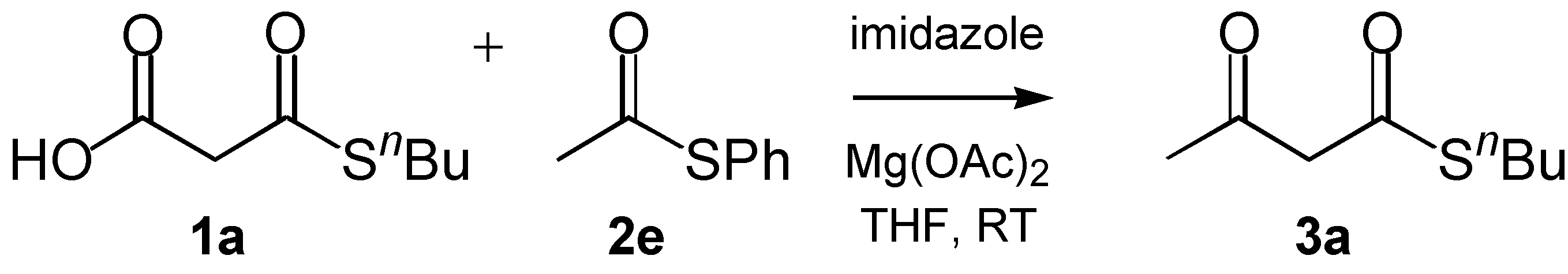
The Claisen condensation of malonyl thioesters is one of the central processes in the biosynthesis of polyketide natural products [
1]. Although the diverse enzymes that catalyze this remarkable biooligomerization are increasingly well understood and extensively exploited in modern biotechnology [
2,
3,
4], synthetic catalysts for similarly controlled oligomerization of malonyl thioesters in enzyme-free systems do not exist. Kobuke and Yoshida have, however, demonstrated more than twenty years ago that Claisen condensation of
n-butyl thiomalonate
1a and phenyl thioacetate
2e can be catalyzed by imidazole and magnesium cations in THF at room temperature to give
n-butyl thioacetoactate
3a in 87 h and 60% yield (
Scheme 1) [
5]. These Kobuke-Yoshida (KY) conditions [
5,
6] contrast sharply with the harsher conditions required in other model systems for polyketide synthesis [
7,
8,
9] but not polyketide cyclization [
10,
11,
12].
Results and Discussion
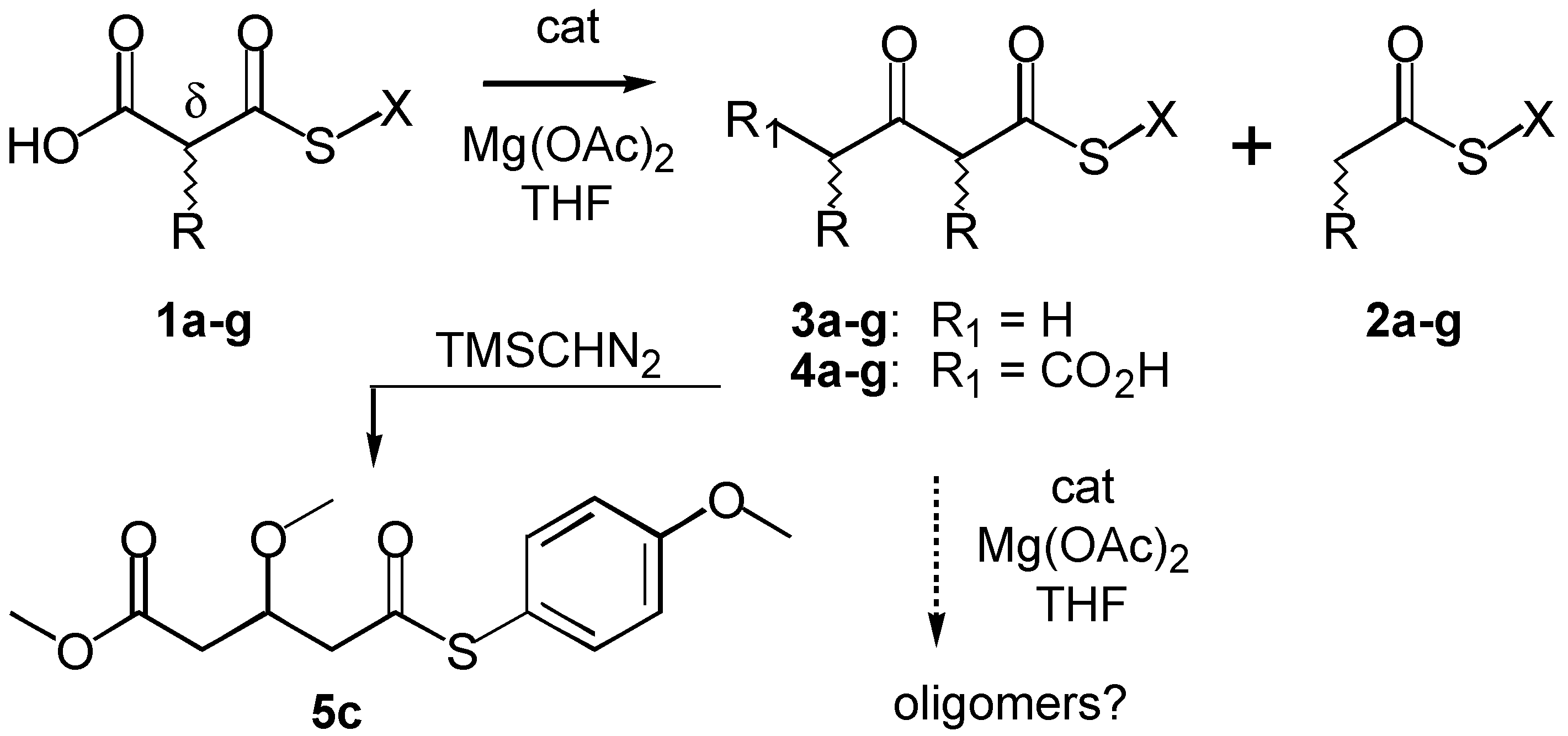
The original KY-conditions are of potential significance for the construction of artificial polyketide synthases because many histidine-rich organic architectonics with esterase and/or phosphatase activity have been elegantly devised over the past two decades [
13,
14,
15]. Original KY-conditions are, however, incompatible with the Claisen self-condensation required for oligomerizations leading to polyketides (
Scheme 2). Specifically, self-condensation of
n-butyl thiomalonate
1a or phenyl thioacetate
2e was impossible because of poor leaving group (LG) ability of
n-butylthiolate or lack of access to activated carbanion intermediates
via decarboxylation, respectively [
5]. Here we report that Mg
2+-imidazole-catalyzed thiomalonate dimerization is possible in up to 71% yield under refined conditions by precise fine tuning of the properties of carbanion intermediate, thiolate leaving group, and catalyst, and show that future applicability of this approach toward biomimetic polyketide synthesis is nevertheless problematic.
To elucidate the subtle balance between leaving group and carbanion activation needed for thiomalonate dimerization under KY-conditions, we prepared substrates
1a-f with systematically varied leaving groups (LG) (
Scheme 2,
Table 1). Reduced Hammett σ
p [
16] (corresponding with one exception to the p
Ka of the employed thiophenols [
17,
18]) should
reduce LG-activation and
increase the reactivity of carbanion intermediates, which can be seen as up-field shifts of the α-hydrogens (
Table 1). Indeed the rate of substrate consumption was inversely related to the Hammett σ
p. However, highly stabilized carbanions did not further react with the electrophiles, thus dominant formation of decarboxylation products
2e-
2g resulted. Thiomalonate self-condensation giving rise to thioacetoacetates
3b-3f in up to 37% yield for
3c occurred only at intermediate activation of both carbanion and LG.
Table 1.
Claisen seilf-condensation of thio(methyl)malonates 1a-g in THF at room temperature in presence of Mg(OAc)2 and imidazole.
Table 1.
Claisen seilf-condensation of thio(methyl)malonates 1a-g in THF at room temperature in presence of Mg(OAc)2 and imidazole.
![Molecules 06 00845 i001]() |
The most promising thiomalonate
1c was studied in more detail. Separation of the product mixture by reverse-phase HPLC was possible on analytical YMC-Pro-C8-columns applying a linear solvent gradient of water / CH
3CN (0.1% TFA) = 20% - 100% over 20 min. Only one additional product with a retention time
Rt = 5.22 min similar to that of substrate
1c (
Rt = 4.94 min) was observed besides the expected acetoacetate
3c (
Rt = 5.90 min), 4-thioanisole (
Rt = 6.31 min) and 4-methoxyphenyl thioacetate
2c (
Rt = 6.31 min). This new product decomposed easily and could not be fully purified. However,
in-situ methylation with trimethylsilyl diazomethane (TMSCHN
2, [
19]) in toluene-methanol gave a stable product with
m/z = 319 in the ESI-MS that was consistent with [
M + Na]
+ expected for methyl ester
5c. The
1H-NMR spectrum of
5c revealed about 50%-conjugation of the enol ether with both carbonyl groups. This derivatization demonstrated that the new unstable product formed from
1c under KY-conditions is carboxylate
4c. Identification of
4c allowed us to assign
all new resonances appearing in
1H-NMR spectra recorded during the course of a reaction in THF-
d8 to either
3c, 4c,
2c or acetylimidazole.
The satisfactory outcome with substrate
1c encouraged us to study the influence of additional parameters. Thiomethylmalonate
1g gave decarboxylation product
2g only, probably due to the steric effect of the methyl group on the α-position (
Table 1). Replacement of Mg
2+ by other divalent cations such as Zn
2+, Cu
2+, Ca
2+, or Ba
2+ under otherwise original KY conditions led to an increase in acetate (
2a-2f) and/or hydrolyzed products rather than Claisen self-condensation. The rate of substrate consumption decreased with decreasing p
Ka of the imidazole catalyst [
20] (
Table 2). The yield of Claisen products
3c and
4c, however, increased from imidazole (36%) over benzimidazole (47%) to 71% with 8-nitrobenzimidazole. Further decrease in catalyst p
Ka gave 51% for 4(5)-nitroimidazole. This identified p
Ka ≈ 3 as optimum catalyst p
Ka for Claisen self-condensation under these conditions (
Table 2).
Table 2.
Claisen seilf-condensation of thiomalonate 3 in THFd at room temperature in presence of Mg(OAc)2 and the indicated imidazole catalyst.
Table 2.
Claisen seilf-condensation of thiomalonate 3 in THFd at room temperature in presence of Mg(OAc)2 and the indicated imidazole catalyst.
![Molecules 06 00845 i002]() |
Replacement of THF with other solvents except dioxane and addition of more than 10% water inhibited self-condensation. Kobuke and Yoshida’s original implication that ether-coordination to Mg
2+ is essential for catalysis was further partially supported by slow appearance of up to 35% Claisen products
3c and
4c in CHCl
3 containing 10% 18-crown-6 with 8-nitrobenzimidazole as catalyst (
Table 2, entry 4). The reaction did, however, not proceed in the “ether-rich” micelles formed by 10% Triton X-100 in water, also when
N-acetylhistidine was used instead of imidazole or 8-nitrobenzimidazole.
No indications for further reaction of thiomalonate
1c with Claisen products
3c or
4c were found. This suggests that Aldol and Claisen reaction with β-ketone and thioester for formal continuation along the terpenoid and polyketide pathway [
1], respectively, are not possible under these conditions. Novel approaches toward transient deactivation of the β-ketone in acetoacetates required for polyketide synthesis under these conditions are under investigation. Preliminary studies with “additives” such as TMSCHN
2, TES-Cl,
n-butylamine,
p-methoxyaniline and phenylhydrazine [
21] were not successful.
Experimental
General
Synthetic reagents were purchased from Fluka or Acros. Column chromatography was carried out on silica gel 60 (Fluka, 40-63 mm). Analytical thin layer chromatography (TLC) and preparative thin layer chromatography were performed on silica gel 60 (Fluka, 0.2 mm) and silica gel GF-2 (Aldrich, 1 mm), respectively. Reverse Phase column chromatography was performed with Silicagel 100 C18-Reverse Phase. 1H- and 13C-NMR spectra were recorded on Bruker 400 MHz Spectrometer. ESI-MS were performed on a Finnigan MAT SSQ 7000. HPLC was carried out using a YMC Pro-C8 (4 x 50 mm) prepacked column. Anhydrous THF and ethyl ether were distilled over Na and benzophenone.
Thiomalonate 1c: General procedure for preparation of thiomalonate esters
Thiomalonate esters were prepared following the procedure in reference [
5]. Namely, to a solution of malonylchloride (1.6 mL, 16 mmol) in dry ether (20 mL) 4-methoxybenzenethiol (2.0 mL, 16 mmol) was added at room temperature (rt) under nitrogen atmosphere. After stirring for 3h, saturated aqueous NaHCO
3 solution was added to the reaction mixture. The ether layer was discarded, and the aqueous layer was further washed with EtOAc. After acidification of the aqueous layer, the product was obtained by extraction (EtOAc), washing (brine), drying (Na
2SO
4) and evaporation under reduced pressure. The crude product was purified by silica gel column chromatography (acetone) then by recrystalization (dichloromethane) to give pure title compound as colorless crystals (1.7 g, 45 %): mp 89-90 ºC;
1H-NMR (CDCl
3) δ 9.80 (br.s, 1 H), 7.36 (d,
J = 6.7 Hz, 2 H), 6.95 (d,
J = 6.7 Hz, 2 H), 3.83 (s, 3 H), 3.69 (s, 2 H);
13C-NMR (CDCl
3) δ 191.0 (s), 171.0 (s), 161.0 (s), 136.1 (2 x d), 117.0 (s), 115.1 (2 x d), 55.4 (q), 48.0 (t).
Acetoacetate 3c: General procedure for Claisen condensation
To a solution of 4-methoxyphenyl thiomalonate (1c, 20 mg, 0.088 mmol) in THF (1 mL), Mg(OAc)2•4H2O (10 mg, 0.047 mmol) and imidazole (6 mg, 0.088 mmol) were successively added. The mixture was stirred for 17h at rt, and then it was diluted with CH2Cl2, washed with 1M HCl and brine, dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography (1:1 to 0:1 petroleum-ether/CH2Cl2) to give pure acetoacetate 3c as a colorless oil (3.7 mg, 37 %): 1H-NMR (CDCl3) δ 7.37 (d, J = 8.8 Hz, 0.6 H), 7.32 (d, J = 8.8 Hz, 1.4 H), 6.94 (d, J = 8.8 Hz, 2 H), 5.47 (s, 0.3 H), 3.82 (s, 3 H), 3.73 (s, 1.4 H), 2.27 (s, 2.1 H), 1.94 (s, 0.9 H).
Methyl ester 5c
The crude product obtained following the general procedure for Claisen condensation starting from 1c (213 mg) was fractionated by column chromatography (ODS, 40 to 100 % CH3CN in 0.1 % aqueous TFA). The fractions containing 4c were concentrated briefly and then extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated under reduced pressure to give crude product (32 mg) consisting of 1c and 4c. The product mixture obtained was dissolved in toluene (0.5 mL) and methanol (0.5 mL), and TMSCHN2 (2 M in hexane, 0.3 mL) was added. After stirring for 30 min, the mixture was concentrated under reduced pressure and purified by preparative TLC (CH2Cl2, Rf 0.26) to give pure 5c (3.5 mg, 1.2%): 1H-NMR (CDCl3) δ 7.36 (d, J = 8.9 Hz, 2 H), 6.94 (d, J = 8.9 Hz, 0.8 H), 6.93 (d, J = 8.9 Hz, 1.2 H), 5.62 (s, 0.4 H), 5.23 (s, 0.6 H), 4.18 (s, 1.2 H), 3.83 (s, 1.2 H), 3.82 (s, 1.8 H), 3.80 (s, 0.8 H), 3.75 (s, 1.2 H), 3.71 (s, 1.8 H), 3.70 (s, 1.8 H), 3.67 (s, 1.2 H); ESI-MS (CH2Cl2 + TFA) m/z 334.9 (8.25) [M + K]+, 319.0 (100) [M + Na]+, 157.4 (31.9) [M - MeOC6H4S]+.




{kind=link}
{kind=link}

