Behaviour of Some Activated Nitriles Toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl-1-Phenylpyrazol-5-one

Abstract
:Introduction
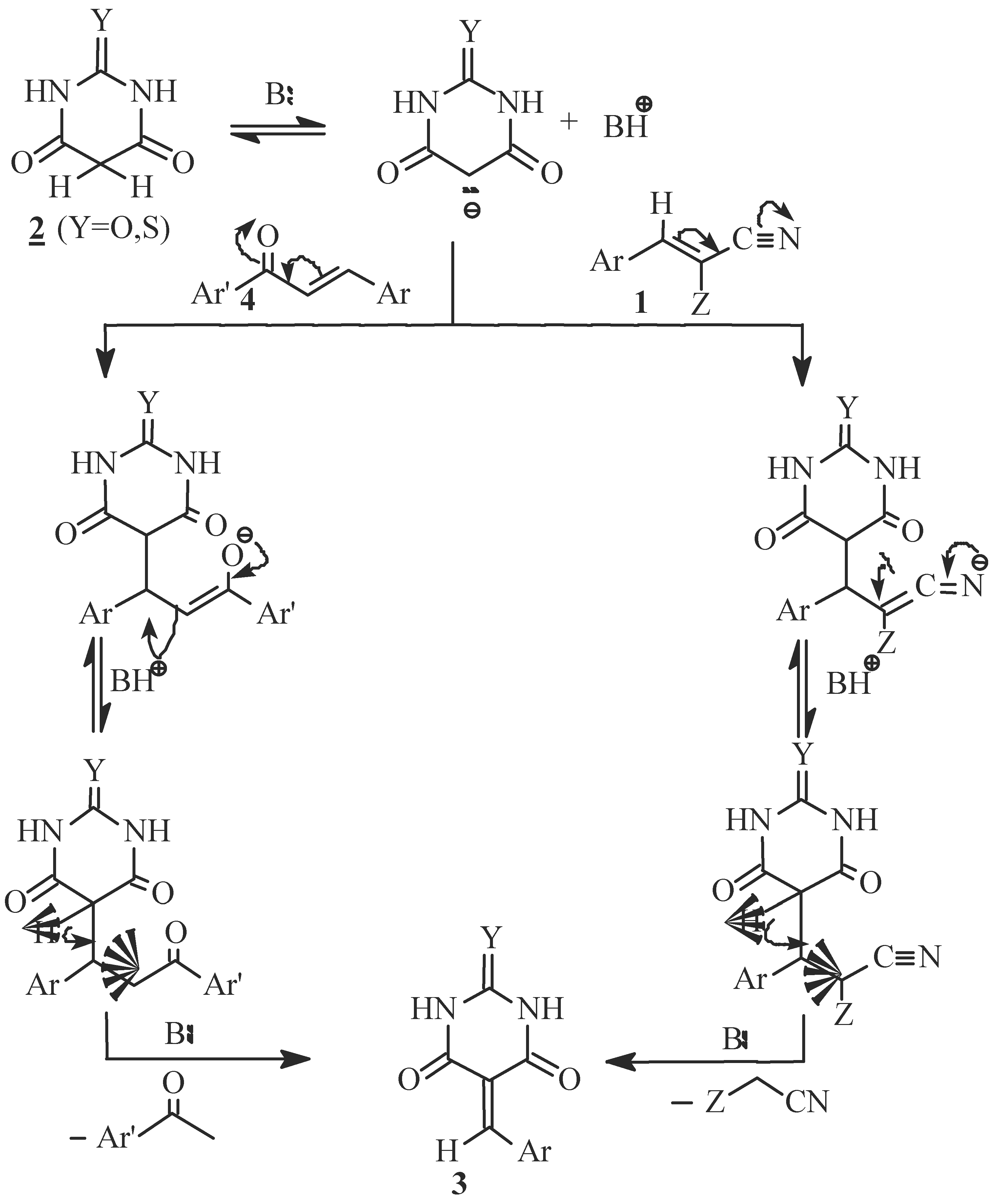
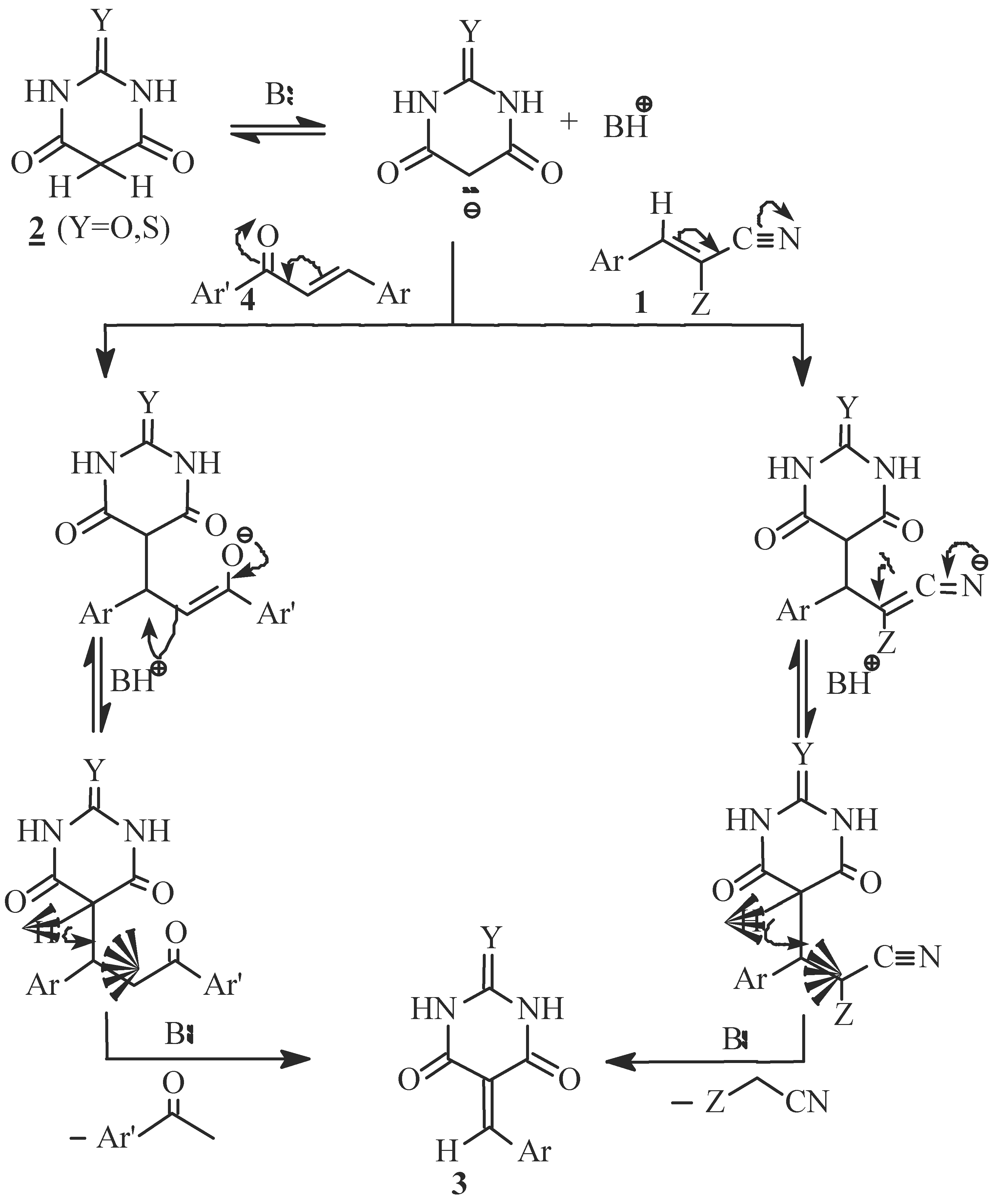
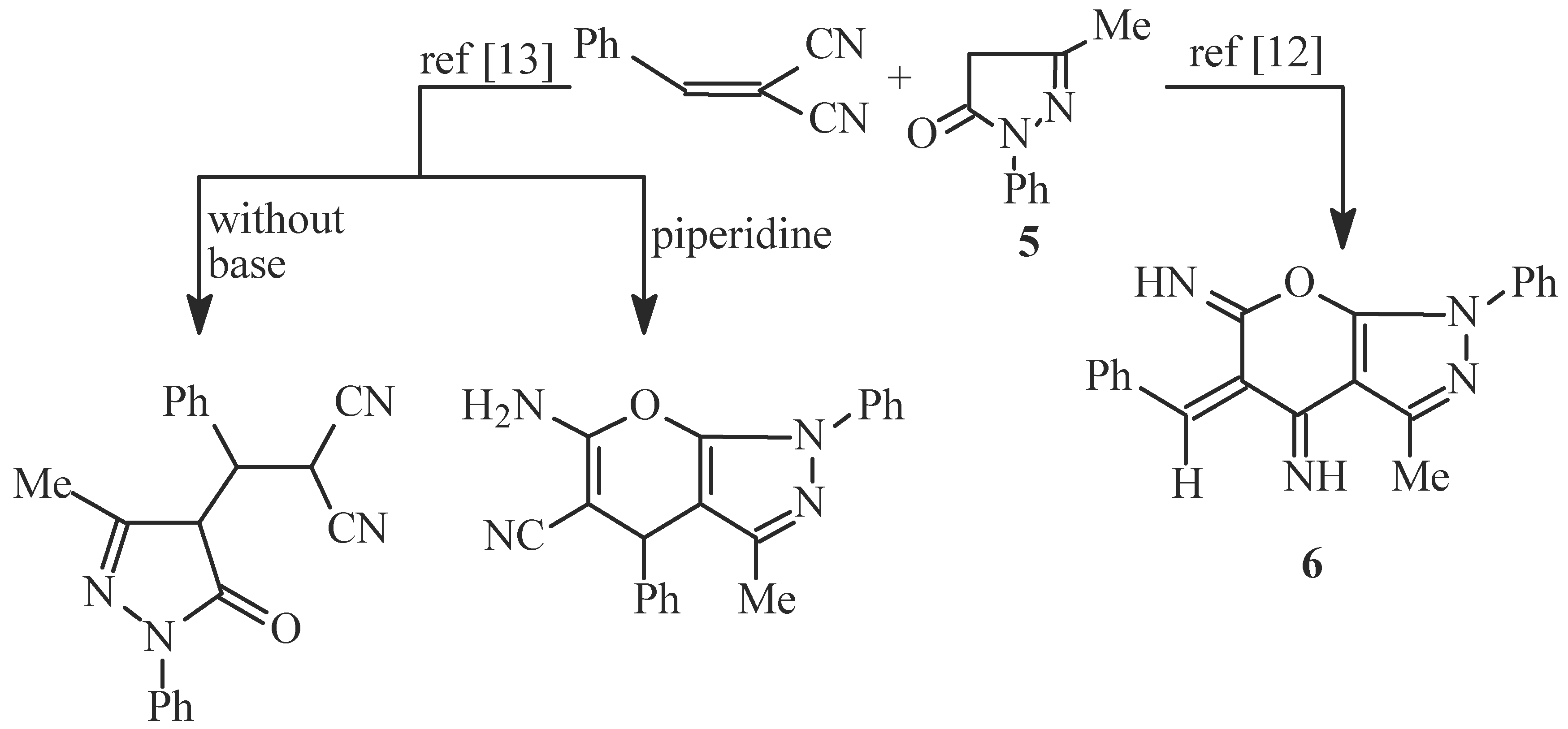
Results and Discussion
- i)
- A satisfactory elemental analysis.
- ii)
- The IR displayed νNH2 at 3485 , 3373 and 3223 cm-1, νCN at 2205 cm-1 and νC=N at 1622 cm-1.
- iii)
- The 1H-NMR spectrum (DMSO-d6) exhibits signals at δ(ppm) 7.9-7.3 (m, 5H, Ph), 6.8 (s, 2H, arom. protons), 5.0 (s, 1H, CHAr), 3.90-3.75 (two s, 9H ; 3OMe) and 2.3 (s, 3H, Me).
- iv)
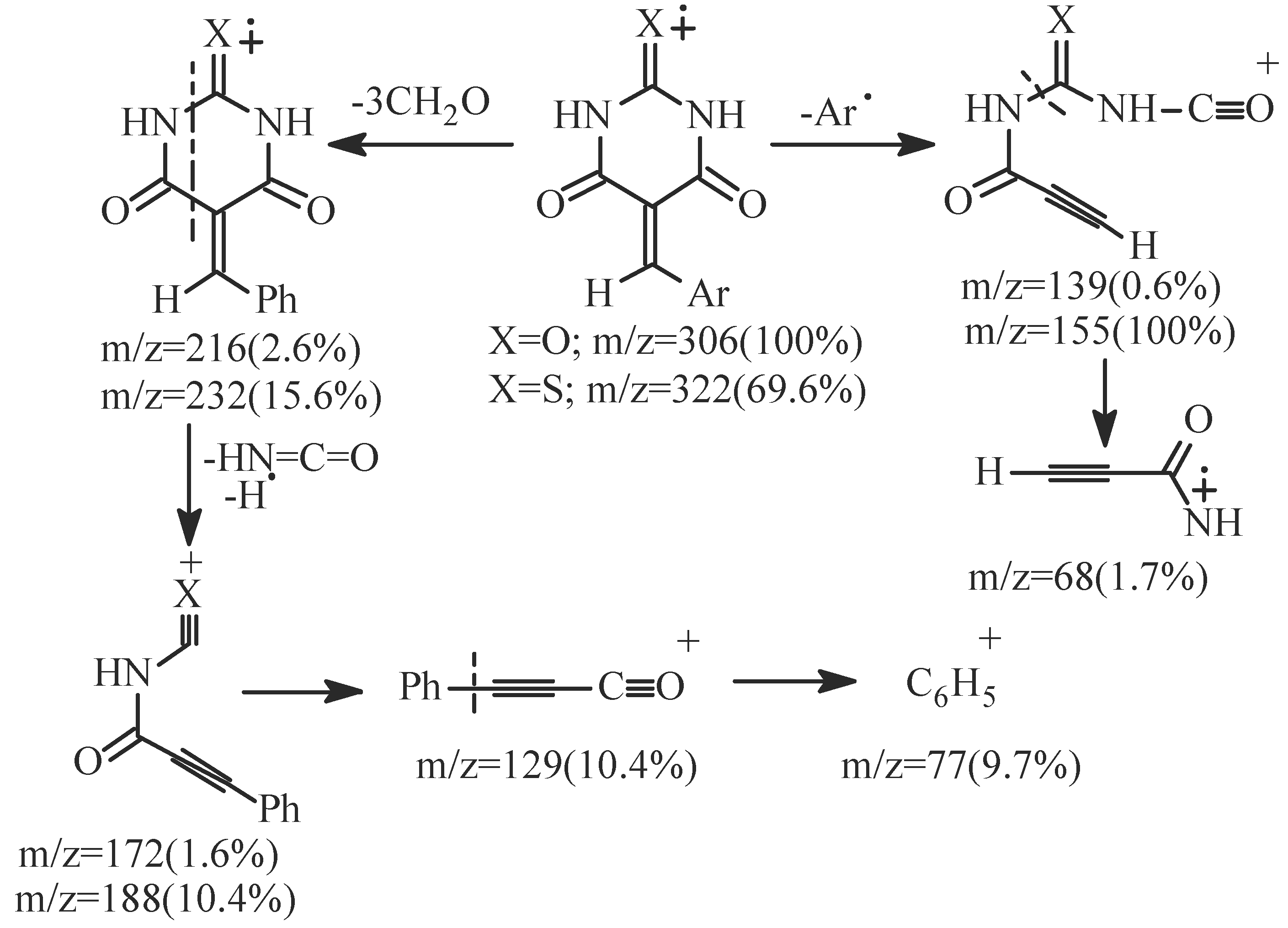
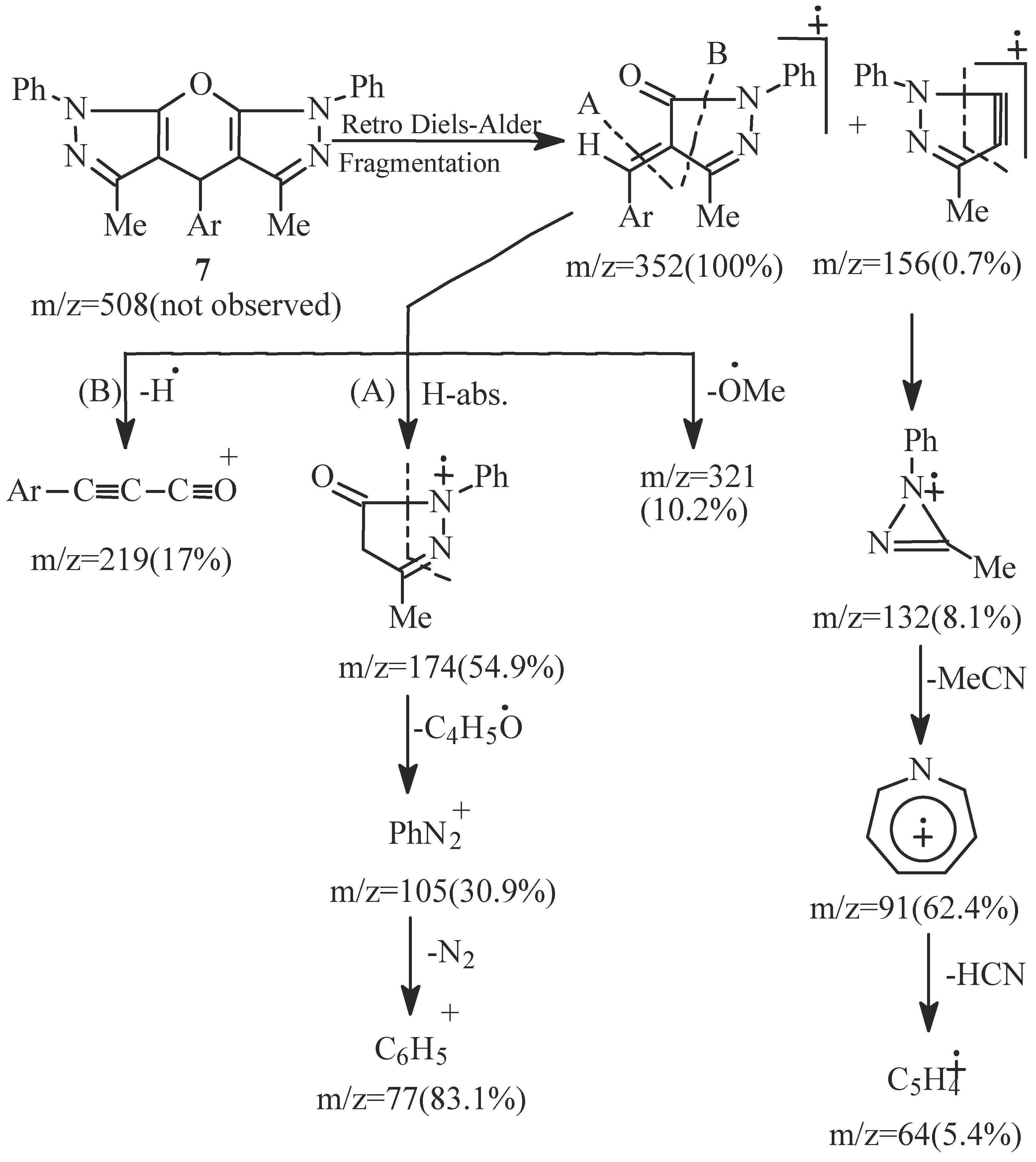
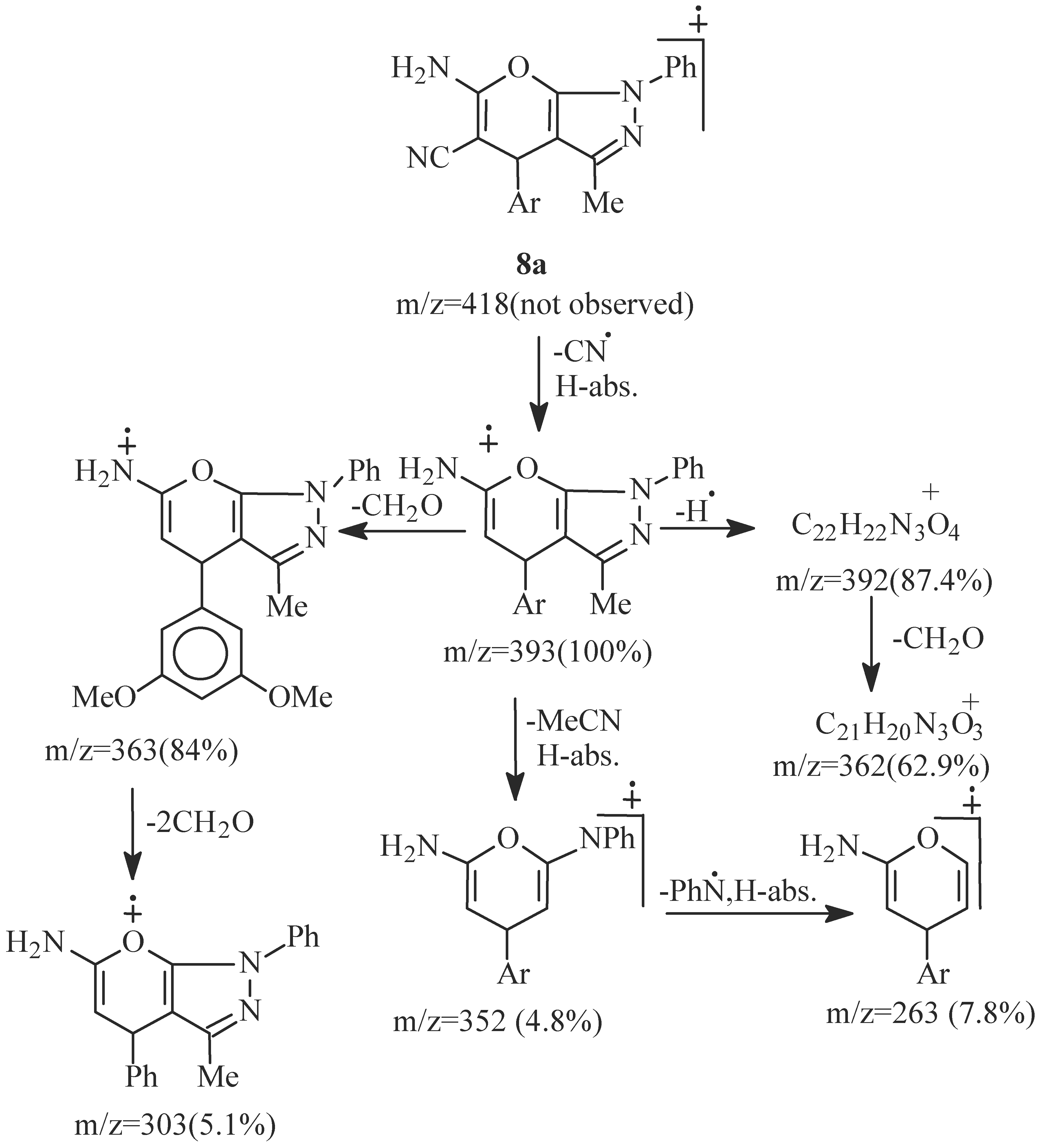
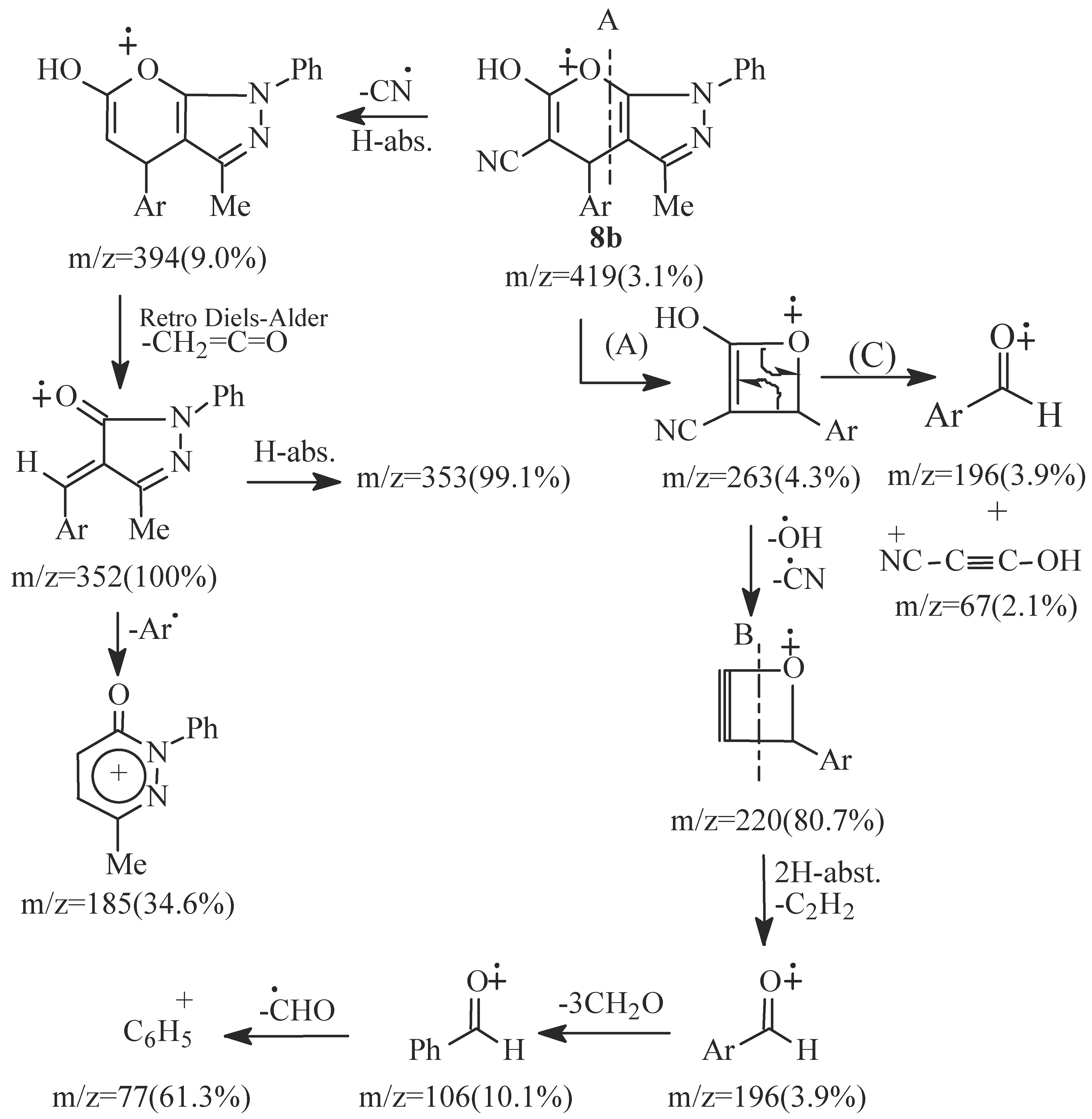
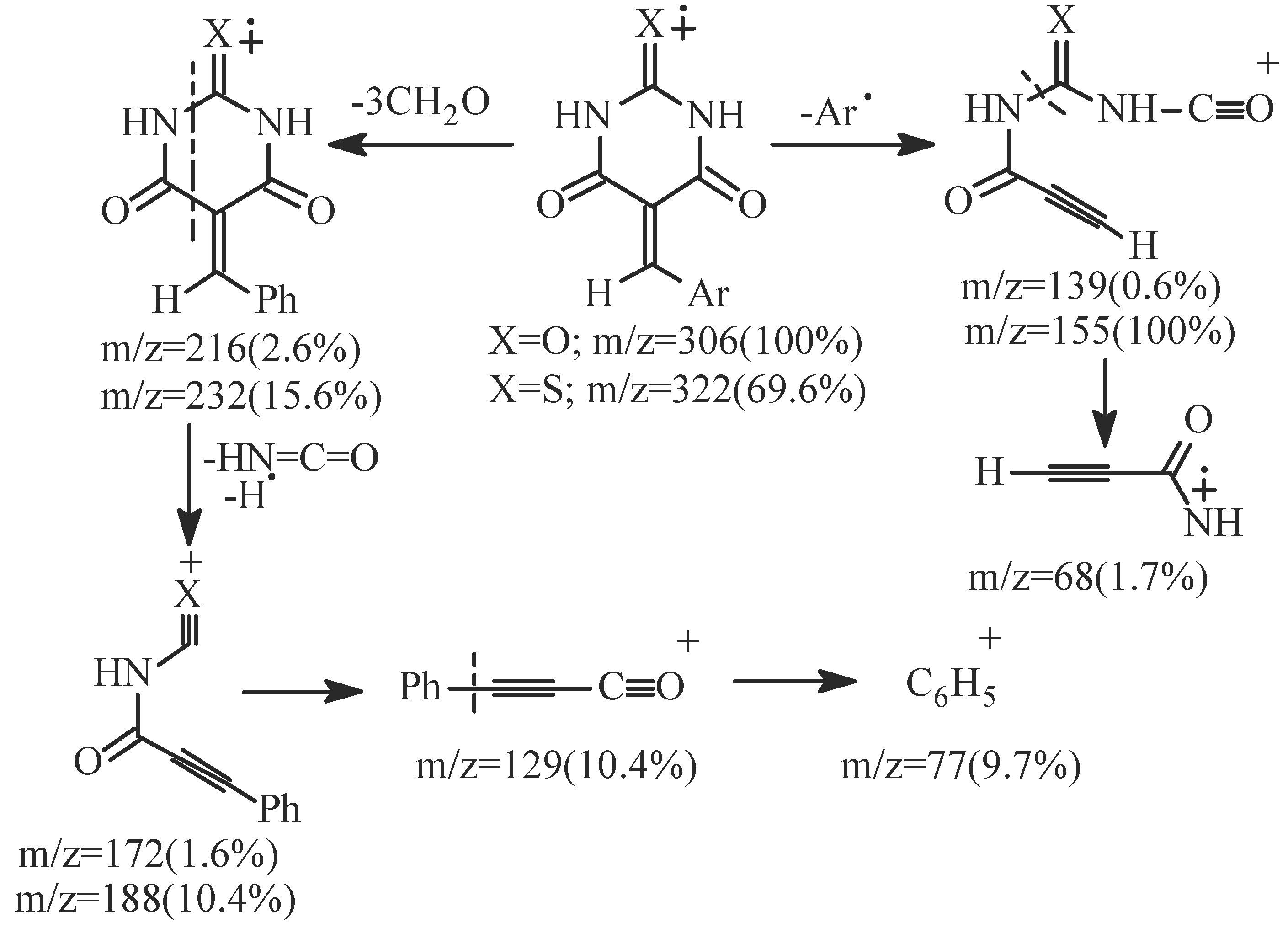
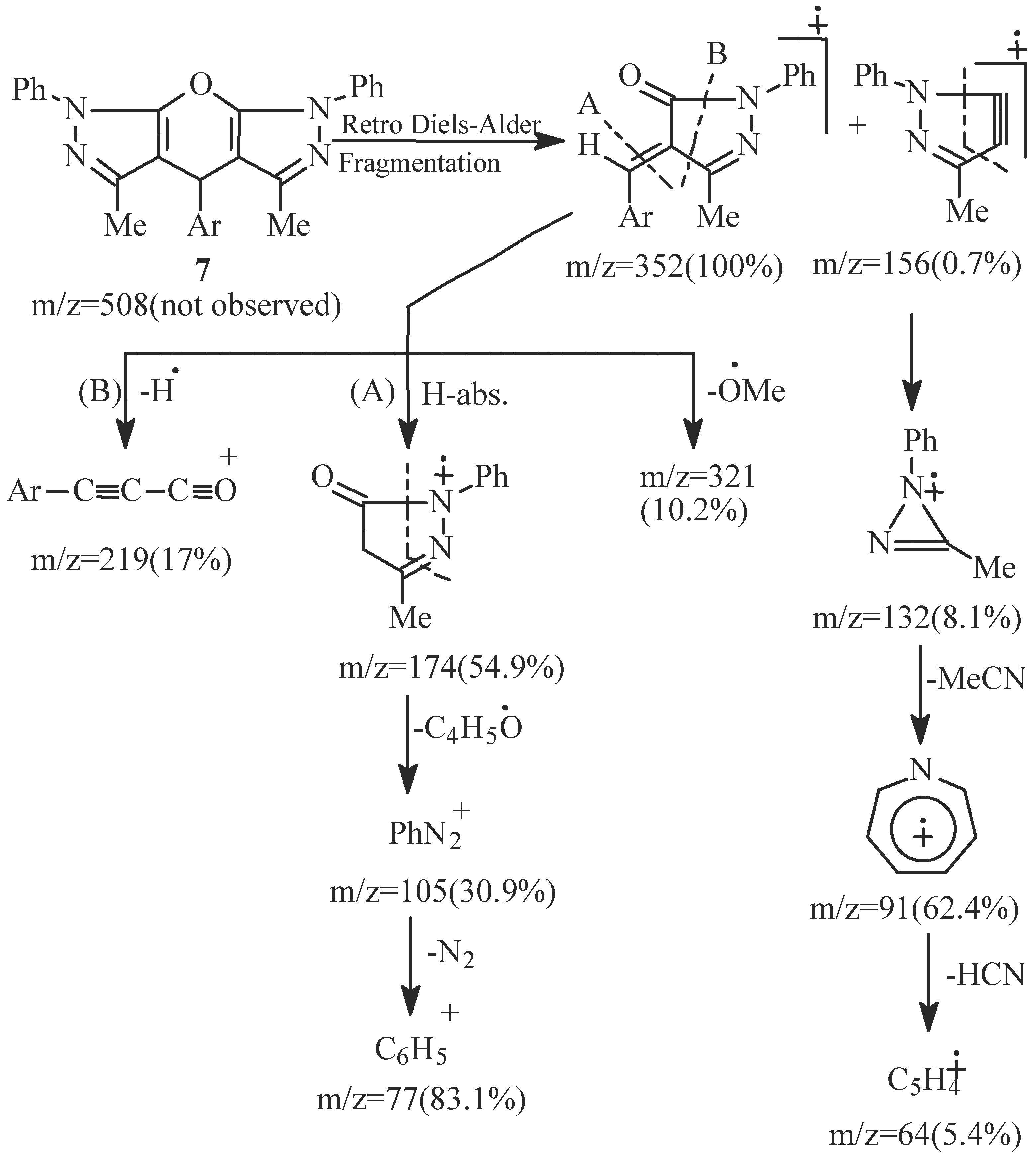
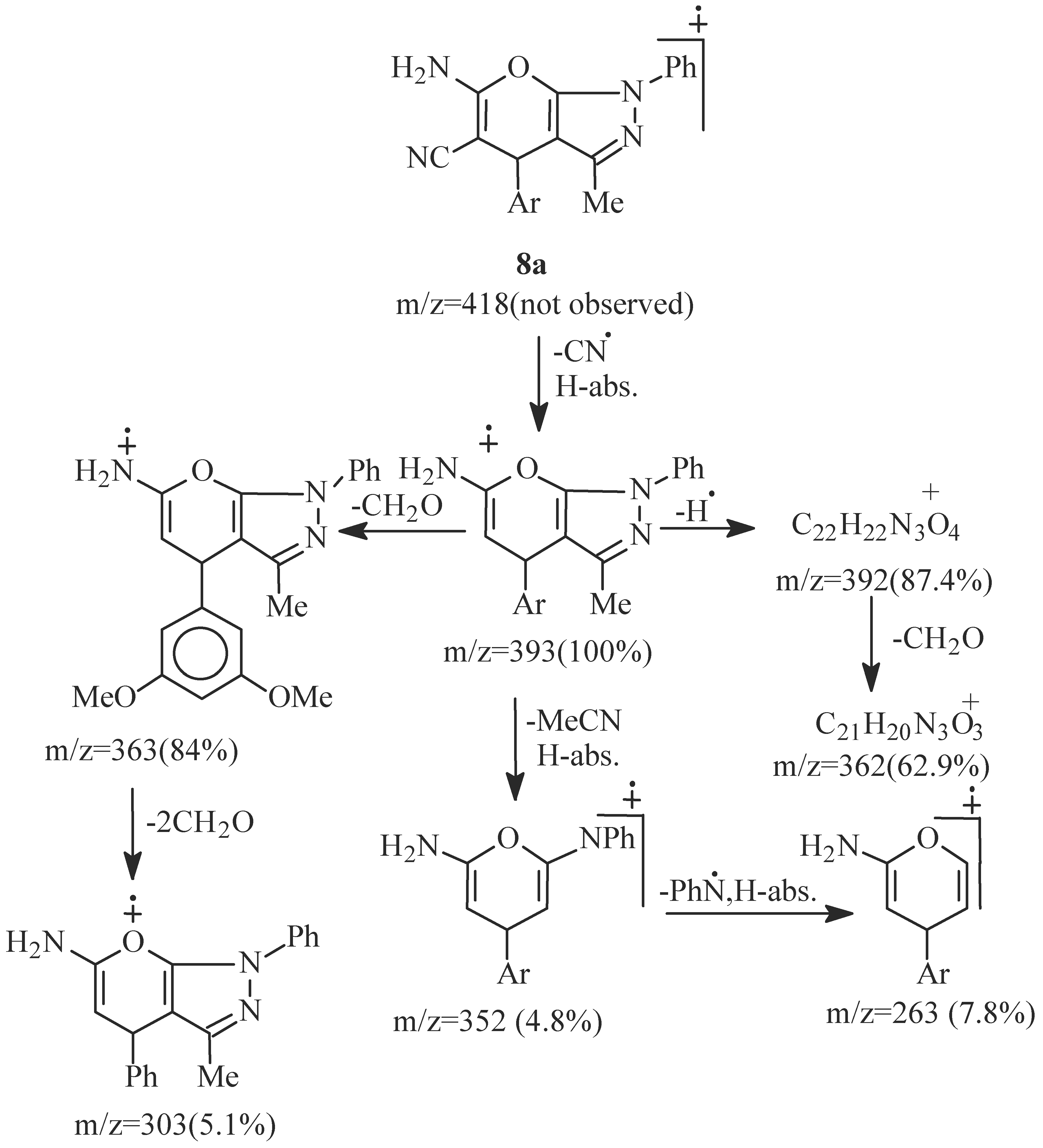
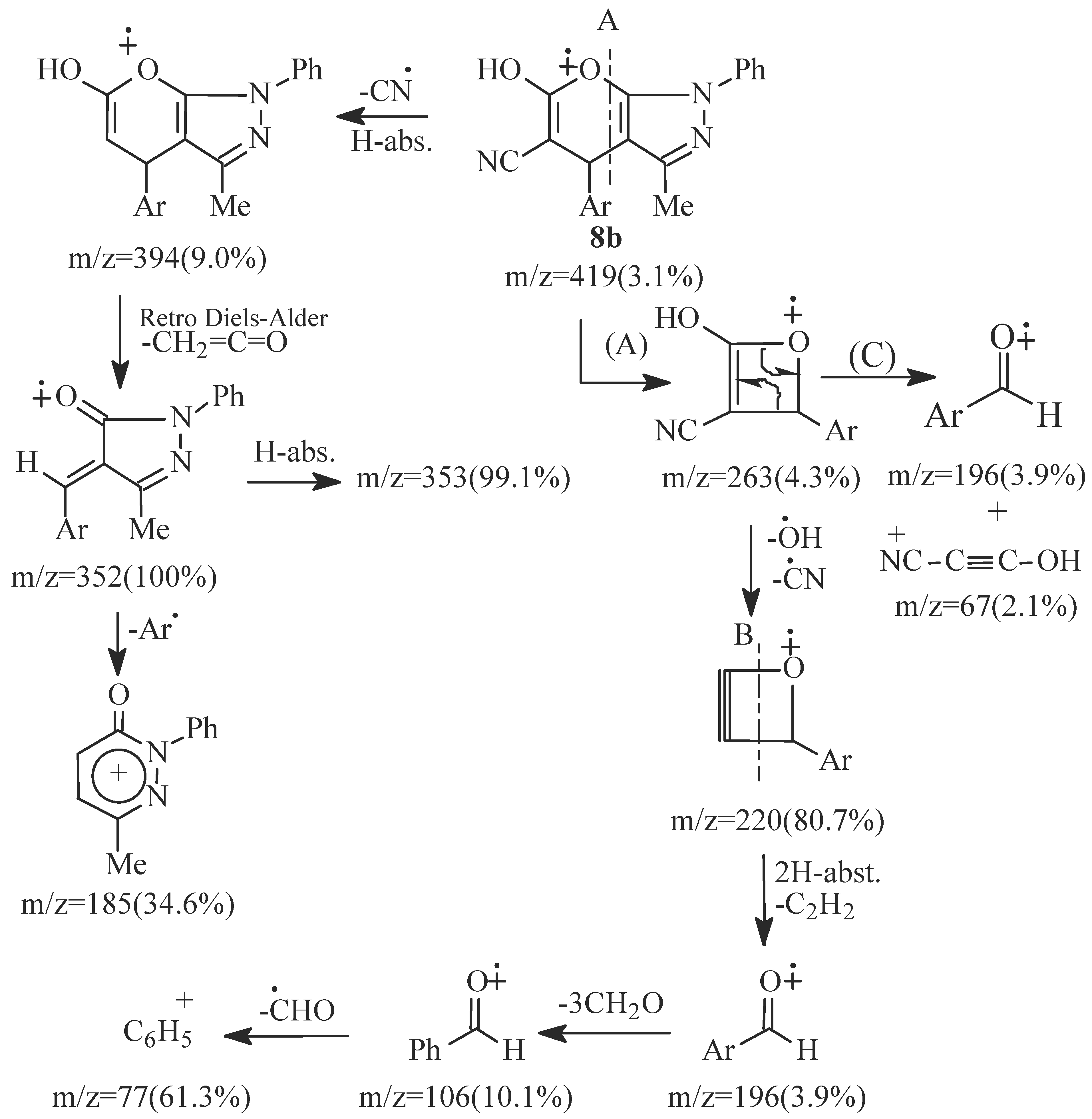
- EI fragmentation of 8a involves primary loss of CN• followed by H-abstraction to give the radi-cal cation of m/z=393/100%; base peak). There is also a loss of formaldehyde molecule to give the ion of m/z=363 (84.0%) which is the major daughter ion. Successive losses of two molecules of formalde-hyde resulted in the radical cation of m/z = 303 (5.1%). The tentative fragmentation pattern of 8a is represented in Figure 3.
Experimental
General
Reaction of α-cyano-3,4,5-trimethoxycinnamonitrile (1a) or ethyl α-cyano-3,4,5-trimethoxycinnamate (1b) with barbituric acid or thiobarbituric acid; Formation of 5-(3,4,5-trimethoxybenzylidene)barbi-turic or thiobarbituric acid (3a or 3b)
Reaction of chalcone 4 with barbituric or thiobarbituric acid, Formation of 3a or 3b
Reaction of barbituric or thiobarbituric acid with 3,4,5-trimethoxybenzaldehyde; Formation of an authentic sample of 3a
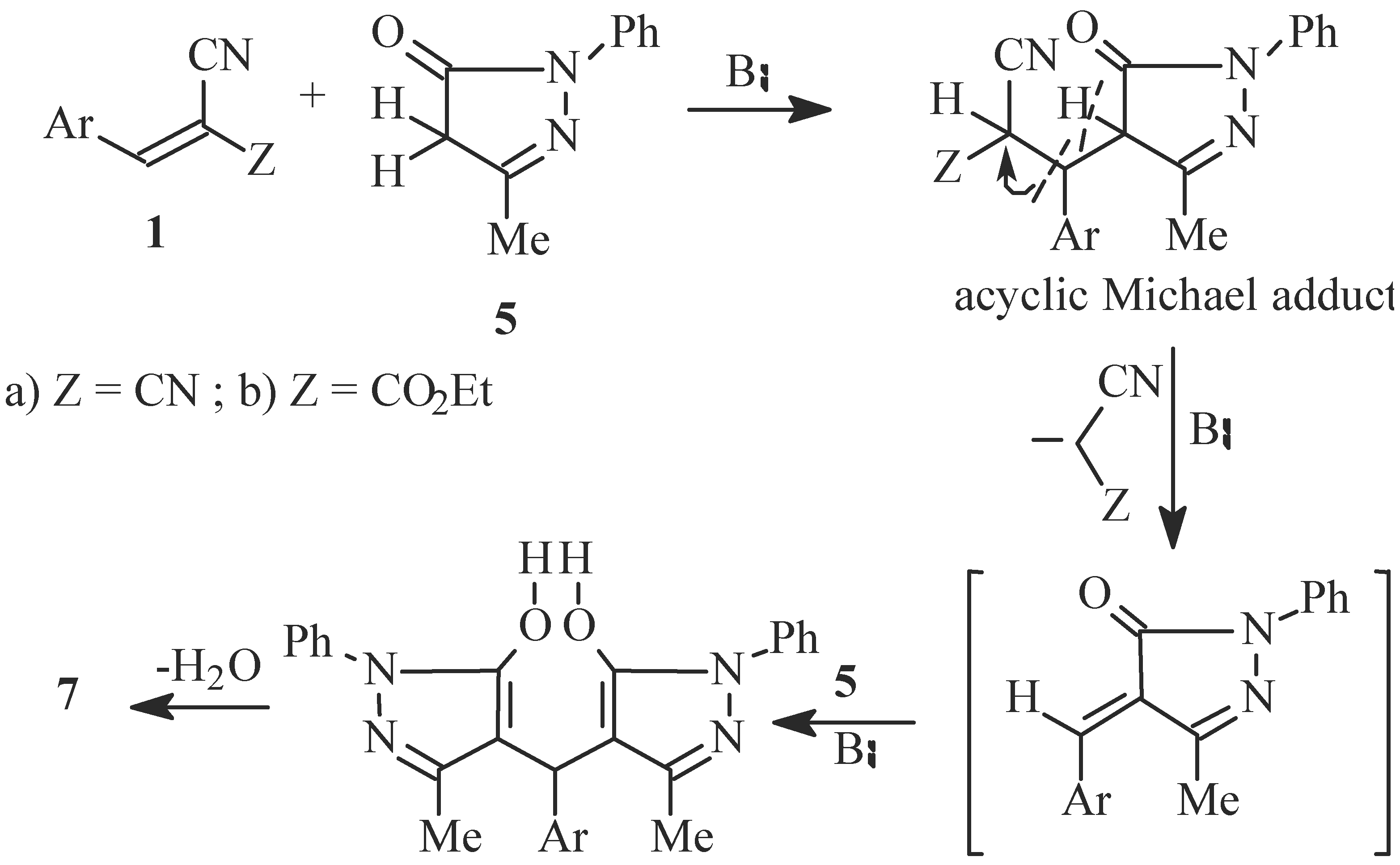
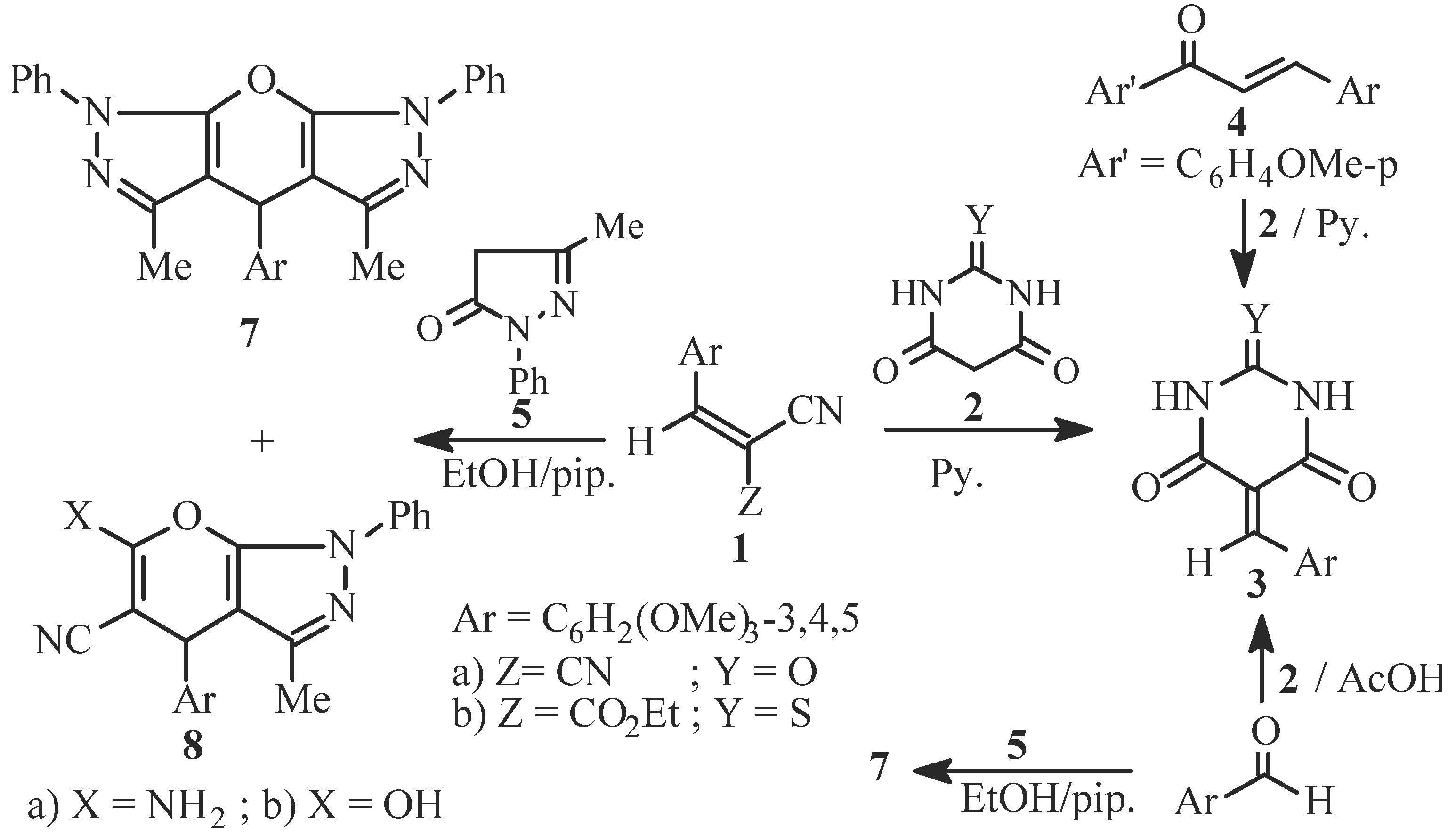
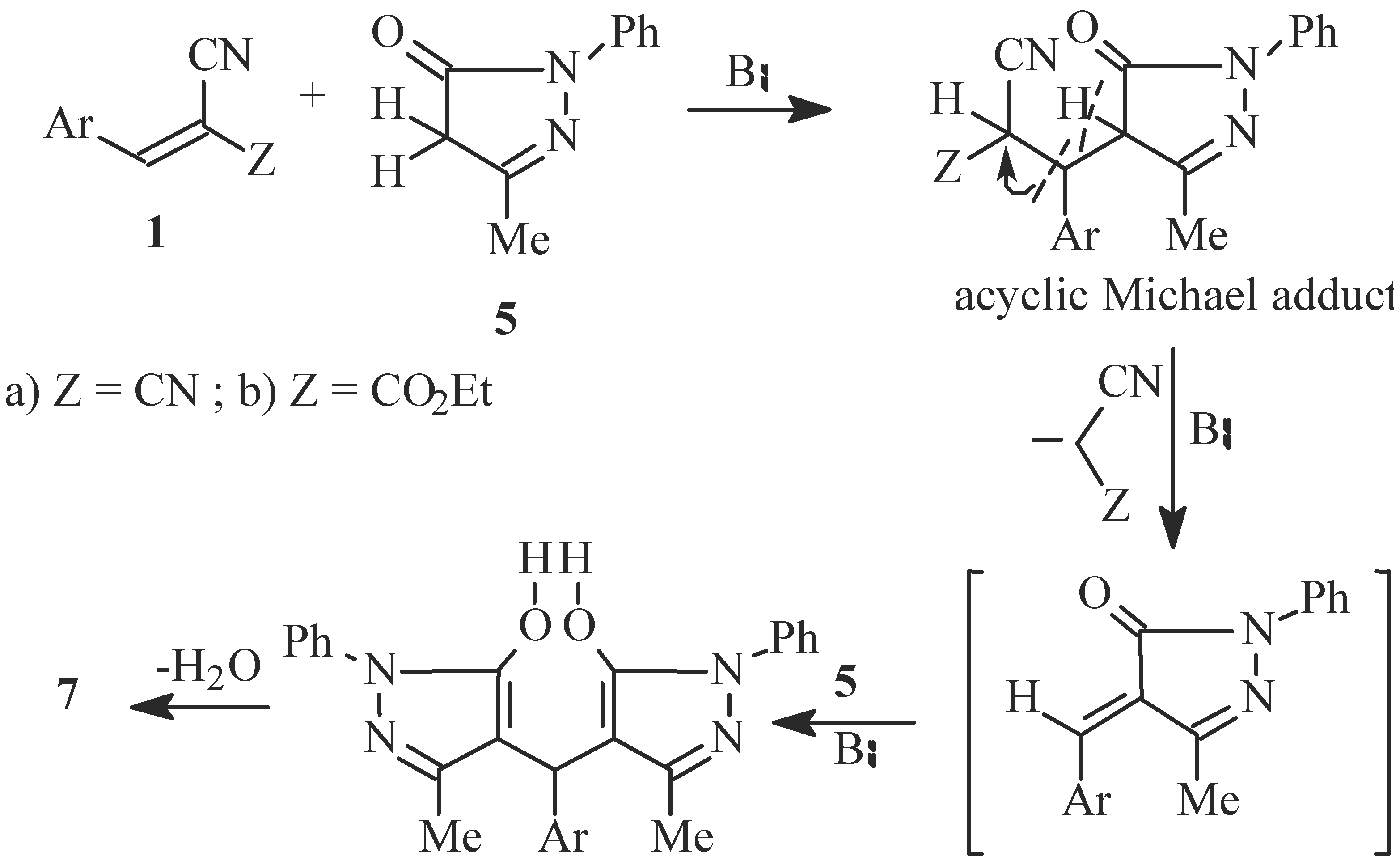
Reaction of 1a or 1b with 3-methyl-1-phenylpyrazol-5-one (5); Formation of 4H-3,5-dimethyl-1,7-diphenyl-4-(3,4,5-trimethoxy-phenyl)oxino[2,3-c:6,5-'c] bis-pyrazole (7) and 4H-6-amino-5-cyano-3-methyl-1-phenyl-4-(3,4,5-trimethoxyphenyl)-pyrano[2,3-c]pyrazole (8a) or 4H-5-cyano-6-hydroxy-3-methyl-1-phenyl-4-(3,4,5-trimethoxyphenyl)pyrano[2,3-c]-pyrazole (8b)
Reaction of 5 with 3,4,5-trimethoxybenzaldehyde; Formation of an authentic sample of 7
References and Notes
- Ahluwalia, V.; Kumar, K.; Alauddin, M.; Khandi, C.; Mallika, N. Synth. Commun. 1990, 20(9), 1265. [CrossRef]
- Ahluwalia, V.K.; Aggarwal, R.; Alauddin, M.; Gill, G.; Khanduri, C.H. Heterocycles 1990, 31(1), 129–137.
- Sharamin, Yu.A.; Klokol, G.V. J. Org. Chem. (USSR) 1984, 20, 2230–2233.
- Abdel-Latif, F.F. Indian J. Chem. Sect. B. 1991, 30(3), 363–365.
- Ibrahim, M.K.A.; El-Moghayar, M.R.H.; Sharaf, M.A.F. Indian J. Chem. Sect.B.. 1987, 26, 216–219.
- Koyama, G.; Umezawa, H. J. Antibiot. 1965, 18A, 175, (Chem. Abstr., 63, 15158d (1965)).
- Robins, R. K.; Townsend, L. B.; Cassidy, F. C.; Geroter, J. F.; Lewis, A. F.; Miller, R.L. J. Heterocycl. Chem. 1963, 3, 110. [CrossRef]
- El-Nagdi, M.H.; El-Moghayer, M.R.H.; El-Fahham, H.A.; Sallam, M.M.M.; Alnima, H.H. J. Heterocycl.Chem. 1980, 17, 209.
- El-Nagdi, M.H.; Fahmy, S.M.; Hafez, E.A.A.; El-Moghayer, M. R. H.; Amer, S.A.R. J. Heterocycl. Chem. 1979, 16, 1109.
- El-Nagdi, M.H.; Hafez, E.A.A.; El-Fahham, H.A.; Kandeel, E.M. J. Heterocycl. Chem. 1980, 17, 73.
- El-Nagdi, M.H.; El-Moghayer, M.R.H.; Fleita, D.H.; Hafez, E.A.A.; Fahmy, S.M. J. Org. Chem. 1976, 41, 3781.
- Abdou, S.; Fahmy, S.M.; Sadek, K.U.; El-Nagdi, M.H. Heterocycles 1981, 16(12), 2177.
- Sharamin, Yu.A.; Promonenkov, V. K. J. Org. Chem. (USSR) 1982, 18, 544–548.
- Samples Availability: Available from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Madkour, H.M.F.; Mahmoud, M.R.; Nassar, M.H.; Habashy, M.M. Behaviour of Some Activated Nitriles Toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl-1-Phenylpyrazol-5-one. Molecules 2000, 5, 746-755. https://doi.org/10.3390/50500746
Madkour HMF, Mahmoud MR, Nassar MH, Habashy MM. Behaviour of Some Activated Nitriles Toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl-1-Phenylpyrazol-5-one. Molecules. 2000; 5(5):746-755. https://doi.org/10.3390/50500746
Chicago/Turabian StyleMadkour, H. M.F., M. R. Mahmoud, M. H. Nassar, and M. M. Habashy. 2000. "Behaviour of Some Activated Nitriles Toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl-1-Phenylpyrazol-5-one" Molecules 5, no. 5: 746-755. https://doi.org/10.3390/50500746
APA StyleMadkour, H. M. F., Mahmoud, M. R., Nassar, M. H., & Habashy, M. M. (2000). Behaviour of Some Activated Nitriles Toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl-1-Phenylpyrazol-5-one. Molecules, 5(5), 746-755. https://doi.org/10.3390/50500746