Introduction
We have previously reported [
1,
2] a synthetic approach to 5-aryl-3-furfurylidene-2-(3
H)-furanones (
1)
via the condensation reactions of furan-2-carboxaldehyde with 3-aroylpropionic acids under Perkin conditions, which yield the corresponding
E-lactones as the only product, with no detectable amount of the
Z-isomers being identified by TLC and
1H NMR [
3]. This result was consistent with the reports of Awad
et al. [
5] and others' [
6,
7] concerning the condensation reaction of 5-methyl-furan-2-carbox-aldehyde and/or 5-methyl thiophene-2-carboxaldehyde with 3-aroylpropionic acids under Perkin condi-tions. The conversion of these 2(3
H) furanones into benzofuran derivatives [
1] and 5-oxo-2-pyrrolines [
4] have also been described by one of us.
Dihydropyridazinone rings are of chemical and biological interest, having been reported as having antihypertensive activity [
8]. In addition, Nannini et al. [
9] has established that the pyridazinones display analgesic and anti-inflammatory activities. On the other hand, 1,3,4-oxadiazoles and
N2(acyl)-benzoic hydrazides are reported to exhibit carcinostatic activity against several types of tumors [
10] and antimi-crobial effects against
Mycobacterium tuberculosis and
Mycobacterium lapreae [
11]. The amino-3-substituted propyl functionality is found in many compounds having vasodilator, antispasmodic, blood platelet aggregation inhibition [
12], antiarrhythmic [
13] and anticholesterolesmic [
14] activities.
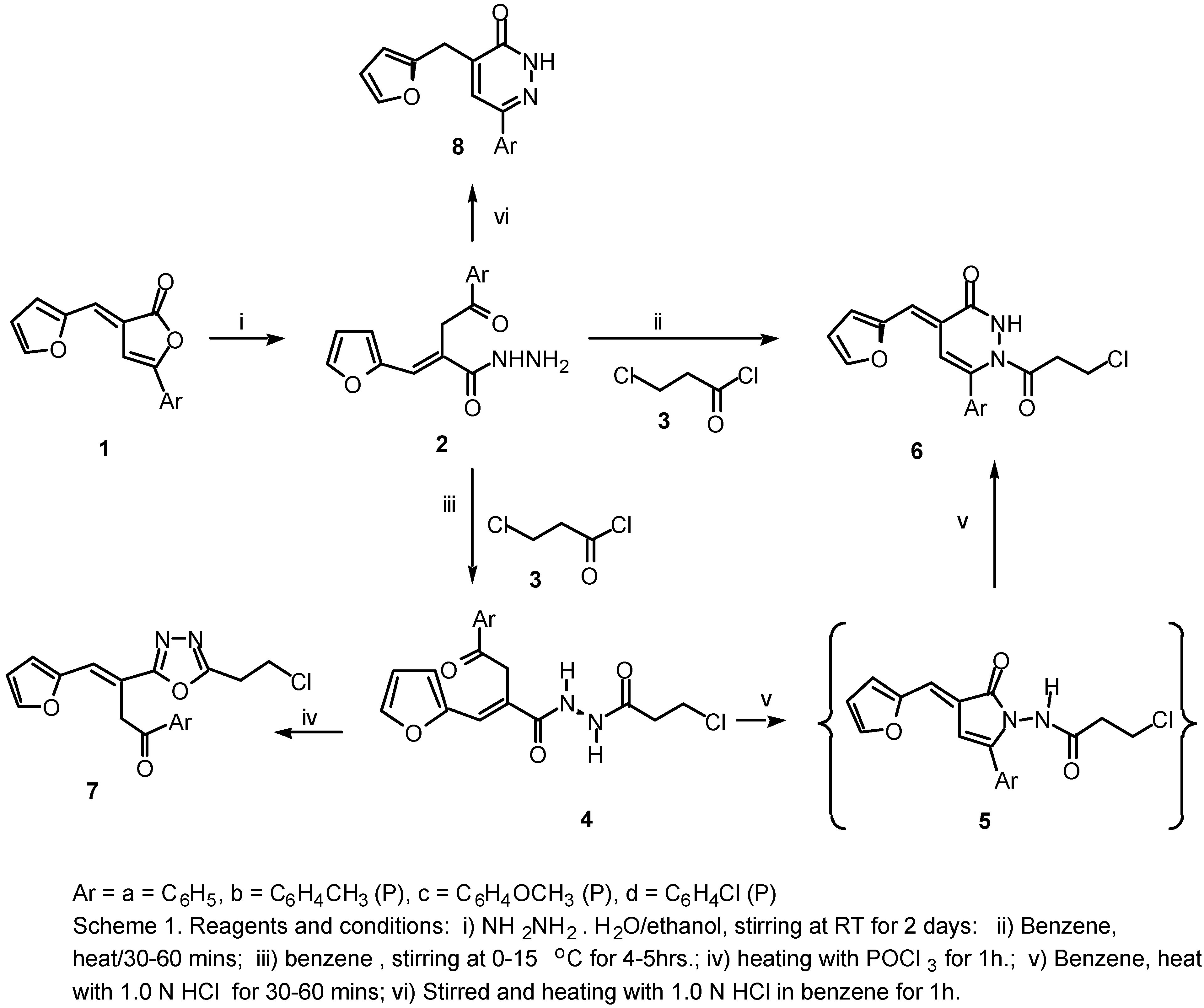
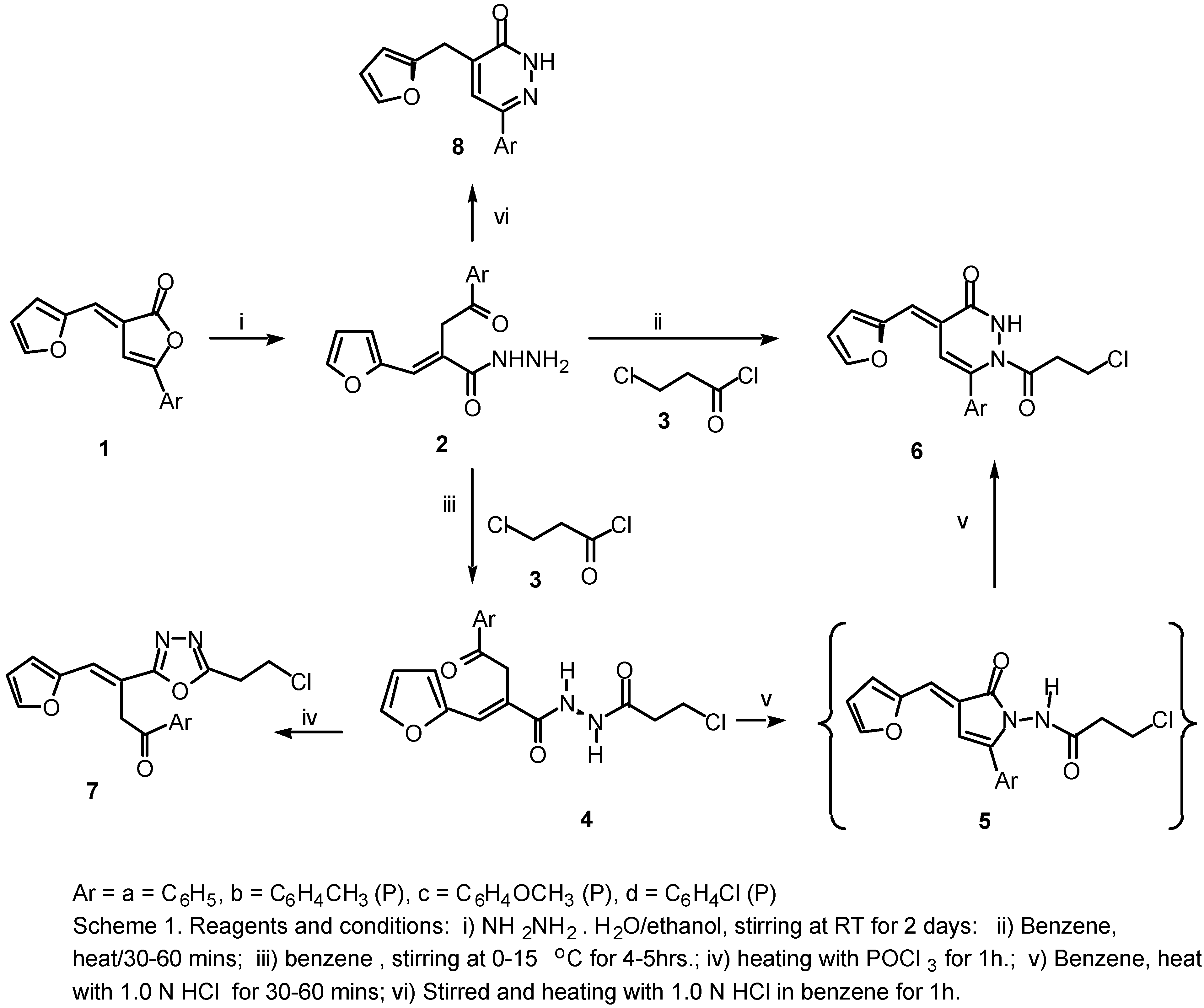
This reported biological importance of 2(3H)-furanones, 4,5-dihydropyridazinones and 1,3,4-oxadiazoles prompted us to attempt the conversion of 2(3H)-furanones (1a-d) into heterocyclic systems of potential synthetic and biological importance, for example, the acrylic acid hydrazides (2a-d), 3-pyridazinones (6a-d), N1[α-aracyl-β−(2-furyl)acroyl-N2[3-chloro propanoyl]hydrazine (4a-d) and 1,3,4-oxadiazole derivatives (7a-d), dihydropyridazinones (8a-d).
Results and Discussion
We report here a simple method for the synthesis of 6-aryl-1-(3-chloropropanoyl)-4-[(E)-1-(2-furyl)methylidene)]-1,2,3,4-tetrahydro-3-pyridazinones (6a-d) and 2-(2-chloroethyl)-5-[α-aracyl-β-(2-furyl)]-(E)-vinyl-1,3,4-oxadiazoles (7a-d) in good yields via the interaction of hydrazides (2a-d) with acid chloride (3) and POCl3 in a one pot reaction.
In this investigation, hydrazides (
2a-d) proved to be useful precursors in the synthesis of several het-erocyclic systems. When the 2(3
H)furanones (
1a-d) were allowed to react with hydrazine hydrate in ethanol, ring opening occurs with the formation of the
E-isomers of
α-aracyl-
β−(2-furyl)acrylic acid hydrazides (
2a-d) as the only products. There was no detectable amount of the corresponding
Z-isomers according to
1H NMR [
3]. The configurational structures of the 2(3
H)-furanones (
1a-d) as
E-isomers were confirmed by
1H and
13C NMR, as illustrated in
Table 2 and
Table 4. The infrared spectra of these products (
2a-d) (
cf. Table 1) show an absorption band at 3199.7-3104.2-3331 cm
-1 (broad band) characteristic of the NH group, and bands at 1648-55 cm
-1 and 1668-97 cm
-1 for υC=O, characteristic of the amide carbonyl and ketone groups, respectively. On heating the hydrazides (
2a-d) in 1.0 N HCl for 30-60 min, ring closure occurs, leading to the formation of the corresponding 3(2
H)-pyridazinones, namely the 6-aryl-4-furylmethyl-3(2
H)-pyridazinones (
8a-d). The infrared spectra of compounds (
8a-d) (
cf. Table 1) show a broad absorption band at 3150-3300 cm
-1 characteristic of the NH group and an absorption band at 1654-60 cm
-1 for υC=O, characteristic of the amide carbonyl group. The infrared spectra of these compounds are similar to those reported for other pyridazinone derivatives [
15,
16,
17,
18].
The
1H NMR spectra of compounds (
2a-d) exhibit singlet signals at 4.41 for (C
H2COAr), a singlet at δ 6.67 for olefinic protons, triplet signals at δ 7.07 for (-N
H-NH
2) and two signals dd for (-NH-N
H2) due to the strong electrical quadrupole moment effect [
19]. In compounds (
8a-d) the CH
2 adjacent to the furan ring exhibited a singlet at δ 3.92, the olefinic proton of the unsaturated double bond showed a doublet signal at δ 7.56 and the (-NH-) group a singlet at δ13.23 ppm.
The interaction of
α-aracyl-
β-(2-furyl)acrylic acid hydrazides (
2a-d) with 3-chloropropanoyl chloride was studied also under different conditions at 0-15°C and 80°C. When the reaction took place at 0-15°C over 4-5 h, it gave a 85% yield of
N1[
α-aracyl-
β−(2-furyl)acroyl-
N2[3-chloropropanoyl]-hydrazine compounds (
4a-d), but when the reaction took place at 80°C over 30-60 min, HCl was liberated to give an 80-92% yield of 6-aryl-1-(3-chloropropanoyl)-4-[(
E)-1-(2-furyl)methylidene)]-1,2,3,4-tetrahydro-3-pyridazinones (
6a-d) as illustrated in
Scheme 1.
In a parallel experiment using benzene solvent in the presence of acid catalyst (1.0 N HCl) at 80°C the hydrazine compounds (4a-d) cyclised and only formation of products (6a-d) was noted, as evi-denced by TLC after 1h. We believe the liberation of HCl aided the ring closure of the hydrazine de-rivatives compounds (4a-d) under the reaction conditions at 80°C to provide compounds (6a-d). These compounds were shown by direct comparison (m.p and mixed m.p) and also by TLC to be identical in all aspects with the authentic products obtained by the action of 1.0 N HCl in benzene solvent with N1[α-aracyl-β−(2-furyl)acroyl-N2[3-chloropropionyl]hydrazine compounds (4a-d). Acid catalysis proved to be essential and reaction was essentially complete after 30-60 min at 80°C (ca. 92% yield). The 1H NMR spectra of (4a-d) exhibited singlet signals for the CH2 protons at δ 4.41 ppm and also show two doublet signals at 9.8 and 10.11 ppm for the hydrazine (-NH-NH-) group.
The alternative mode of ring closure of
N1[
α-aracyl-
β−(2-furyl)acroyl-
N2[3-chloropropionyl]-hydrazine compounds (
4a-d), that is attack by the
N1, would lead to the formation of the corresponding
N-(3-chloropropionyl)aminopyrrolines (
5a-d) as intermediates. But these
N-aminopyrrolines (
5) are not formed, and the pyridazinones (
6a-d) are the only isolable products obtained from this reaction. We be-lieve that the
N-aminopyrrolines (
5a-d) are unstable under the acid catalyzed reaction conditions and if formed, they might undergo ring expansion [
1b] yielding the corresponding 3(2
H) pyridazinones (
6a-d). This behavior is in accordance with the reported rearrangements of
N-aminophthalimides in acid me-dium to the corresponding phthalaza-1,4-diones [
15]. The structure of compounds (
6a-d) was con-firmed by
1H NMR (300 MHz), microanalysis and infrared spectral data. The IR spectra show an ab-sorption band for the υC=O stretching vibrations of cyclic carboxamides at 1666-1678 cm
-1, character-istic of the 3(2
H) pyridazinone ring, and in the 1725-1733 cm
-1 region for the C=O of the open alkyl-carboximide, as well as a broad band at 3227-3337 cm
-1 characteristic of the NH group. The spectra of compounds (
6a-d) (
cf, Table 1) are similar to those of 1-benzoyl-6-aryl-4-thienylidene-1,6-dihydropyridazin-3-(2
H)ones [
20]. The
1H and
13C NMR data showed evidence for the formation of a bicyclic structure as shown in
Table 2 and
Table 3. In compounds (
6a-d), as expected, the two olefins (=C-H) have different chemical shifts both in the
1H and
13C NMR spectra. These highly conjugated struc-tures show a typical two singlet signal for the C-H in the
1H NMR spectrum at δ 6.6-6.7 and 7.08-7.13 for CH=C-Ar and Fur-CH=C-respectively, because in the case of Fur-CH=C-there is a long range coupling with the furan-H and this evidence can be obtained from the coupling constants between H(7) and furan-H(9) as illustrated in
Table 2. Further evidence to support our assignments was obtained from the
13C NMR spectrum of compound (
6b) that showed the carbons of the highly conjugated system at C(5) and C(7) to be at δ 118.7 and 126.25 respectively as shown in
Table 3. It is clear also from the
1H NMR spectrum of the compounds (
6a-d) that in the ClCH
2CH
2CO-system, the CH
2 adjacent to the carbonyl appears as a triplet at δ 3.722 ppm and the other CH
2 adjacent to the chlorine nuclei are com-pletely decoupled from directly attached protons, and appear as two triplet signals at 2.54-2.77 ppm, due to the strong electrical quadrupole moment effect [
19].
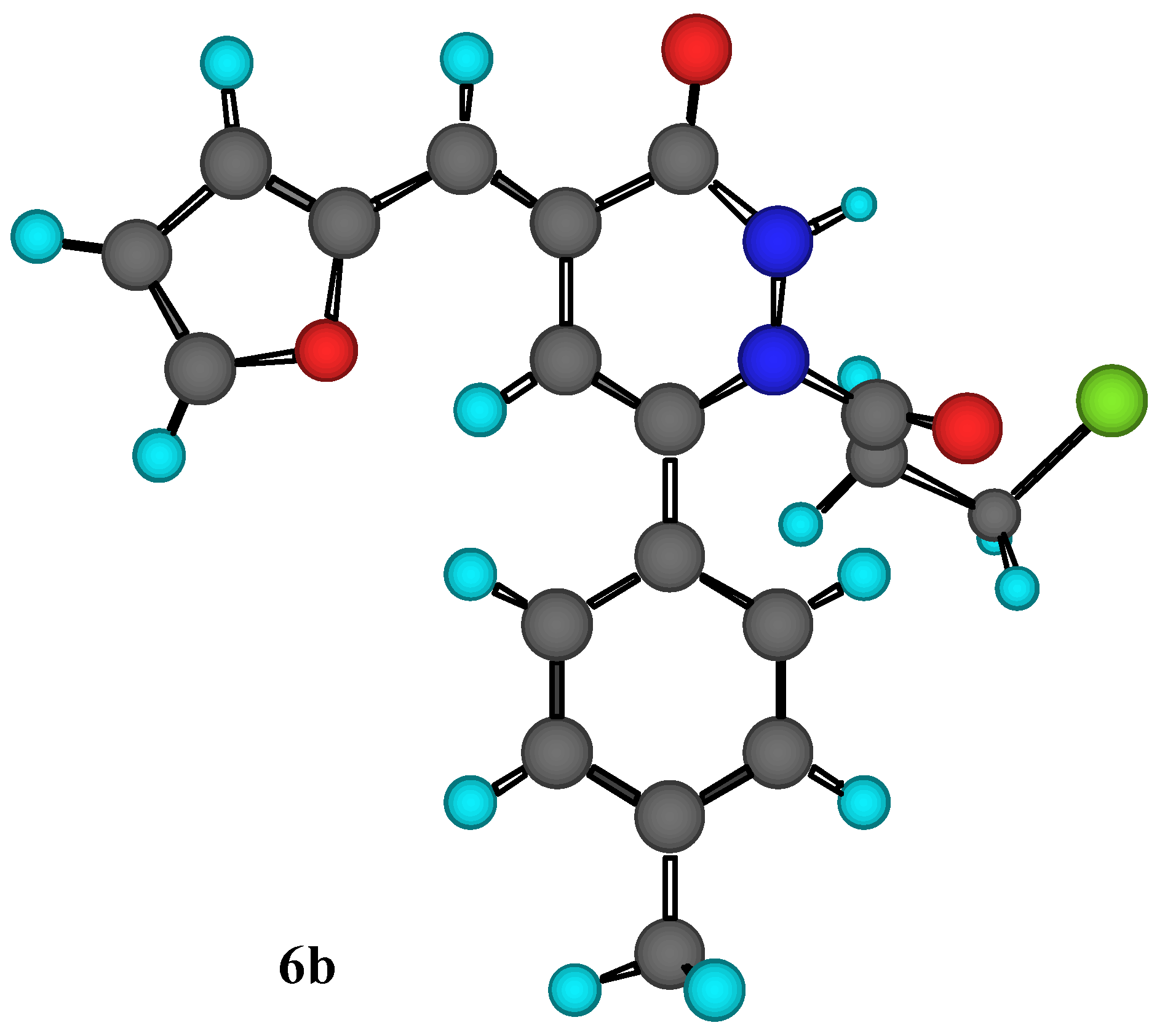
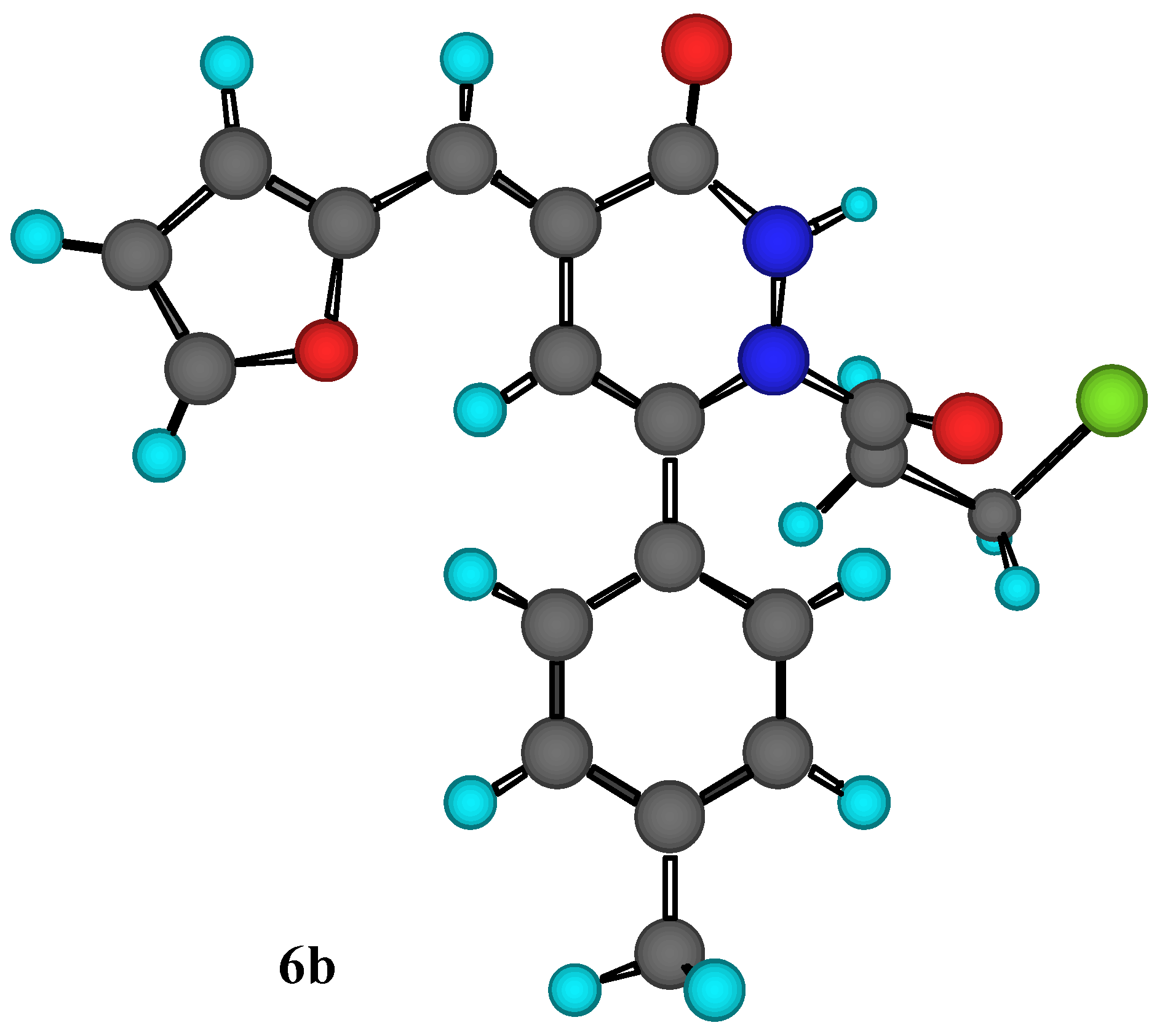
Figure 1.
MM2 Calculations for compound 6b.
Figure 1.
MM2 Calculations for compound 6b.
Another piece of evidence to support our assignment the structure of compound (
6b) was obtained through MM2 calculations for the minimum and total steric energy, as illustrated in
Figure 1. It is im-portant to point out that we have examined steric energies from 68.479 kcal/mole to the lowest energy structure at 8..86 kcal/mole over 392 iterations. Minimization terminated normally because the gradient norm was less than the minimum gradient norm.
The interaction of hydrazides (
2a-d) with POCl
3 provided 2-(
β-chloroethyl)-5-[
α-aracyl-
β-(2-furyl)](
E)-vinyl-1,3,4-oxadiazoles (
7a-d) as the only products in 75-83% yield.. The infrared spectra (IR) of these products (
cf. Table 1) show an absorption band at 1695-1698 cm
-1 for υC=O, character-istic of the ketone carbonyl group, and 1578-1617 cm
-1 for υC=N. The
1H NMR spectrum of com-pounds (
7a-d) exhibit singlet signals at δ 4.37 for (C
H2COAr) and a singlet signal at δ 6.65 for the ole-finic proton and the disappearance the signals of the hydrazo group as illustrated in
Table 2.
Table 1.
Infrared (IR) and 1H NMR (300 MHz) spectral data for compounds 1a-8d.







{kind=link}
{kind=link}


