treated with a few drops of water and stirred for 5h, to hydrolyze the formed mixed anhydride.
Bismaleimide (7a)
It was obtained following the general procedure in 83% yield: mp: >360°C; IR (KBr, cm-1) : 1773 (s, C=O), 1714 (s, C=O), 1630 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.56 (s, 4H, 9,10-H), 7.00-7.07 (m, 8H, 2,3,6,7-H), 7.19-7.24 (m, 3H, 1,3-disubstituted phenyl-H), 7.36-7.44 (m, 9H, 1,4,5,8-H and NCHN); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.2, 123.4, 124.5, 124.7, 125.7, 129.1, 132.4, 143.9, 156.3, 164.8; MS (EI, m/z): 621 MH+, EA(%C,%H,%N): Calcd.: 81.28, 3.90, 4.51, Found: 81.7, 3.9, 4.1.
Bismaleimide (7b)
It was obtained following the general procedure in 81% yield: mp: >360°C; 1H-NMR (250MHz/CDCl3) δ (ppm) : 5.58 (s, 4H, 9,10-H), 6.98-7.02 (m, 8H, 2,3,5,6-H), 7.38-7.42 (m, 8H, 1,4,5,8-H), 7.48 (t, J=1Hz, 1H, 4-H phenyl), 7.93 (d, J=1Hz, 2H, 2,6-H phenyl); 13C-NMR (62.5MHz/CDCl3) δ (ppm): 47.27, 124.63, 125.76, 126.06, 127.94, 130.77, 132.72, 143.75, 156.46, 164.50, 169.31; MS (EI, m/z): 665 M+.
Bismaleimide (8)
It was obtained following the general procedure in 93% yield: mp: >360°C; IR (KBr, cm-1) : 1769 (s, C=O), 1709 (s br, C=O, s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.52 (s, 4H, 9,10-H), 7.06- 7.14 (m, 8H, 2,3,5,6-H), 7.35 (s, 4H, 1,4-disubstituted phenyl-H), 7.46-7.53(m, 8H, 1,4,5,8-H); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.3, 124.6, 125.7, 126.7, 130.9, 143.9, 156.3, 165.0; MS (EI, m/z): 620 M+; The compound was recrystallized from acetone for X-ray analysis. Crystal data (acetone) : C42H24N204.2C3H60, Mr = 736.79, light yellow plates (0.40 x 0.35 x 0.04 mm), triclinic, space group P- 1 (no. 2) with a = 8.1320(10), b = 10.742(2), c = 12.175(2) Å, α = 106.92(1), β = 105.11(1), γ = 98.65(1)°, V = 952.4(3) Å3, Z = 1, Dc = 1.285g cm-3, F(000) = 386, μ(Mo-Kα) = 0.085 mm-1, 4012 reflections measured, 3253 independent, Rint = 0.0344, 1.8° < θ < 24.7°, ω scan, T = 289K, Mo-Kα radiation, graphite monochromator, λ = 0.71073 Å on a Siemens P4 diffractometer. Data were corrected for Lp effects, but not for absorption. The structure was solved by direct methods (SHELXTL). Refinement on F2 was carried out by full-matrix least-squares techniques (SHELXTL) with anisotropic displacement parameters for non-H atoms and riding isotropic H-atoms. Refinement converged at a final wR2 value of 0.1217, R1 = 0.0471 for 2161 reflections with Fo > 4σ(Fo), S = 10016, 269 parameters. A final difference Fourier showed no residual density outside -0.20 and 0.15 e Å-3.
Trismaleimide 11
A solution of the anhydride (2) (500mg; 1.8 mmol) and tris(2-aminoethyl)amine (66mg; 0.45 mmol) in dry THF (30 ml) was stirred for 3 h. The solvent was evaporated in vacuo and acetic anhydride (10 ml) and sodium acetate (0.1 g) were added and the mixture heated for 30 minutes at 80°C. The solvent was evaporated and the compound was obtained as a viscous oil, after purification by column chromatography (silica) with dichloromethane/diethyl ether (2:1) as the eluent, in a 61% yield: IR (CCl4, cm-1): 1707 (s, C=O), 1636 (m, C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : 2.46 (t, J=6Hz, 6H, CH2N), 3.12 (t, J=6Hz, 6H, CH2N(CO)2), 5.45 (s, 6H, 9,10-H), 6.87-6.91 (m, 12H, 2,3,5,6-H), 7.32-7.36 (m, 12H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 36.1, 47.1, 52.2, 124.4, 125.4, 144.1, 156.2, 166.1; (EI, m/z): 914 M+.
Tetrakismaleimide 12
A suspension of the anhydride (2) (220mg; 0.8mmol) and 5,10,15,20-tetrakis(4-aminophenyl)- porphyrin (80mg; 0.118mmol) was refluxed in toluene (40 ml) for 12 h. The solvent was evaporated and acetic anhydride (20ml) and sodium acetate (0.1mg) were added and the mixture heated for 2 h at 80°C. The solvent was stripped off in vacuo and the compound 12 was obtained in 59% yield after purification by column chromatography (silica) with dichloromethane as the eluent: mp >360°C; IR (KBr, cm-1) : 3500 (w; NH), 3370 (w, NH), 1723 (s, C=O), 1638 (m, C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : -2.86 (sbr, 2H, NH), 5.80 (s, 8H, 9,10-H (anthracene)), 7.07-7.19 (m, 16H, 2,3,6,7-H), 7.62 (d, J=8Hz, 8H, 3,5-H (phenyl)), 8.20 (d, J=8Hz, 8H, 2,6-H (phenyl)), 8.82 (s, 8H, 2,3,7,8,12,13,17,18-H (porphyrin)); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.4, 119.3, 124.3, 124.7, 125.8, 131.6, 135.0, 141.1, 144.0, 156.5, 165.5.
Acknowledgement
We thank the University of Leuven, the FWO-Vlaanderen and the Ministerie voor Wetenschapsbeleid for financial support. L. V. M. is a research director of the FWO Vaanderen. M.S. and D.C. thank the FWO Vlaanderen and the IWT, respectively, for a predoctoral fellowship.
References and Notes
- Kearney, P.C.; Mizoue, L.S.; Kumpf, R.A.; Forman, J. E.; Mc Curdy, A.; Dougherty, D. A. “Hydrophobic” binding of water-soluble guests by high-symmetry, chiral hosts. An electron-rich receptor site with a general affinity for quaternary ammonium compounds and electron-deficient π systems. J. Am. Chem. Soc. 1993, 115, 9907. [Google Scholar] [CrossRef]
- See for instance : Csöregh, I.; Weber, E.; Hens, T.; Czugler, M. J. Chem. Soc., Perkin Trans. 2 1996, 2733. Weber, E.; Finge, S.; Csöregh, I. Modular design of hosts involving a rigid succinimide framework and N-bonded lateral groups. Crystalline inclusion properties and crystal structures of inclusion compounds with dioxane, MeOH and DMF. J. Org. Chem. 1991, 56, 7281. [Google Scholar] [CrossRef]
- Shahlai, K.; Hart, H. Synthesis of supertriptycene and two related iptycenes. J. Org. Chem. 1991, 56, 6905, and references included therein. [Google Scholar] [CrossRef]
- Jye-Shane, Y.; Swager, T.M. Fluorescent porous polymer films as TNT chemosensors: electronic and structural effects. J. Am. Chem. Soc. 1998, 120, 11864–11873. [Google Scholar]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Liebigs Ann. Chem. 1931, 486, 191. [Google Scholar] [CrossRef] Chaturvedi, C.; Verma, S. M. Effect of α-substituents on conformation about the N-N and N-C bonds in N-amino- and N-aryl-succinimidyl systems: a PMR study. Indian J. Chem. 1990, 29B, 9. [Google Scholar] Smith, W. B.; Shoulders, B. A. The nuclear magnetic resonance spectra of some 9,10-bridged 9,10-dihydroanthracenes. J. Phys. Chem. 1965, 69, 2022. [Google Scholar] [CrossRef]
- Treibs, A.; Häberle, N. Liebigs Ann. Chem. 1968, 718, 183. Semeikin, A. S.; Koifman, O. T.; Berezin, B. D. Khim. Geterosikl. Soedin. 1982, 1354, CA 98: 71769s.
- Samples Availability: Available from the authors.
Scheme 1.
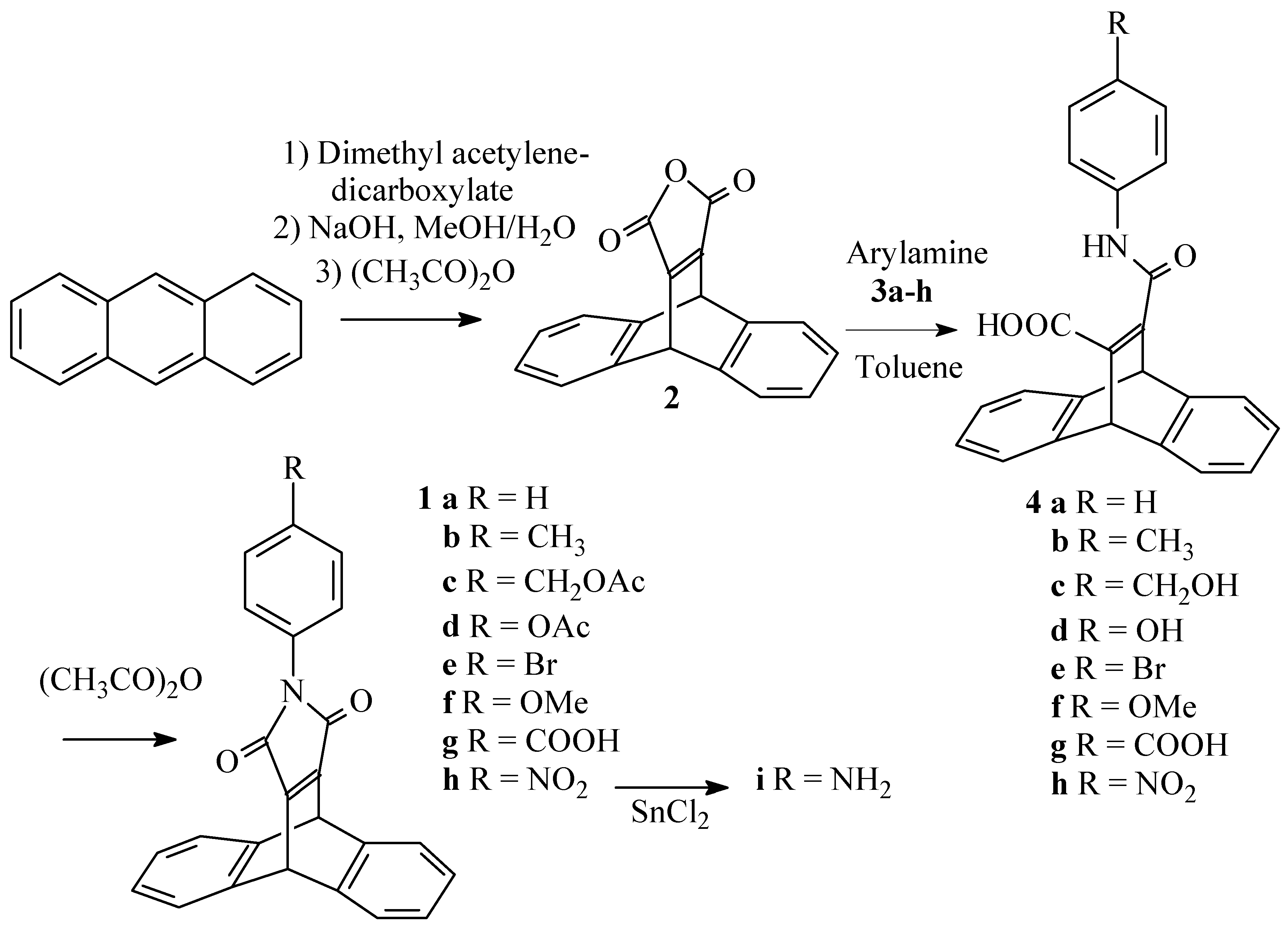
Figure 1.
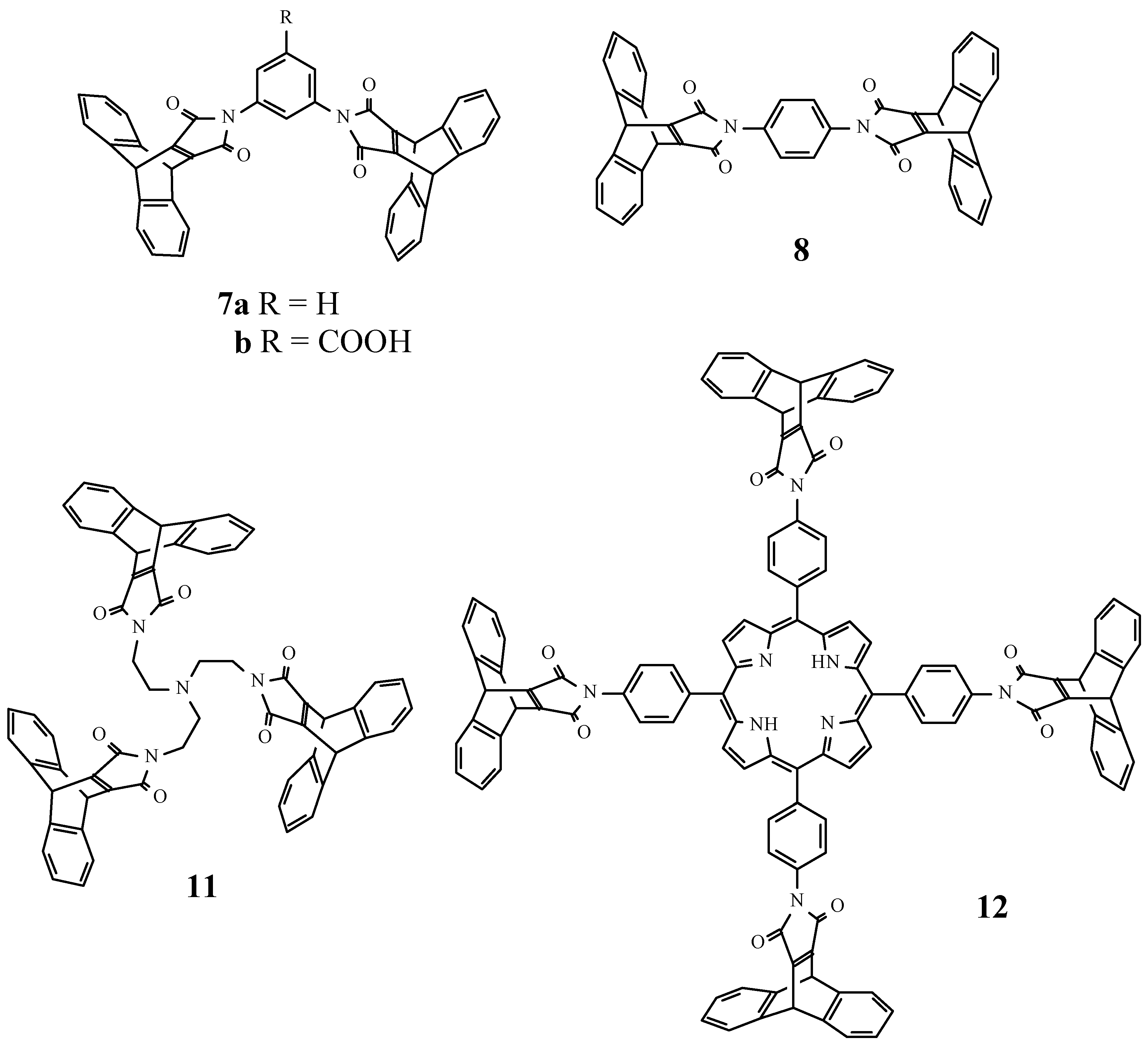
Figure 2.
View of the crystal packing of 8.

© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Smet, M.; Corens, D.; Van Meervelt, L.; Dehaen, W. Synthesis of the Formal Diels-Alder Adducts of N-substituted Dehydromaleimides and Anthracene. Molecules 2000, 5, 179-188. https://doi.org/10.3390/50200179
AMA Style
Smet M, Corens D, Van Meervelt L, Dehaen W. Synthesis of the Formal Diels-Alder Adducts of N-substituted Dehydromaleimides and Anthracene. Molecules. 2000; 5(2):179-188. https://doi.org/10.3390/50200179
Chicago/Turabian StyleSmet, Mario, David Corens, Luc Van Meervelt, and Wim Dehaen. 2000. "Synthesis of the Formal Diels-Alder Adducts of N-substituted Dehydromaleimides and Anthracene" Molecules 5, no. 2: 179-188. https://doi.org/10.3390/50200179