Cohalogenation of Allyl and Vinylsilanes using Polymer-bound Haloate(I)-Reagents

Abstract
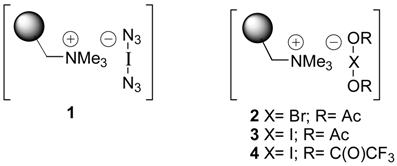
:Introduction
Results and Discussion

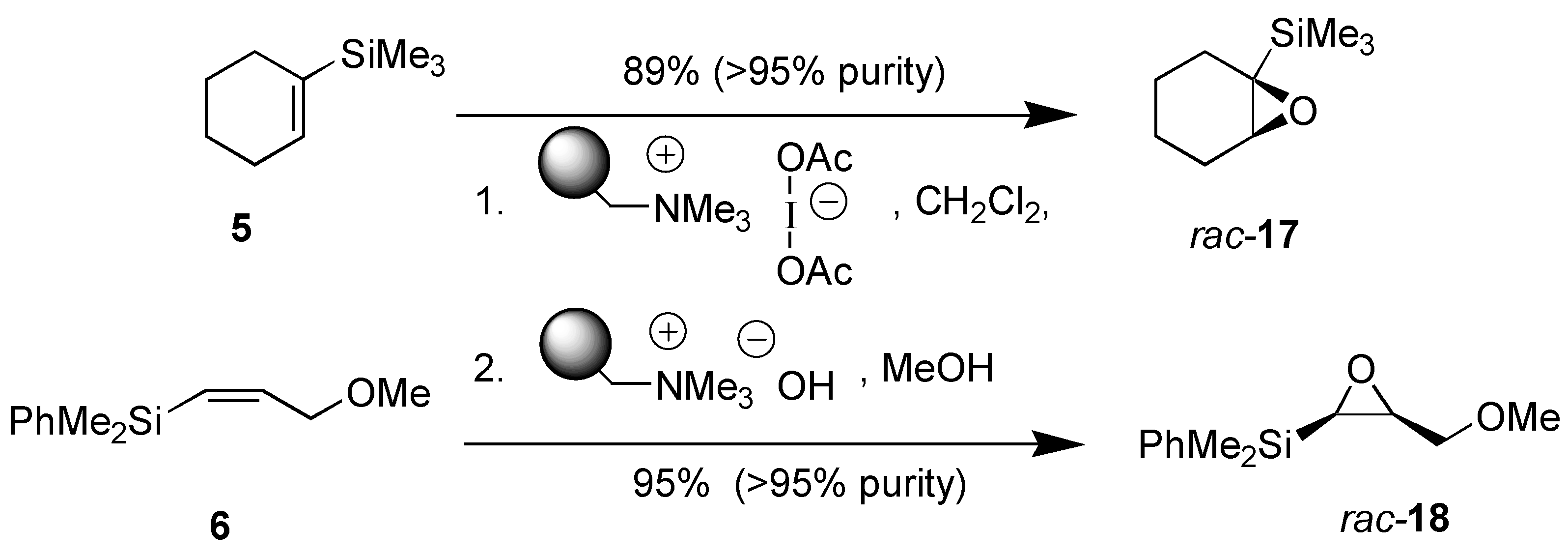{kind=link}
| Alkene | Reagenta (eq.)b | Product | Yield %, c Purity % | ||
|---|---|---|---|---|---|
| 5 | 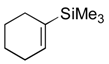 |  | 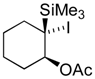 | 11 | 76 (80), >99 |
| 3 (4), 5h | |||||
| 6 |  | | 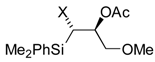 | 12 | 84 (98), >99 |
| 3 (6), 60h | X= I | 13 | 77 (90)d | ||
| 2 (4), 40h | X= Br | ||||
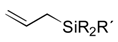 | | 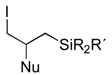 | |||
| 7 | R= Me, R´= Ph | 3 (4), 6h | Nu= OAc | 14 | 62 (75), >95 |
| 8 | R= Ph, R´= Me | 3 (3), 6h | Nu= OAc | 15 | 67 (80), >95 |
| 9 | 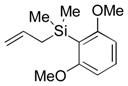 | |  | 16 | 83, >95 |
| 3 (5), 2.5h | |||||
| 10 | 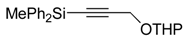 | | -------- | no reaction | |
| 3 (8), 8d |

Experimental
General Methods
General Procedure for the Preparation of Polymer-Bound Reagents 2 and 3
General procedure for the 1,2-cohalogenation of silylated alkenes
Acknowledgements
References and Notes
- Reviews: Shuttleworth, S. J.; Allin, S. M.; Sharma, P. K. Synthesis 1998, 1217–1239. . Kaldor, S. W.; Siegel, M. G. Curr. Opin. Chem. Biol. 1997, 1, 101–106. [PubMed]. Hodge, P.; Sherringtom, D. C. Polymer-Supported Reaction in Synthesis; Wiley: New York, 1990. [Google Scholar] . Laszlo, P. (Ed.) Preparative Chemistry using Supported Reagents; Academic: San Diego, 1987.
- Flynn, D. L.; Devraj, J. J.; Parlow, S.S. Curr. Opin. Drug Discovery Dev. 1998, 1, 41–50. Suto, M. J.; Gayo-Fung, L. M.; Palanki, M. S. S.; Sullivan, R. Tetrahedron 1998, 54, 4141–4150. Parlow, S. S. Tetrahedron Lett 1996, 37, 5257–5260. Habermann, J.; Ley, S. V.; Scott, J. S. J. Chem. Soc., Perkin Trans. I 1998, 3127–3130. Caldarelli, M.; Habermann, J.; Ley, S. V. J. Chem. Soc., Perkin Trans. I 1999, 107–110.
- Hodge, P. Chem. Soc. Rev. 1997, 26, 417–424.
- Yaroslavsky, C.; Patchornik, A.; Katchalski, E. Tetrahedron Lett. 1970, 3629–3632.
- Kirschning, A.; Plumeier, C.; Rose, L. Chem. Commun. 1998, 33–34. Hashem, Md. A.; Jung, A.; Ries, M.; Kirschning, A. Synlett 1998, 195–197. Kirschning, A.; Hashem, Md. A.; Monenschein, H.; Rose, L.; Schöning, K.-U. J. Org. Chem. 1999, 64, 2720–2722.
- Kirschning, A; Monenschein, H.; Schmeck, C. Angew. Chem 1999, 111, 2720–2722, Angew. Chem. Int. Ed, 1999, 38, 2594-2596.
- Kirschning, A.; Jesberger, M.; Monenschein, H. Tetrahedron Lett. 1999, 40, 8999–9002. Monenschein, H.; Sourkouni-Argirusi, G.; Schubothe, K. M.; O´Hare, T.; Kirschning, A. Org. Lett. 1999, 1, 2101–2104.
- For a recent example for iodate(I)-promoted iodination of arenes refer to S. Tripathy, S.; LeBlanc, R.; Durst, T. Org. Lett 1999, 1, 1973–1975.
- Recently, an interesting new method for 1,2-coiodination of alkenes using dimethyldioxirane- promoted oxidation of iodomethane was disclosed by G. Asensio, G.; Andrea, C.; Boix-Bernardini, C.; Mello, R.; González-Nuñez, M. E. Org. Lett. 1999, 1, 2125–2128.
- Chan, T. H.; Lau, P. W.; Mychajlowskij, W. Tetrahedron Lett. 1977, 18, 3317–3320. Miller, R. B.; Reichenbach, J. Tetrahedron Lett. 1974, 15, 543–546. Miller, R. B.; McGarvey, G. J. Org. Chem. 1978, 43, 4424–4431. Chan, T. H.; Fleming, I. Synthesis 1979, 761–786. Huynh, C.; Linstrumentelle, G. Tetrahedron Lett. 1979, 20, 1073–1076. Tamao, K.; Akita, M.; Maeda, K.; Kumuda, M. J. Org. Chem. 1995, 36, 2153–2156. Barluenga, J.; Alvarez-Garcia, L. J.; Gonzaléz, J. M. Tetrahedron Lett 1995, 36, 2153–2156.
- Stanos, D. P.; Taylor, A. G.; Kishi, Y. Tetrahedron Lett. 1996, 37, 8647–8650, and references cited therein.
- We collected evidence that the binary compound [I-OAc] is the active electrophilic reagent, which is slowly released from the resin 3: Monenschein, H.; Kirschning, A. unpublished results.
- This fact, may either be rationalized by assuming that only a proportional amount of immobilized halide was transformed into the haloate(I)-species or that only the most accessible haloate(I) anions are involved in the cohalogenation process. If acylated hypohalites are the active species after release from the polymer (see also reference [12]), their degradation prior to the reaction with alkenes may also contribute to the need for a formal excess of reagent.
- The functionlized polymers were recycled to the halide form without loss of activity by treatment with concentrated aqueous HBr or HI for 1 h at rt.
- Sample Availability: Samples of compounds 1, 2, 3, 4, 11, 12, 17 are available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Domann, S.; Sourkouni-Argirusi, G.; Merayo, N.; Schönberger, A.; Kirschning, A. Cohalogenation of Allyl and Vinylsilanes using Polymer-bound Haloate(I)-Reagents. Molecules 2001, 6, 61-66. https://doi.org/10.3390/60100061
Domann S, Sourkouni-Argirusi G, Merayo N, Schönberger A, Kirschning A. Cohalogenation of Allyl and Vinylsilanes using Polymer-bound Haloate(I)-Reagents. Molecules. 2001; 6(1):61-66. https://doi.org/10.3390/60100061
Chicago/Turabian StyleDomann, Silvie, Georgia Sourkouni-Argirusi, Nuria Merayo, Andreas Schönberger, and Andreas Kirschning. 2001. "Cohalogenation of Allyl and Vinylsilanes using Polymer-bound Haloate(I)-Reagents" Molecules 6, no. 1: 61-66. https://doi.org/10.3390/60100061
APA StyleDomann, S., Sourkouni-Argirusi, G., Merayo, N., Schönberger, A., & Kirschning, A. (2001). Cohalogenation of Allyl and Vinylsilanes using Polymer-bound Haloate(I)-Reagents. Molecules, 6(1), 61-66. https://doi.org/10.3390/60100061