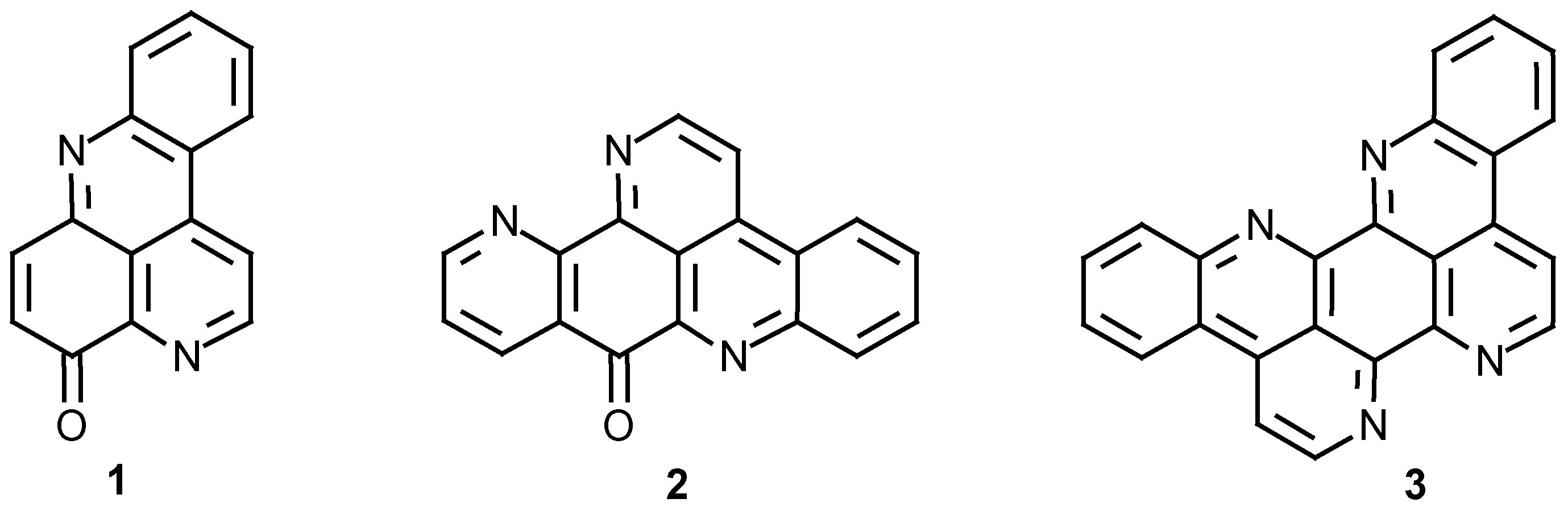
Experimental
General
Commercially available reagents were purchased from standard chemical suppliers and were used without further purification. 2(N)-(4-methoxyaniline)-1,4-naphthoquinone (
16) was synthesized by a literature method [
13]. IR spectra (KBr disks) were recorded on a Nicolet 205 FT-IR spectrophotometer. MS and HRMS spectra were recorded on a Fisons Autospec Q instrument.
1H-NMR and
13C-NMR spectra were recorded on a Bruker ARX-500 or a Bruker AMX-360 spectrometer. NOE experiments and 2D NMR spectra (COSY, HMQC and HMBC) were recorded on a Bruker ARX-500 instrument using standard pulse sequences. TLC was performed on Merck precoated Kieselgel 60 F
254 plates. Column chromatography was performed using Silica gel 60 H (Merck) unless otherwise stated. The silica was washed with methanol, before use, in a Soxhlet apparatus. In all cases silica gel chromatography was performed with vacuum.
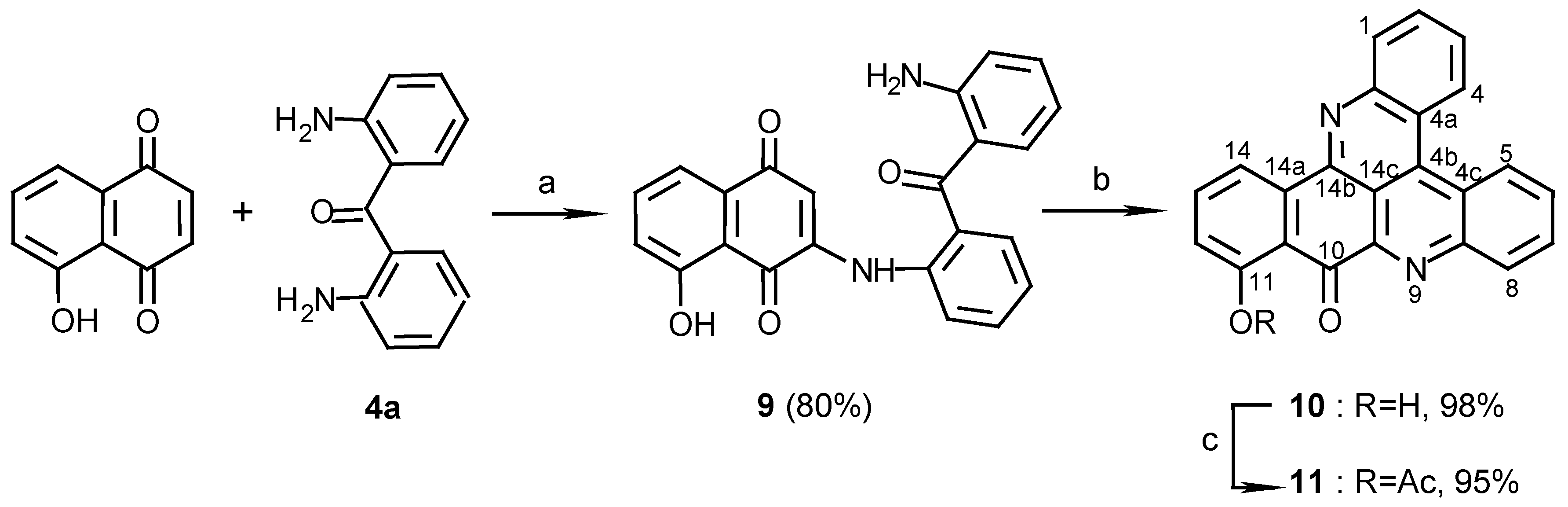
2,2’-Diaminobenzophenone (4a):
Compound
4a was performed from 2,2’-dinitrobenzophenone by hydrogenation, instead of the literature method of reduction with iron powder [
19]. 2,2’-Dinitrobenzophenone [
19] (250 mg, 0.92 mmol) was dissolved in dichloromethane (40 mL), 5% Pd/C (100 mg) was added and the solution was shaken in a Parr apparatus under H
2 (3 atm) for 1 hour. The catalyst was filtered off and the solvent evaporated to afford
4a (195 mg, quantitative yield), yellow crystals (from 80% aqueous methanol), m.p. 134°C (lit [
19], 134-135 °C).
2,2’-Diamino-4,4’-dimethoxybenzophenone (4b):
4b was prepared from 2,2’-Dinitro-4,4’-dimethoxybenzophenone [
20] (250 mg, 0.75 mmol) under the same conditions as described for the synthesis of
4a. The product was recrystallized from methanol to afford
4b (185 mg, 90%): m.p. 138°C (lit [
20], 137-138°C).
General procedure for the reaction between arylamines and 1,4-naphthoquinones
The procedure of Pratt [
7] was adopted. The corresponding amine (1 equiv.) was dissolved in ethanol, CeCl
3·7H
2O (0.05 equiv.) was added followed by the naphthoquinone (1.5 equiv.). The resulting red solution was refluxed for 9 h. During this time air saturated with ethanol (prepared by passage of air through hot ethanol to avoid evaporation of the ethanol) was bubbled through the reaction mixture. After cooling, the ethanol was evaporated and the residue purified by chromatography on silica gel (eluting with chloroform/methanol) to afford the desired amination product.
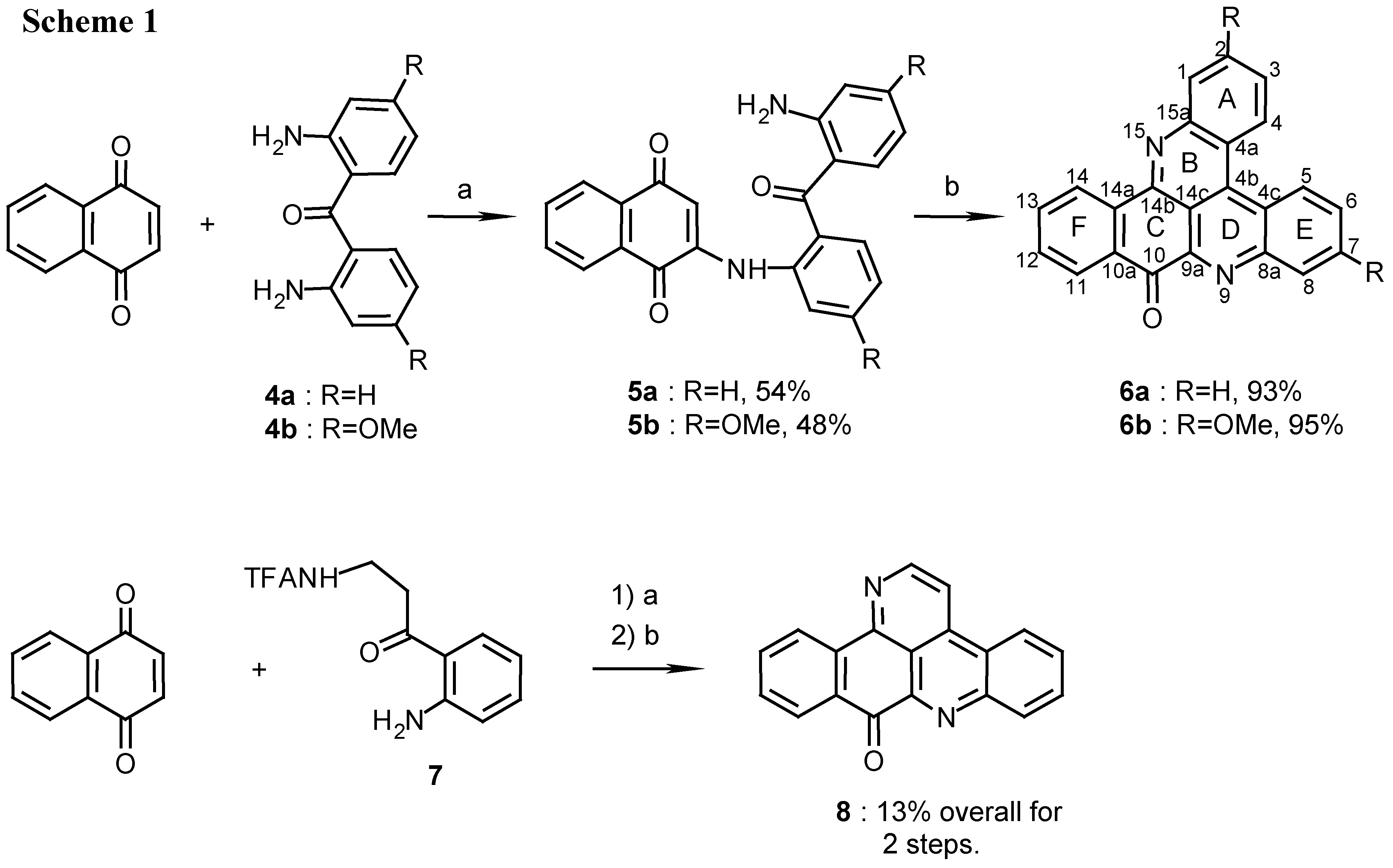
2(N)-(2,2’-diaminobenzophenone)-1,4-naphthoquinone (5a):
4a (450 mg, 2.1 mmol) was dissolved in ethanol (20 mL) and reacted with naphthoquinone (500 mg, 3.2 mmol) by the above described general procedure. The crude mixture containing the product and starting materials was chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH, 50:1) to afford 5a (420 mg, 54%), red prisms (from EtOH), m.p. 208°C; MS (EI); m/z: 368 (100) [M+, C23H16N2O3]; IR: = 1667, 1611, 1568, 1512, 1292, 1246 cm-1; 1H-NMR (CDCl3): δ = 6.56 (s, 1 H, 3-H), 6.59 (t, J=7.5 Hz, 1 H, 12’-H), 6.72 (d, J=7.5 Hz, 1 H, 10’-H), 7.21 (t, J=7.5 Hz, 1 H, 11’-H), 7.29 (t, J=7.5 Hz, 1 H, 4’-H), 7.40 (d, J=7.5 Hz, 1 H, 6’-H), 7.52 (d, J=7.5Hz, 1 H, 13’-H), 7.55 (t, J=7.5 Hz, 1 H, 5’-H), 7.63 (d, J=7.5 Hz, 1 H, 3’-H), 7.65 (t, J=7.6 Hz, 1 H, 7-H), 7.75 (t, J=7.6 Hz, 1 H, 6-H), 8.08 (d, J=7.6 Hz, 1 H, 5-H), 8.09 (d, J=7.6 Hz, 1 H, 8-H), 9.30 (br s, 1 H, NH).
2(N)-(2,2’-diamino-4,4’-dimethoxybenzophenone)-1,4-naphthoquinone (5b):
4b (200 mg, 0.74 mmol) was dissolved in ethanol (10 mL) and reacted with naphthoquinone (170 mg, 1.1 mmol) by the general procedure described above. After two subsequent silica gel columns (eluting with CHCl3) compound 5b was isolated (150 mg, 48%): red prisms (from EtOH), m.p. 214°C. – MS (EI); m/z: 428 (100) [M+, C25H20N2O5]; IR: = 1658, 1624, 1576, 1521, 1448, 1244 cm-1; 1H-NMR (CDCl3): δ = 3.81 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 6.14 (s, 1 H, 10’-H), 6.17 (d, J=8.5 Hz, 1 H, 12’-H), 6.62 (s, 1 H, 3-H) , 6.68 (dd, J=8.5, 2.5 Hz, 1 H, 4’-H), 7.11 (d, J=2.5Hz, 1 H, 6’-H), 7.36 (d, J=8.5 Hz, 1 H, 13’-H), 7.44 (d, J=8.5 Hz, 1 H, 3’-H), 7.65 (t, J=7.0 Hz, 1 H, 7-H), 7.74 (t, J=7.0 Hz, 1 H, 6-H), 8.10 (d, J=7.0 Hz, 1 H, 5-H), 8.11 (d, J=7.0 Hz, 1 H, 8-H), 9.90 (br s, 1 H, NH).
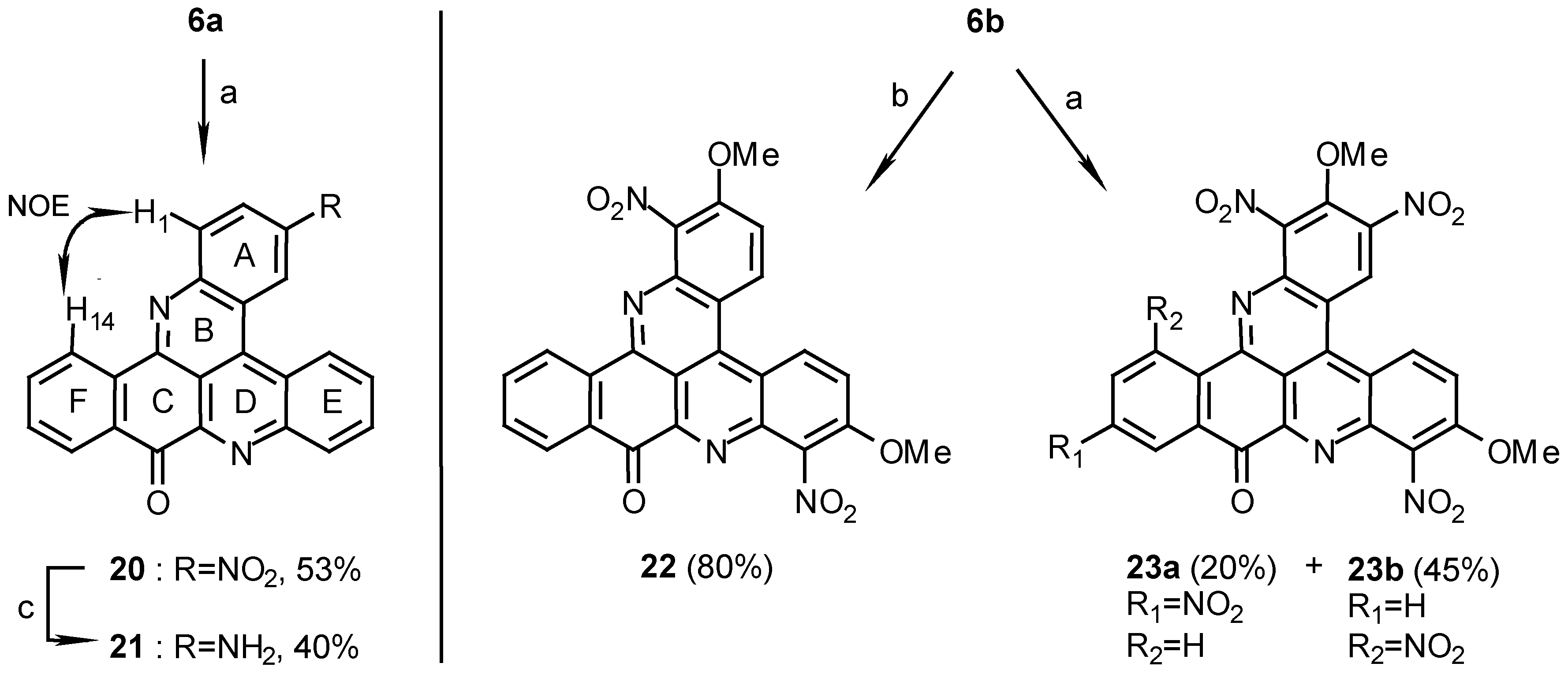
10H-benzo[i]quino[2,3,4-kl]acridin-10-one (6a):
5a (400 mg, 1.1 mmol) was added to a solution of 25% aq. ammonia (10mL) in methanol (100mL) and stirred for 7 days at room temperature. During this time the reaction was monitored by TLC, Rf=0.7 and 0.5 for 5a and 6a respectively (petroleum ether/ethyl acetate, 1:1). The reaction mixture was evaporated and the crude product was chromatographed (eluting with CHCl3/MeOH 30:1) to afford 6a (335 mg, 93%), amorphous powder (CHCl3/MeOH, 9:1), m.p. 255°C; Analysis: C23H12N2O (332.1): calcd. C 83.1, H 3.64, N 8.43, found C 82.4, H 3.53, N 8.70; HRMS calcd. for C23H12N2O [M+] 332.0950, found 332.0947; IR: = 1678, 1654, 1562, 1396, 1203 cm-1; 1H-NMR (CDCl3): δ = 7.73 (t, J=7.5 Hz, 1 H, 12-H), 7.83 (t, J=8.0 Hz, 1 H, 3-H), 7.90 (t, J=7.5 Hz, 1 H, 13-H), 7.95 (t, J=8.0 Hz, 1 H, 6-H), 7.95 (t, J=8.0 Hz, 1 H, 2-H), 8.03 (t, J=8.0 Hz, 1 H, 7-H), 8.42 (d, J=8.0 Hz, 1 H, 1-H), 8.52 (d, J=7.5 Hz, 1 H, 11-H), 8.73 (d, J=8.0 Hz, 1 H, 8-H), 9.09 (d, J=8.0 Hz, 1 H, 4-H), 9.11 (d, J=7.5 Hz, 1 H, 14-H), 9.18 (d, J=8.0 Hz, 1 H, 5-H); 13C-NMR (CDCl3): δ = 115.8 (s, C-14c), 122.4 (s, C-4a), 124.2 (s, C-4c), 125.9 (d, C-14) , 127.0 (d, C-5), 127.1 (d, C-4), 127.9 (d, C-11), 128.1 (d, C-3), 129.9 (d, C-6), 130.8 (d, C-2), 130.9 (d, C-7), 131.0 (d, C-1), 131.0 (d, C-12), 132.2 (s, C-10a), 132.4 (d, C-8), 134.7 (d, C-13), 136.0 (s, C-14a), 136.1 (s, C-4b), 145.4 (s, C-9a), 147.2 (s, C-15a), 147.3 (s, C-8a), 147.7 (s, C-14b), 182.2 (s, C-10).
2,7-Dimethoxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (6b):
5b (150 mg, 0.35 mmol) was treated with ammonia in methanol by the same procedure described for the synthesis of 6a. The product (6b) was obtained after chromatography (eluting with chloroform/methanol, 30:1) (130 mg, 95%): amorphous powder (CHCl3/MeOH, 8:2), m.p. 296°C; Analysis: C25H16N2O3 (392.4): calcd. C 76.5, H 4.11, N 7.14, found C 76.4, H 3.90, N, 7.84; MS (EI); m/z: 392 (100) [M+], 377 (5) [M+- CH3], 361 (14) [(M+- CH3O]; IR: = 1676, 1611, 1587, 1564, 1415, 1239, 1218 cm-1; 1H-NMR (CDCl3): δ = 4.08 (s, 3 H, OCH3), 4.10 (s, 3 H, OCH3), 7.40 (dd, 1 H, J=9.5, 2.5 Hz, 3-H), 7.54 (dd, 1 H, J=9.5, 2.5 Hz, 6-H), 7.64 (t, 1 H, J=8.0 Hz, 12-H), 7.77 (d, 1 H, J=2.5 Hz, 1-H), 7.88 (t, 1 H, J=8.0 Hz, 13-H), 8.04 (d, 1 H, J=2.5 Hz, 8-H), 8.49 (d, 1 H, J=8.0 Hz, 11-H), 8.90 (d, 1 H, J=9.5 Hz, 4-H), 8.98 (d, 1 H, J=9.5 Hz, 5-H), 9.07 (d, 1 H, J=8.0 Hz, 14-H); 13C-NMR (CDCl3): δ = 55.5 (OCH3), 55.9 (OCH3), 108.7 (d, C-8), 110.3 (d, C-1), 113.1 (s, C-14c), 116.0 (s, C-4a), 118.7 (s, C-4c), 119.8 (d, C-3), 122.5 (d, C-6), 126.0 (d, C-14), 128.0 (d, C-11), 128.2 (d, C-5), 128.4 (d, C-4), 131.4 (d, C-12), 131.4 (s, C-10a), 135.0 (d, C-13), 135.3 (s, C-14a), 137.0 (s, C-4b), 143.7 (s, C-9a), 147.6 (s, C-14b), 148.0 (s, C-8a), 149.0 (s, C-15a), 159.0 (s, C-7), 159.4 (s, C-2), 182.3 (s, C-10).
9H-benzo[i]pyrido[2,3,4-kl]acridin-9-one (8):
TFA-kynuramine (
7)[
4] (500 mg, 1.9 mmol), naphthoquinone (330 mg, 2.1 mmol) and CeCl
3·7H
2O (35 mg, 0.094 mmol) were dissolved in ethanol (25 mL). The reaction mixture was refluxed for 9 h. while air was bubbled through it as described above in the general procedure. After evaporation of the ethanol and chromatography (eluting with CHCl
3/MeOH, 50:1) the crude product was added to a solution of 25% aq. ammonia (10 mL) in methanol (100 mL) and was stirred at room temperature for 7 days. The reaction mixture was evaporated and chromatographed (eluting with chloroform/methanol, 30:1) to afford
8 (70mg, 13%), amorphous powder (chloroform/methanol, 9:1), mp 258°C (lit [
8], 260-262
0C).
3(N)-(2,2’-diaminobenzophenone)-5-hydroxy-1,4-naphthoquinone (9):
4a (100 mg, 0.47 mmol) and CeCl3 ·7H2O (186 mg, 0.50 mmol) were dissolved in ethanol (10 mL) and 5-Hydroxy-1,4-naphthoquinone (juglone) (87 mg, 0.50 mmol) was added. The color of the solution changed immediately from yellow to red. The reaction mixture was stirred at room temperature, with bubbled air, for 3 days. The solvent was then evaporated and the red product purified by chromatography (eluting with CHCl3/MeOH, 100:1) (144mg, 80%): red prisms (from EtOH), m.p. 221°C; MS (EI); m/z: 384 (100) [M+, C23H16N2O4]; IR: = 3450, 1625, 1606, 1572, 1513, 1448, 1273, 1242 cm-1; 1H-NMR (CDCl3): δ = 6.2 (br s, 2 H, NH2), 6.52 (s, 1 H, H-2), 6.58 (t, J=7.5 Hz, 1 H, 12’-H), 6.72 (d, J=7.5Hz, 1 H, 10’-H), 7.16 (dd, J=7.5, 2.0 Hz, 1 H, 6-H), 7.21 (t, J=7.5 Hz, 1 H, 11’-H), 7.30 (t, J=7.5 Hz, 1 H, 4’-H), 7.40 (d, J=7.5 Hz, 1 H, 6’-H), 7.51 (d, J=7.5 Hz, 1 H, 13’-H), 7.54 (t, J=7.5 Hz, 1 H, 5’-H), 7.60 (d, J=7.5 Hz, 1 H, 8-H), 7.61 (d, J=7.5 Hz, 1 H, 3’-H), 7.62 (t, J=7.5 Hz, 1 H, 7-H), 9.45 (br s, 1 H, NH), 11.60 (s, 1 H, OH).
11-Hydroxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (10):
9 (70 mg, 0.18 mmol) was added to a solution of Et3N (2 mL) in methanol (20 mL) and the reaction mixture was stirred at room temperature for 10 days. The solution was evaporated and the residual solid chromatographed (eluting with CHCl3/MeOH, 100:1) to afford 10 (61 mg, 98%), yellow needles (CHCl3-MeOH 50:1), m.p. 292°C; HRMS calcd. for C23H12N2O2 [M+] 348.0899, found 348.0899; IR: = 3441, 2924, 1639, 1611, 1567, 1458, 1402, 1277 cm-1; 1H-NMR (CDCl3): δ = 7.30 (d, J=8.0 Hz, 1 H, 12-H), 7.83 (t, J=8.0 Hz, 1 H, 13-H), 7.88 (t, J=8.5 Hz, 1 H, 3-H), 8.00 (t, J=8.5 Hz, 1 H, 6-H), 8.00 (t, J=8.5 Hz, 1 H, 2-H), 8.08 (t, J=8.5 Hz, 1 H, 7-H), 8.67 (d, J=8.5 Hz, 1 H, 1-H), 8.73 (d, J=8.0 Hz, 1 H, 14-H), 8.83 (d, J=8.5 Hz, 1 H, 8-H), 9.08 (d, J=8.5 Hz, 1 H, 4-H), 9.17 (d, J=8.5 Hz, 1 H, 5-H), 12.90 (s, 1 H, OH); 13C-NMR (CDCl3/CD3OD, 50:1): δ = 116.9 (s, C-10a), 117.7 (d, C-14), 120.3 (d, C-12), 122.7 (s, C-4a), 124.6 (s, C-4c), 127.2 (d, C-5), 127.3 (d, C-4), 128.4 (d, C-3), 130.4 (d, C-6), 131.1 (d, C-7), 131.2 (d, C-2), 131.3 (d, C-1), 132.7 (d, C-8), 136.1 (s, C-4b), 136.4 (s, C-14a), 137.8 (d, C-13), 145.3 (s, C-9a), 147.5 (s, C-14b), 147.5 (s, C-8a), 147.5 (s, C-15a), 163.9 (s, C-11), 183.6 (s, C-10); C-14c could not be seen due to a long relaxation time.
11-Acetoxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (11):
10 (10 mg, 0.029 mmol) was acetylated with acetic anhydride-pyridine, 1:1 (1 mL), at room temperature for 24 h. The reaction mixture was evaporated and chromatographed (eluting with CHCl3/MeOH, 40:1) to give 11 (11 mg, 95%), yellow needles (CHCl3/MeOH, 100:1), m.p. 222oC; MS (EI); m/z: 390 (5) [M+, C25H14N2O3], 348 (100) [M+-CH2CO]; IR: = 2924, 1765, 1677, 1563, 1401, 1193 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.52 (s, 3 H, COCH3), 7.28 (d, J=8.0 Hz, 1 H, 12-H), 7.69 (t, J=8.0 Hz, 1 H, 13-H), 7.80 (t, J=8.0 Hz, 1 H, 3-H), 7.80 (t, J=8.0 Hz, 1 H, 2-H), 7.85 (t, J=8.0 Hz, 1 H, 6-H), 7.93 (t, J=8.0 Hz, 1 H, 7-H), 8.23 (d, J=8.0 Hz, 1 H, 1-H), 8.57 (d, J=8.0 Hz, 1 H, 14-H), 8.89 (d, J=8.0 Hz, 1 H, 8-H), 8.96 (d, J=8.0 Hz, 1 H, 4-H), 9.00 (d, J=8.0 Hz, 1 H, 5-H).
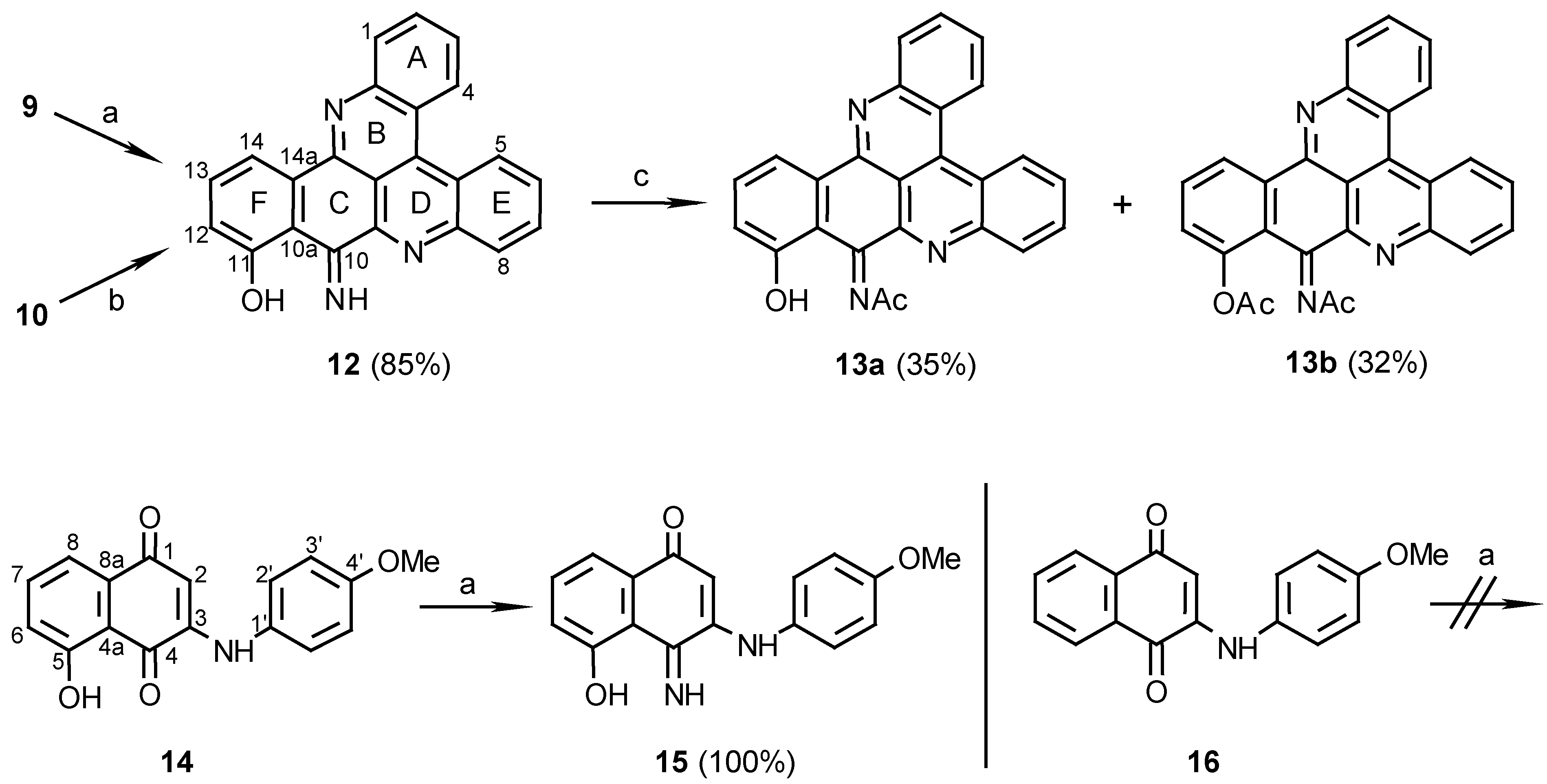
11-Hydroxy-10-imino-10H-benzo[i]quino[2,3,4-kl]acridine (12):
Method A: 9 (110 mg, 0.28 mmol), was added to a mixture of 25% aq. ammonia (2 ml) and methanol (20 mL) and stirred at room temperature for 7 days. The reaction mixture was then evaporated and the residue chromatographed (eluting with CHCl3/MeOH, 30:1) to afford 12 (83 mg, 85%).
Method B: 10 (10 mg, 0.029 mmol) was stirred in a saturated ammonia/methanol solution (2 mL) for 14 days. The solvent was then evaporated and the residue chromatographed using CHCl3/MeOH, 30:1, as eluant to afford 12 (7 mg, 70%), dark-green needles (CHCl3/MeOH, 20:1), m.p. 266 C; HRMS calcd. for C23H13N3O [M+] 347.10586, found 347.10590; IR: = 3384, 1615, 1565, 1488, 1472, 1405, 1124, 1048 cm-1; 1H-NMR (CDCl3): δ = 7.11 (d, J=8.0 Hz, 1 H, 12-H), 7.57 (d, J=8.0Hz, 1 H, 13-H), 7.77 (t, J=8.0 Hz, 1 H, 3-H), 7.88 (t, J=8.0 Hz, 1 H, 6-H), 7.89 (t, J=8.0 Hz, 1 H, 2-H), 7.97 (t, J=8.0 Hz, 1 H, 7-H), 8.32 (d, J=8.0 Hz, 1 H, 1-H), 8.37 (d, J=7.5 Hz, 1 H, 14-H), 8.40 (d, J=8.0 Hz, 1 H, 8-H), 9.00 (d, J=8.0 Hz, 1 H, 4-H), 9.10 (d, J=8.0 Hz, 1 H, 5-H), 10.81 (br s, 1 H), 14.8 (br s, 1 H); 13C-NMR (CDCl3): δ = 113.5 (s, C-10a), 115.9 (d, C-14), 122.4 (s, C-4a), 123.6 (d, C-12), 124.3 (s, C-4c), 127.1 (d, C-4), 127.3 (d, C-5), 127.6 (d, C-3), 129.2 (d, C-6), 130.8 (d, C-7), 130.8 (d, C-2), 131.2 (d, C-8), 131.3 (d, C-1), 133.3 (s, C-14a), 135.2 (d, C-13), 136.1 (s, C-4b), 143.2 (s, C-9a), 146.7 (s, C-8a), 147.9 (s, C-15a), 148.8 (s, C-14b), 162.1 (s, C-10), 171.0 (s, C-11); C-14c could not be seen due to a long relaxation time.
Acetylation of compound 12 to afford compounds 13a and 13b:
12 (10 mg, 0.029 mmol) was added to a mixture of acetic anhydride/pyridine, 1:1 (1 mL), and the solution was stirred at room temperature for 24 h. The reaction mixture was evaporated and chromatographed. Elution with CHCl3/MeOH, 200:1, afforded the mono N-acetylated product 13a (4 mg, 35%) and further elution with CHCl3/MeOH, 100:1, afforded the diacetylated product 13b (4 mg, 32%). Acetylation of 13a (1 mg, 2.6 µmol) by the same procedure gave the O,N-diacetate derivative 13b (1 mg). 13a: MS (EI); m/z: 389 (14) [M+, C25H15N3O2], 373 (66) [M+- O], 347 (100) [M+- CH2CO]; IR: = 3430, 1698, 1615, 1568, 1404, 1241, 1175 cm-1; 1H-NMR (CDCl3): δ = 2.72 (s, 3 H, NCOCH3), 7.22 (d, J=8.5 Hz, 1 H, 12-H), 7.66 (t, J=8.5 Hz, 1 H, 13-H), 7.76 (t, J=8.5 Hz, 1 H, 3-H), 7.89 (t, J=8.5 Hz, 1 H, 2-H), 7.91 (t, J=8.0 Hz, 1 H, 6-H), 7.97 (t, J=8.0 Hz, 1 H, 7-H), 8.31 (d, J=8.5 Hz, 1 H, 1-H), 8.37 (d, J=8.0 Hz, 1 H, 8-H), 8.61 (d, J=8.5 Hz, 1 H, 14-H), 8.98 (d, J=8.5 Hz, 1 H, 4-H), 9.09 (d, J=8.5 Hz, 1 H, 5-H); 13C-NMR (CDCl3): δ = 26.6 (NCOCH3), 114.4 (s, C-14c), 114.8 (s, C-10a), 117.8 (d, C-14), 120.3 (d, C-12), 122.5 (s, C-4a), 123.8 (s, C-4c), 127.1 (d, C-4), 127.3 (d, C-5), 128.0 (d, C-3), 129.8 (d, C-6), 131.0 (d, C-7), 131.0 (d, C-2), 131.2 (d, C-8), 131.2 (d, C-1), 134.6 (d, C-13), 135.0 (s, C-14a), 136.5 (s, C-4b), 141.8 (s, C-9a), 146.1 (s, C-8a), 147.8 (s, C-15a), 148.4 (s, C-14b), 153.8 (s, C-10), 161.6 (s, C-11), 183.8 (NCOCH3). 13b: MS (EI); m/z: 433 (17) [(M+2)+, C27H19N3O3], 389 (15) [M+-CH2CO], 373 (100) [(M+2)+-CH3CO2H], 347 (82) [M+- 2CH2CO]; IR: = 3430, 1767, 1676, 1640, 1569, 1204 cm-1; 1H-NMR (CDCl3): δ = 2.47 (s, 3 H, OCOCH3), 2.60 (s, 3 H, NCOCH3), 7.35 (dd, J=8.0, 1.5 Hz, 1 H, 12-H), 7.77 (t, J=8.0 Hz, 1 H, 13-H), 7.79 (t, J=8.0 Hz, 1 H, 6-H), 7.88 (t, J=8.0 Hz, 1 H, 3-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 7.95 (dt, J=8.0, 1.5 Hz, 1 H, 2-H), 8.34 (d, J=8.0 Hz, 1 H, 8-H), 8.38 (dd, J=8.0, 1.5 Hz, 1 H, 1-H), 8.98 (d, J=8.0 Hz, 1 H, 14-H), 9.08 (d, J=8.0 Hz, 1 H, 4-H), 9.10 (dd, J=8.0, 1.5 Hz, 1 H, 5-H).
3N-(4-methoxyaniline)-5-hydroxy-1,4-naphthoquinone (14):
p-Anisidine (40 mg, 0.32 mmol) was reacted with juglone (57 mg, 0.33 mmol) in the present of CeCl3 ·7H2O (122 mg, 0.33 mmol) by the procedure described for the preparation of compound 9. The red product was purified by chromatography (eluting with CHCl3/MeOH, 100:1) (65 mg, 69%): red crystals (ethanol), m.p. 211°C. – MS (EI); m/z: 295 (100) [M+, C17H13NO4]; IR: = 3274, 1627, 1590, 1572, 1516, 1240 cm-1; 1H-NMR (CD3SOCD3): δ = 3.78 (s, 3 H, OCH3), 5.87 (s, 1 H, 2-H), 7.01 (d, J=8.5 Hz, 2 H, 3’-H), 7.24 (d, J=8.5 Hz, 1 H, 6-H), 7.28 (d, J=8.5 Hz, 2 H, 2’-H), 7.44 (d, J=8.5 Hz, 1 H, 8-H), 7.72 (t, J=8.5 Hz, 1 H, 7-H), 9.18 (s, 1 H, NH), 11.54 (s, 1 H, OH); 13C-NMR (CD3SOCD3): δ = 55.3 (q, OCH3), 101.3 (d, C-2), 114.3 (s, C-4a), 114.6 (d, C-3’), 117.6 (d, C-8), 122.1 (d, C-6), 125.8 (d, C-2’), 130.5 (s, C-1’), 133.1 (s, C-8a), 137.6 (d, C-7), 146.9 (s, C-3), 157.1 (s, C-4’), 160.5 (s, C-5), 181.6 (s, C-1), 185.7 (s, C-4).
3(N)-(4-methoxyaniline)-5-hydroxy-1,4-naphthoquinon-4-imine (15):
14 (20 mg, 0.068 mmol) was added to a mixture of 25% aq. ammonia (2 mL) and methanol (20 mL) and stirred at room temperature for 7 days. Evaporation of the solvent afforded compound 15 (20 mg, 100%); HRMS calcd. for C17H14N2O3 [M+] 394.1004, found 294.1008; IR: = 3300, 1571, 1535, 1513, 1257 cm-1; 1H-NMR (CD3SOCD3): δ = 3.77 (s, 3 H, OCH3), 5.66 (s, 1 H, 2-H), 7.04 (d, J=9.0 Hz, 2 H, 3’-H), 7.14 (d, J=8.0 Hz, 1 H, 6-H), 7.26 (d, J=9.0 Hz, 2 H, 2’-H), 7.39 (d, J=8.0 Hz, 1 H, 8-H), 7.49 (t, J=8.0 Hz, 1 H, 7-H); 13C NMR (CD3SOCD3): δ = 54.9 (q, OCH3), 100.5 (d, C-2), 113.4 (s, C-4a), 114.3 (d, C-3’), 115.5 (d, C-8), 121.8 (d, C-6), 125.5 (d, C-2’), 130.2 (s, C-1’), 131.6 (s, C-8a), 132.7 (d, C-7), 147.1 (s, C-3), 156.6 (s, C-4’), 161.5 (s, C-5), 161.6 (s, C-4), 181.6 (s, C-1).
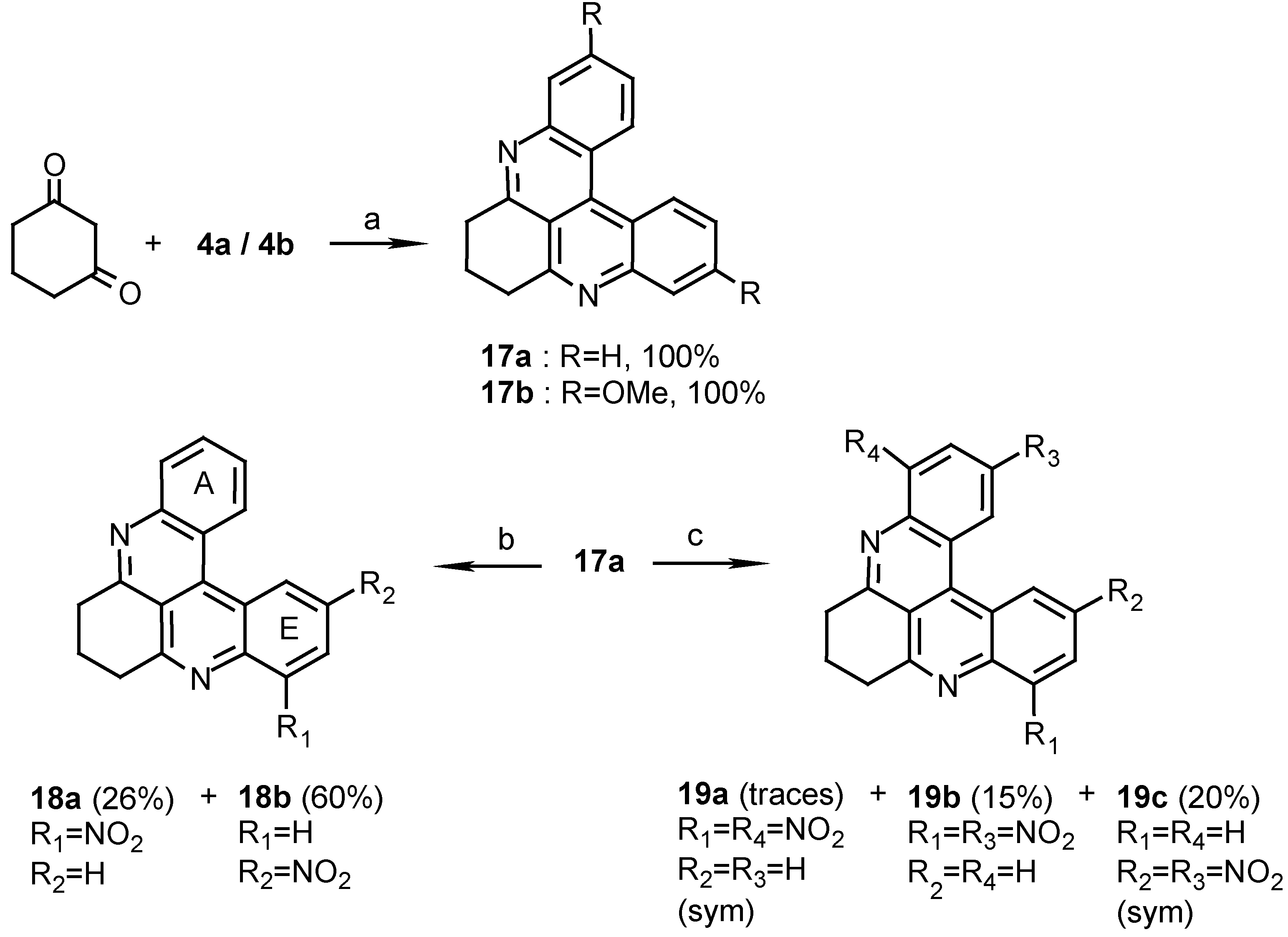
10H,11H,12H-dihydroquino[2,3,4-kl]acridine (17a):
To a stirred solution of 4a (400 mg, 1.9 mmol) in AcOH (50 mL) and conc.HCl (0.25 mL), 1,3- cyclohexanedione (425 mg, 3.8 mmol) and sodium m-nitrophenylsulfonate (1.3 g, 5.7 mmol) were added and the reaction mixture was refluxed for 2 h. After cooling, the mixture was poured onto ice (100 g), the solution brought to pH 8 with 25% ammonia and then extracted with chloroform (3 x 30 mL). The chloroform solution was washed with water (2 x 50 mL) and evaporated to afford 17a (515 mg, quantitative), amorphous powder (CHCl /MeOH, 20:1), m.p. 186°C; HRMS calcd. for C H N [M+] 270.1157, found 270.1157; IR: = 2940, 1585, 1572, 1488, 1400, 1389 cm-1; 1H-NMR (CDCl3): δ = 2.42 (quintet, J=6.0 Hz, 2 H, 11-H), 3.50 (t, J=6.0 Hz, 4 H, 10-, 12-H), 7.76 (t, J=7.0 Hz, 2 H, 2-, 7-H), 7.89 (t, J=7.0 Hz, 2 H, 3-, 6-H), 8.28 (d, J=7.0 Hz, 2 H, 1-, 8-H), 9.06 (d, J=7.0 Hz, 2 H, 4-, 5-H); 13C-NMR (CDCl3): δ = 22.3 (t, C-11), 34.4 (t, C-10), 116.5 (s, C-12b), 122.4 (s, C-4a), 126.1 (d, C-3), 127.0 (d, C-4), 129.2 (d, C-1), 130.0 (d, C-2), 135.9 (s, C-4b), 146.6 (s, C-8a), 159.5 (s, C- 9a).
2,7-Dimethoxy-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (17b):
Reacting 4b (520 mg, 1.9 mmol) with 1,3-cyclohexanedione (425 mg, 3.8 mmol) by the same procedure described for the synthesis of 17a afforded 17b (625mg, quantitative), amorphous powder (CHCl3/MeOH, 20:1), mp 218°C; HRMS calcd. for C21H18N2O2 [M+] 330.1368, found 330.1368; IR: = 2950, 1612, 1583, 1413, 1219, 1033 cm-1; 1H-NMR (CDCl3): δ = 2.57 (quintet, J=6.5 Hz, 2 H, 11-H), 3.90 (t, J=6.5 Hz, 4 H, 10-, 12-H), 4.16 (s, 6 H, OMe), 7.65 (dd, J=9.0, 2.5 Hz, 2 H, 3-, 6-H), 8.15 (d, J=2.5 Hz, 2 H, 1-, 8-H), 8.95 (d, J=9.0Hz, 2 H, 4-, 5-H).
General procedure for nitration of compounds 6a, 6b and 17a:
The pyridoacridine (10 mg) was added to a 1:1 mixture of conc.H2SO4/fuming HNO3 (2 mL) at 0°C. The reaction mixture was then allowed to warm up to room temperature and after 1 h. or 12 h. it was poured onto ice (10 g). The solution was neutralized with 10% NaOH and extracted with chloroform (4 x 10 mL). The chloroform solution was evaporated and the residue was chromatographed.
8-Nitro-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (18a) and 6-Nitro-10H,11H,12H- dihydroquino [2,3,4-kl]acridine (18b):
Reaction of 17a (10 mg, 0.037 mmol) by the above described procedure for 1 h. gave two products that were separated by chromatography. Elution with dichloromethane afforded the less polar isomer 18a (3 mg, 26%) and further elution with CH2Cl2/MeOH, 30:1, afforded the more polar isomer 18b (7 mg, 60%). 18a: MS (EI); m/z: 315 (100) [M+, C19H13N3O2], 285 (37) [M+- NO], 269 (30) [M+- NO2]; IR: = 2925, 1591, 1374, 1139 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.43 (quintet, J=6.0 Hz, 2 H, 11-H), 3.51 (t, J=6.0 Hz, 2 H, 12a-H), 3.75 (t, J=6.0 Hz, 2 H, 10a-H), 7.94 (t, J=8.0 Hz, 1 H, 6-H), 8.05 (t, J=8.0 Hz, 1 H, 2-H), 8.18 (t, J=8.0 Hz, 1 H, 3-H), 8.25 (d, J=8.0 Hz, 1 H, 7-H), 9.06 (d, J=8.0 Hz, 1 H, 1-H), 9.07 (d, J=8.0 Hz, 1 H, 4-H), 9.21 (d, J=8.0 Hz, 1 H, 5-H). 18b: HRMS calcd. for C19H13N3O2 [M+] 315.1008, found 315.1006; MS (EI); m/z: 315 (100) [M+], 269 (33) [M+- NO2]; IR: = 2927, 1588, 1339 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.42 (quintet, J=6.0 Hz, 2 H, 11-H), 3.50 (t, J=6.0 Hz, 2 H, 12-H, interchangeable), 3.54 (t, J=6.0 Hz, 2 H, 10a-H), 7.87 (t, J=7.5 Hz, 1 H, 3-H), 7.97 (t, J=7.5 Hz, 1 H, 2-H), 8.32 (d, J=7.5 Hz, 1 H, 1-H), 8.36 (d, J=7.5 Hz, 1 H, 8-H), 8.63 (dd, J=7.5, 2.5 Hz, 1 H, 7-H), 8.95 (d, J=7.5 Hz, 1 H, 4-H), 9.93 (d, J=2.5 Hz, 1 H, 5-H).
1,8-Dinitro-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (19a), 3,8-Dinitro-10H,11H, 12H-dihydro- quino[2,3,4-kl]acridine (19b) and 3,6-Dinitro-10H,11H,12H-dihydroquino [2,3,4-kl]acridine (19c):
Reaction of 17a (10 mg, 0.037mmol) in conc.H2SO4/fuming HNO3 , 1:1 (2ml), by the general above procedure for 12 h. gave three products that were separated by chromatography. Elution with dichloromethane/petroleum ether, 4:1, afforded isomer 19a (0.5 mg). Elution with dichloromethane afforded isomer 19b (2 mg, 15%) and isomer 19c (3 mg, 20%). 19a: MS (EI); m/z: 360 (47) [M+, C19H12N4O4], 330 (60) [M+- NO], 300 (100) [M+- 2NO]; IR: = °2925, 1620, 1583, 1534 cm-1; 1H-NMR (CDCl3): δ = 2.39 (quintet, J=7.0 Hz, 2 H, 11-H), 3.50 (t, J=7.0 Hz, 4 H, 10-, 12-H), 7.83 (t, J=7.5 Hz, 2 H, 3-, 6-H), 8.13 (d, J=7.5 Hz, 2 H, 2-, 7-H), 9.10 (d, J=7.5 Hz, 2 H, 4-, 5-H). 19b: HRMS calcd. for C19H12N4O4 [M+] 360.0859, found 360.0858; MS (EI); m/z: 360 (100) [M+], 330 (23) [M+- NO], 302 (29) [(M+2)+- 2NO], 267 (31) [(M-1)+- 2NO2]; IR: = 2925, 1618, 1589, 1535, 1341 cm-1; 1H-NMR (CDCl3): δ = 2.43 (quintet, J=7.0 Hz, 2 H, 11-H), 3.52 (t, J=7.0 Hz, 2 H, 12 -H), 3.59 (t, J=7.0 Hz, 2 H, 10-H, interchangeable), 7.95 (t, J=8.0 Hz, 1 H, 6-H), 8.19 (d, J=8.0 Hz, 1 H, 7-H), 8.55 (d, J=8.0 Hz, 1 H, 1-H), 8.73 (dd, J=8.0, 2.0 Hz, 1 H, 2-H), 9.16 (d, J=8.0 Hz, 1 H, 5-H), 9.90 (d, J=2.0 Hz, 1 H, 4-H). 19c: MS (EI); m/z: 360 (100) [M+, C19H12N4O4], 315 (49) [(M+1)+- NO2], 267 (53) [(M-1)+- 2NO2]; IR: = 2925, 1600, 1588, 1506, 1346 cm-1; 1H-NMR (CDCl3): δ = 2.54 (quintet, J=6.0 Hz, 2 H, 11-H), 3.67 (t, J=6.0 Hz, 4 H, 10-, 12-H), 8.60 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.79 (dd, J=8.0, 2.0 Hz, 2 H, 2-, 7-H), 9.96 (d, J=2.0 Hz, 2 H, 4-, 5-H).
3-Nitro-10H-benzo[i]quino [2,3,4-kl]acridin-10-one (20):
Reaction of 6a (80 mg, 0.24 mmol) in conc.H2SO4/fuming HNO3, 1:1 (5mL), for 12 h. by the above general procedure followed by crystallization of the crude product from pyridine afforded 20 (48 mg, 53%), yellow needles, m.p. 339°C; MS (EI); m/z: 377 (100) [M+, C23H11N3O3], 347 (36) [(M+- NO], 330 (41) [(M-1)+- NO2]; IR: = 1682, 1513, 1404, 1388, 1340, 1276 cm-1; 1H-NMR (CDCl3): δ =7.79 (t, J=8.0 Hz, 1 H, 12-H), 7.93 (t, J=8.0 Hz, 1 H, 13-H), 8.12 (t, J=8.0 Hz, 1 H, 7-H), 8.12 (t, J=8.0 Hz, 1 H, 6-H), 8.50 (d, J=8.0 Hz, 1 H, 11-H), 8.54 (d, J=9.0 Hz, 1 H, 1-H), 8.71 (d, J=8.0 Hz, 1 H, 8-H), 8.72 (dd, J=9.0, 2.5 Hz, 1 H, 2-H), 9.09 (d, J=8.0 Hz, 1 H, 14-H), 9.11 (d, J=8.0 Hz, 1 H, 5-H), 10.01 (d, J=2.5 Hz, 1 H, 4-H); 13C-NMR (CDCl3/CF3CO2D, 100:1): δ = 116.5 (s, C-14c), 121.6 (s, C-4a), 123.6 (d, C-4),123.7 (s, C-4c), 124.7 (d, C-2), 126.4 (d, C-5), 126.7 (d, C-14), 128.4 (d, C-11), 131.5 (d, C-6), 131.8 (d, C-7), 131.9 (d, C-8), 132.2 (s, C-10a), 132.3 (d, C-12), 132.6 (d, C-1), 134.8 (s, C-14a), 135.3 (d, C-13), 136.8 (s, C-4b), 146.0 (s, C-3), 146.7 (s, C-8a), 149.0 (s, C-9a), 149.8 (s, C-15a), 151.3 (s, C-14b), 181.1 (s, C-10).
3-Amino-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (21):
20 (40 mg, 0.11 mmol) was dissolved in AcOH (3 mL) and TFA (6mL), 5% Pd-C (25 mg) was added and the reaction mixture was shaken in a Parr apparatus under H2 (3 atm) for 1 h. The catalyst was then filtered off, the solution poured onto ice (10 g), brought to pH 8 with 25% ammonia and then extracted with chloroform (3 x 20 mL). After evaporation of the solvent the residue was chromatographed (eluting with CHCl3/MeOH, 30:1) to afford 21 (15 mg, 40%); HRMS calcd. for C23H13N3O [M+] 347.1059, found 347.1058; IR: = 1677, 1646, 1540, 1515 cm-1; 1H-NMR (CD3SOCD3): δ = 7.42 (d, J=8.0 Hz, 1 H, 2-H), 7.71 (t, J=8.0 Hz, 1 H, 12-H), 7.93 (t, J=8.0 Hz, 1 H, 13-H), 8.05 (t, J=8.0 Hz, 1 H, 6-H), 8.09 (t, J=8.0 Hz, 1 H, 7-H), 8.13 (d, J=8.0 Hz, 1 H, 1-H), 8.28 (d, J=2.0 Hz, 1 H, 4-H), 8.30 (d, J=8.0 Hz, 1 H, 11-H), 8.53 (d, J=8.0 Hz, 1 H, 8-H), 8.91 (d, J=8.0 Hz, 1 H, 14-H), 9.31 (d, J=8.0 Hz, 1 H, 5-H).
2,7-Dimethoxy-1,8-dinitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (22):
Reaction of 6b (10 mg, 0.026 mmol) in conc.H2SO4/fuming HNO3, 1:1 (2ml), by the above general procedure for 1 hr afforded 22 (10 mg, 80%); HRMS calcd. for C25H14N4O7 [M+] 482.0862, found 482.0855, 452 (100) [(M+- NO]; MS (EI); m/z: 482 (79) [M+], 452 (100) [M+-NO]; IR: = 1687, 1617, 1540, 1375, 1290 cm-1; 1H-NMR (CD3SOCD3): δ = 4.21 (s, 3 H, OCH3), 4.24 (s, 3H, OCH3), 7.85 (t, J=8.5 Hz, 1 H, 12-H), 7.97 (d, J=10.0 Hz, 1 H, 3-H), 7.99 (t, J=8.5 Hz, 1 H, 13-H), 8.13 (d, J=10.0 Hz, 1 H, 6-H), 8.27 (d, J=8.5 Hz, 1 H, 11-H), 8.66 (d, J=8.5 Hz, 1 H, 14-H), 9.28 (d, J=10.0 Hz, 1 H, 4-H), 9.36 (d, J=10.0 Hz, 1 H, 5-H).
2,7- Dimethoxy-1,3,8,12-tetrainitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (23a) and 2,7-di- methoxy-1,3,8,14-tetranitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (23b):
Reaction of 6b (10 mg, 0.026 mmol) in conc.H2SO4/fuming HNO3, 1:1 (2 ml), by the above general procedure for 12 hr gave two products that were separated by chromatography. Elution with dichloromethane afforded first the less polar isomer 23a (3 mg, 20%) and then the more polar isomer 23b (7 mg, 45%). 23a: MS (EI); m/z: 572 (100) [M+, C25H12N6O11], 542 (45) [M+- NO], 482 (78) [M+-3NO], 452 (32) [M+- 4NO]; IR: = 2925, 1706, 1619, 1547, 1375 cm-1; 1H-NMR (CD3SOCD3/CF3CO2D, 100:1): δ = 4.18 (s, 3 H, OCH3), 4.29 (s, 3 H, OCH3), 8.28 (d, J=10.0 Hz, 1 H, 6-H), 8.82 (dd, J=8.5, 2.5 Hz, 1 H, 13-H), 8.90 (d, J=8.5 Hz, 1 H, 14-H), 8.90 (d, J=2.5 Hz, 1 H, 11-H), 9.48 (d, J=10.0 Hz, 1 H, 5-H), 9.86 (s, 1 H, 4-H); 1H-NMR (CD3CN): δ = 4.30 (s, 3 H, OCH3), 4.32 (s, 3 H, OCH3), 8.15 (d, J=10.0 Hz, 1 H, 6-H), 8.70 (dd, J=8.5, 2.0 Hz, 1 H, 13-H), 8.99 (d, J=8.5 Hz, 1 H, 14-H), 9.05 (d, J=2.0 Hz, 1 H, 11-H), 9.30 (d, J=10.0Hz, 1 H, 5-H), 9.75 (s, 1 H, 4-H). 23b: HRMS calcd. for C25H12N6O11 [M+] 572.0564, found 572.0562; IR: = 2925, 1692, 1629, 1557, 1547, 1376 cm-1; 1H-NMR (CD3SOCD3): δ = 4.14 (s, 3 H, OCH3), 4.27 (s, 3 H, OCH3), 8.06 (t, J=8.0 Hz, 1 H, 12-H), 8.22 (d, J=8.0 Hz, 1 H, 13-H), 8.26 (d, J=9.5 Hz, 1 H, 6-H), 8.54 (d, J=8.0 Hz, 1 H, 11-H), 9.43 (d, J=9.5 Hz, 1 H, 5-H), 9.79 (s, 1 H, 4-H).
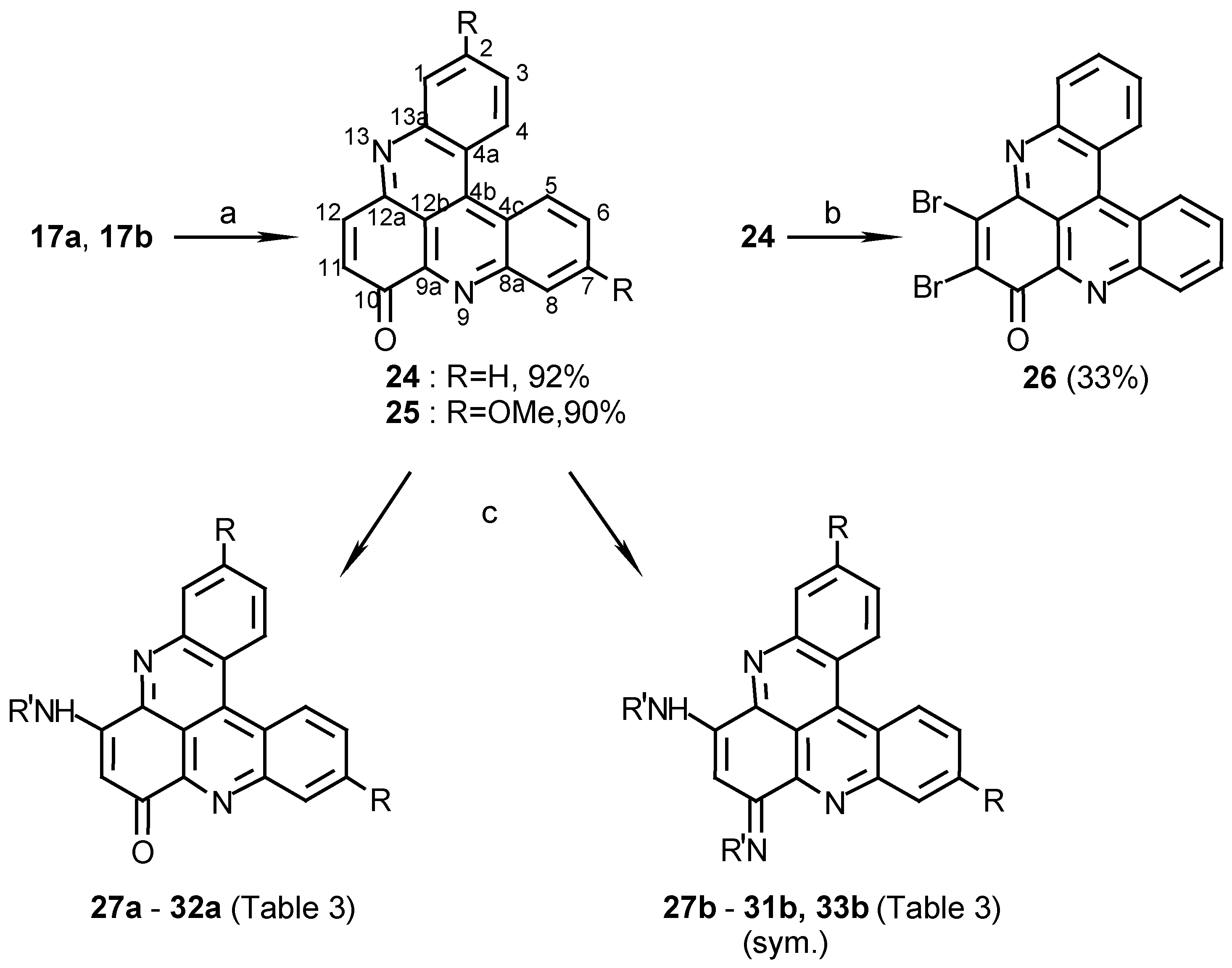
10H-quino[2,3,4-kl]acridin-10-one (24):
To a solution of 17a (300 mg, 1.1 mmol) in acetonitrile (120 mL) cerium ammonium nitrate (2.4 g, 4.4 mmol) was added and the reaction mixture was refluxed for 10 min. The acetonitrile was evaporated and the residue dissolved in chloroform (100 mL), washed with 0.1% aq. ammonia (2 x 100 mL) and evaporated to afford 24 (290 mg, 92%), amorphous powder (CHCl3/MeOH, 20:1), m.p. 254°C; HRMS calcd. for C19H10N2O [M+] 282.0793, found 282.0799; IR: = 1663, 1565, 1493, 1280 cm-1; 1H-NMR (CDCl3): δ = 7.11 (d, J=11.0 Hz, 1 H, 11-H), 7.90 (t, J=8.5 Hz, 1 H, 3-H), 7.98 (t, J=8.5 Hz, 1 H, 2-H), 7.98 (t, J=8.5 Hz, 1 H, 6-H), 7.99 (d, J=11.0 Hz, 1 H, 12-H), 8.05 (t, J=8.5 Hz, 1 H, 7-H), 8.43 (d, J=8.5 Hz, 1 H, 1-H), 8.73 (d, J=8.5 Hz, 1 H, 8-H), 9.14 (d, J=8.5 Hz, 1 H, 4-H), 9.19 (d, J=8.5 Hz, 1 H, 5-H).
2,7-Dimethoxy-10H-quino[2,3,4-kl]acridin-10-one (25):
Oxidation of 17b (360 mg, 1.1 mmol) by the same procedure described for the synthesis of 24 afforded 25 (340 mg, 90%), amorphous powder (CHCl3/MeOH, 20:1), m.p. 291°C; HRMS calcd. for C21H14N2O3 [M+] 342.1004, found 342.1004; IR: = 1666, 1608, 1415, 1259, 1225 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 4.03 (s, 3 H, OCH3), 4.04 (s, 3 H, OCH3), 7.02 (d, J=10 Hz, 1 H, 11-H), 7.43 (dd, J=8.5, 2.5 Hz, 1 H, 3-H), 7.51 (dd, J=8.5, 2.5 Hz, 1 H, 6-H), 7.72 (d, J=2.5 Hz, 1 H, 1-H), 7.93 (d, J=10 Hz, 1 H, 12-H), 7.94 (s, 1 H, 8-H), 8.88 (d, J=8.5 Hz, 1 H, 4-H), 8.92 (d, J=8.5 Hz, 1 H, 5-H).
11,12-Dibromo-10H-quino[2,3,4-kl]acridin-10-one (26):
24 (30 mg, 0.11 mmol) was dissolved in acetic acid (5 mL) and Br2(0.2 mL, 3.9 mmol) was added at room temperature. The mixture was stirred at 80°C for 2 h. and then allowed to cool to room temperature. Water (25 mL) and chloroform (25 mL) were added. The organic phase was extracted with 5% NaHSO3 (10 mL) and evaporated. The crude product was chromatographed (eluting with chloroform) to afford 26 (16 mg, 33%); HRMS calcd. for C19H8Br2N2O [M+] 439.8988, found 439.9000; IR: = 3425, 1669, 1491, 1394, 1242, 767 cm-1; 1H-NMR (CDCl3): δ = 7.86 (t, J=8.0 Hz, 1 H, 3-H), 7.93 (t, J=8.0 Hz, 1 H, 2-H), 7.93 (t, J=8.0 Hz, 1 H, 6-H), 7.99 (t, J=8.0 Hz, 1 H, 7-H), 8.39 (d, J=8.0 Hz, 1 H, 1-H), 8.56 (d, J=8.0 Hz, 1 H, 8-H), 8.97 (d, J=8.0 Hz, 1 H, 4-H), 9.04 (d, J=8.0 Hz, 1 H, 5-H).
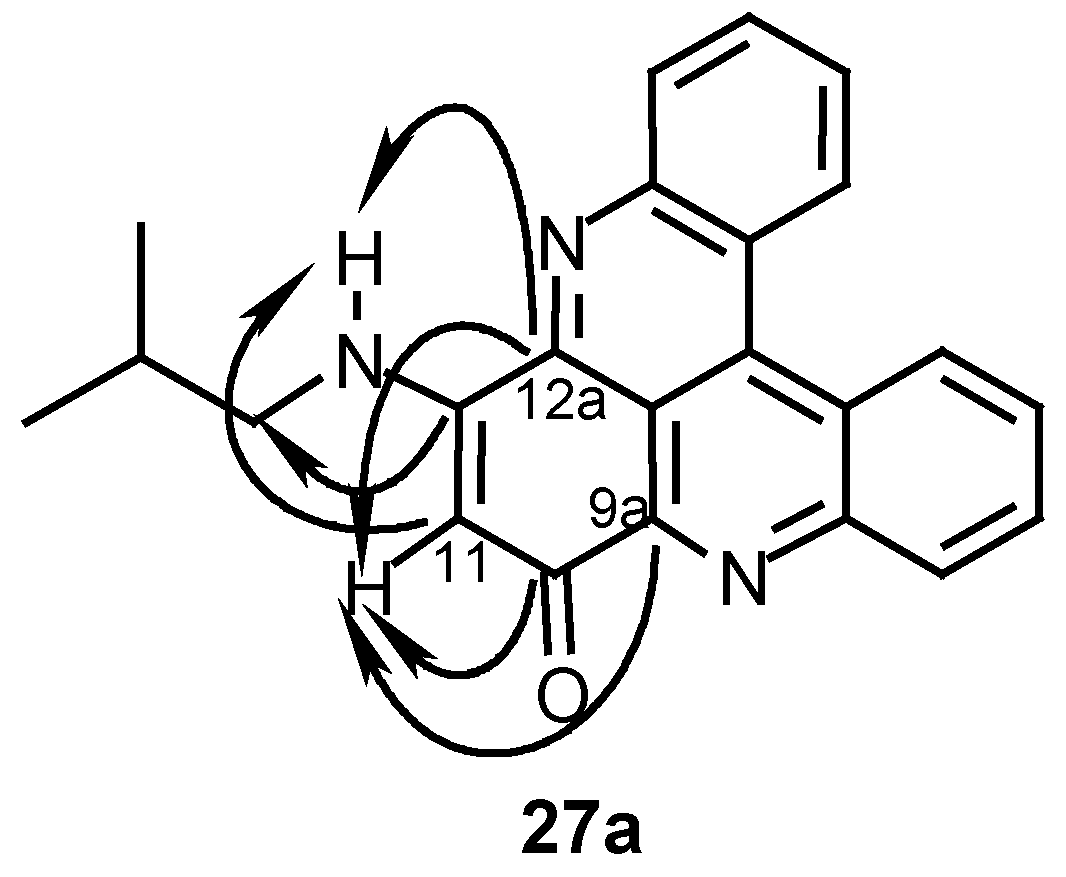
12-Isobutylamino-10H-quino[2,3,4-kl]acridin-10-one (27a):
24 (40 mg, 0.14 mmol) and isobutylamine (0.12 mL, 1.2 mmol) in acetonitrile (10 mL) were stirred at room temperature for 48 h. The reaction mixture was then evaporated and the residue chromatographed by a silica gel column (eluting with CHCl3/MeOH, 30:1) to afford 27a (18 mg, 36%); HRMS calcd. for C23H19N3O [M+] 353.1528, found 353.1525; MS (EI); m/z: 353 (20) [M+], 310 (100) [M+- (CH3)2CH]; IR: = 2925, 1609, 1514, 1466, 1258, 761 cm-1; 1H-NMR (CDCl3): δ = 1.15 (d, J=7.0 Hz, 6 H, 3’-H), 2.20 (quintet, J=7.0 Hz, 1 H, 2’-H), 3.31 (t, J=7.0 Hz, 2 H, 1’-H), 6.09 (s, 1 H, 11-H), 7.49 (br s, 1 H, NH), 7.76 (t, J=8.0 Hz, 1 H, 3-H), 7.79 (t, J=8.0 Hz, 1 H, 6-H), 7.86 (t, J=8.0 Hz, 1 H, 2-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 8.21 (d, J=8.0 Hz, 1 H, 1-H), 8.60 (d, J=8.0 Hz, 1 H, 8-H), 8.91 (d, J=8.0 Hz, 1 H, 5-H), 8.93 (d, J=8.0 Hz, 1 H, 4-H); 13C-NMR (CDCl3): δ = 20.5 (q, C-3’), 28.0 (d, C-2’), 50.5 (t, C-1’), 100.7 (d, C-11), 115.3 (s, C-12b), 123.5 (s, C-4c), 123.8 (s, C-4a), 126.8 (d, C-5), 127.3 (d, C-4), 129.0 (d, C-3), 129.3 (d, C-6), 130.7 (d, C-2), 130.8 (d, C-7), 131.0 (d, C-1), 132.9 (d, C-8), 134.9 (s, C-4b), 144.6 (s, C-9a), 145.7 (s, C-12a), 147.0 (s, C-13a), 147.6 (s, C- 8a), 152.1 (s, C-12), 180.4 (s, C-10).
10,12-Di(isobutylamino)quino[2,3,4-kl]acridine (27b):
24 (100 mg, 0.36 mmol) and isobutylamine (0.30 mL, 3.0 mmol) in ethanol (25 mL) were stirred at room temperature for 18 h. The reaction mixture was evaporated and the residue chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH/trifluoroacetic acid, 20:1:0.01) to afford 27b (56 mg, 38%); HRMS calcd. for C27H26N4 [(M - 2)+] 406.2157, found 406.2156; IR: = 3445, 1611, 1559, 1462, 1386, 1125, 766 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 1.13 (d, J=7.0 Hz, 12 H, 3’-H), 2.25 (quintet, J=7.0 Hz, 2 H, 2’-H), 3.59 (d, J=7.0 Hz, 4 H, 1’-H), 6.19 (s, 1 H, 11-H), 7.99 (t, J=8.0 Hz, 2 H, 3-, 6-H), 8.05 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.46 (d, J=8.0 Hz, 2 H, 1-, 8-H), 9.16 (d, J=8.0 Hz, 2 H, 4-, 5-H), 9.49 (br s, 1 H, NH); 13C-NMR (CDCl3/CD3OD, 20:1): δ = 20.1 (q, C-3’), 28.4 (d, C-2’), 51.1 (t, C-1’), 87.6 (d, C-11), 113.0 (s, C-12b), 124.2 (s, C-4a), 127.4 (d, C-4), 131.1 (d, C-3), 132.0 (d, C-2), 132.2 (d, C-1), 136.1 (s, C-4b), 141.1 (s, C-9a), 146.2 (s, C-8a), 156.0 (s, C-10).
2,7-Dimethoxy-12-isobutylamino-10H-quino[2,3,4-kl]acridin-10-one (28a) and 10,12-Di (isobutyl- amino)-2,7-dimethoxyquino[2,3,4-kl]acridine (28b):
25 (50 mg, 0.15 mmol) was reacted with isobutylamine (0.20 mL, 2.0 mmol) by the described procedure for the synthesis of 27b. Two obtained compounds were separated by chromatography; elution with CHCl3/MeOH, 30:1, afforded the monoamination product (28a) (12 mg, 19%) and further elution with CHCl3/MeOH, 10:1, afforded the diamination product (28b) (25mg, 37%). 28a: MS (EI); m/z: 413 (29) [M+, C25H23O3N3], 370 (100) [M+- (CH3)2CH]; IR: = 2959, 1658, 1612, 1562, 1467, 1450, 1422, 1223, 1134 cm-1; 1H-NMR (CDCl3): δ = 1.11 (d, J=7.0 Hz, 6 H, 3’-H), 2.17 (quintet, J=7.0 Hz, 1 H, 2’-H), 3.25 (t, J=7.0 Hz, 2 H, 1’-H), 4.03 (s, 3 H, OCH3), 4.05 (s, 3 H, OCH3), 6.07 (s, 1 H, 11-H), 7.40 (d, J=10.0 Hz, 1 H, 3-H), 7.42 (d, J=10.0 Hz, 1 H, 6-H), 7.50 (br s, 1 H, NH), 7.58 (s, 1 H, 1-H), 8.00 (s, 1 H, 8-H), 8.83 (d, J=10.0 Hz, 1 H, 5-H), 8.86 (d, J=10.0 Hz, 1 H, 4-H). 28b: MS (EI); m/z: 466 (61) [(M-2)+, C29H30N4O2], 423 (100) [(M-2)+- (CH3)2CH]; IR: = 2924, 1658, 1612, 1564, 1467, 1412, 1252, 1219, 669 cm-1; 1H-NMR (CDCl3): δ = 1.14 (d, J=7.0 Hz, 12 H, 3’-H), 2.28 (quintet, J=7.0 Hz, 2 H, 2’-H), 3.67 (d, J=7.0 Hz, 4 H, 1’-H), 4.06 (s, 6 H, OCH3), 5.89 (s, 1 H, 11-H), 7.36 (dd, J=9.0, 1.5 Hz, 2 H, 3-, 6-H), 7.70 (d, J=1.5 Hz, 2 H, 1-, 8-H), 8.58 (d, J=9.0 Hz, 2 H, 4-, 5-H), 9.78 (br s, 1 H, NH); 13C-NMR (CDCl3): δ = 20.6 (q, C-3’), 28.7 (d, C-2’), 51.4 (t, C-1’), 56.2 (OCH3), 87.3 (d, C-11), 110.4 (s, C-12b), 111.3 (d, C-1), 117.6 (s, C-4a), 121.9 (d, C-3), 128.0 (d, C-4), 134.8 (s, C-4b), 141.5 (s, C-9a), 148.1 (s, C-8a), 155.3 (s, C-10), 161.7 (s, C-2).
12-Methylamino-10H-quino[2,3,4-kl]acridin-10-one (29a):
24 (40 mg, 0.14 mmol), methylamine (0.20 mL of 33% methylamine in ethanol, 1.6 mmol) and CeCl3·7H2O (52 mg, 0.14 mmol) were mixed together in ethanol (10 mL).The reaction mixture was stirred at room temperature for 15 min. then evaporated and chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH, 30:1) to afford 29a (20 mg, 46%); MS (EI); m/z: 311 (100) [M+, C20H13N3O]; IR: = 3430, 1610, 1560, 1419 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.11 (s, 3 H, NCH3), 5.97 (s, 1 H, 11-H), 7.74 (t, J=8.0 Hz, 1 H, 3-H), 7.78 (t, J=8.0 Hz, 1 H, 6-H), 7.82 (t, J=8.0 Hz, 1 H, 2-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 8.14 (d, J=8.0 Hz, 1 H, 1-H), 8.51 (d, J=8.0 Hz, 1 H, 8-H), 8.90 (d, J=8.0 Hz, 1 H, 5-H), 8.92 (d, J=8.0 Hz, 1 H, 4-H).
10,12-Di(methylamino)quino[2,3,4-kl]acridine (29b):
24 (30 mg, 0.11 mmol) was reacted with methylamine (0.20 mL of 33% methylamine in ethanol, 1.6 mmol) in ethanol (10 mL) at room temperature for 24 h. The product (29b) was purified by silica gel column chromatography eluting with CHCl3/MeOH, 10:1 (10 mg, 28%); MS (EI); m/z: 322 (100) [(M-2)+, C21H14N4], 294 (44) [(M-2)+- CH2N]; IR: = 3405, 1618, 1564, 1419 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.38 (s, 6 H, NCH3), 5.83 (s, 1H, 11-H), 7.83 (t, J=8.0 Hz, 2 H, 3-, 6-H), 7.90 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.21 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.79 (d, J=8.0 Hz, 2 H, 4-, 5-H).
12-(4-methoxyanilino)-10H-quino[2,3,4-kl]acridin-10-one (30a):
24 (10 mg, 0.036 mmol) and p-anisidine (5 mg, 0.041 mmol) were refluxed in acetonitrile for 48 h. The solvent was evaporated and the residue chromatographed on a silica gel column (eluting with CHCl3/MeOH, 50:1) to afford 30a (8 mg, 55%); MS (EI); m/z: 403 (100) [M+, C26H17N3O2], 372 (36) [M+- CH3O]; IR: = 3448, 1614, 1556, 1512, 1245 cm-1; 1H-NMR (CDCl3): δ = 3.87 (s, 3 H, OCH3), 6.54 (s, 1 H, 11-H), 7.00 (d, J=9.0 Hz, 2 H, 3’-H), 7.38 (d, J=9.0 Hz, 2 H, 2’-H), 7.88 (t, J=8.5 Hz, 1 H, 3-H), 7.88 (t, J=8.5 Hz, 1 H, 6-H), 7.96 (t, J=8.5 Hz, 1 H, 2-H), 7.96 (t, J=8.5 Hz, 1 H, 7-H), 8.37 (d, J=8.5 Hz, 1 H, 1-H), 8.67 (d, J=8.5 Hz, 1 H, 8-H), 9.07 (d, J=8.5 Hz, 1 H, 5-H), 9.10 (d, J=8.5 Hz, 1 H, 4-H).
10,12-Di(4-methoxyanilino)quino[2,3,4-kl]acridine (30b):
24 (10 mg, 0.036 mmol) and p-anisidine (10 mg, 0.081 mmol) in ethanol (4 mL) were stirred at 50°C for 12 h. The ethanol was then evaporated and the residue chromatographed (eluting with CHCl3/MeOH, 10:1) to afford 30b (7 mg, 38%); MS (EI); m/z: 509 (100) [(M+1)+], 508 (99) [M+, C33H24N4O2]; IR: = 3440, 2925, 1607, 1548, 1506, 1460, 1253, 1171, 1105, 1025 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.89 (s, 6 H, OCH3), 6.71 (s, 1 H, 11-H), 7.00 (d, J=8.0 Hz, 4 H, 3’-H), 7.47 (d, J=8.0 Hz, 4 H, 2’-H), 7.91 (t, J=8.0 Hz, 2 H, 3-, 6-H), 7.99 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.53 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.99 (d, J=8.0 Hz, 2 H, 4-, 5-H).
12-Dodecylamino-10H-quino[2,3,4-kl]acridin-10-one (31a) and 10,12-Di(dodecylamino)quino- [2,3,4-kl]acridine (31b):
24 (10 mg, 0.036 mmol) and dodecylamine (0.030 mL, 0.13 mmol) were stirred in ethanol (4 mL) at 50°C for 12 h. After evaporation of the ethanol, the two products were separated by silica gel chromatography; elution with CHCl3/MeOH, 30:1, to afford the monoamination product (31a) (4 mg, 24%) and by elution with CHCl3/MeOH, 10:1, the diamination product (31b) which was further purified on a Sephadex LH-20 column (eluting with CHCl3/MeOH/petroleum ether, 1:1:2) (4 mg, 18%). 31a: MS (EI); m/z: 465 (64) [M+, C31H35N3O], 310 (100) [M+- (CH2)10CH3]; IR: = 2923, 2852, 1610, 1561, 1466 cm-1; 1H-NMR (CDCl3): δ = 0.90 (t, J=7.5 Hz, 3 H, 12’-H), 1.35 (br m, 14 H, 5’-H - 11’-H), 1.46 (m, 2 H, 4’-H), 1.54 (quintet, J=7.5 Hz, 2 H, 3’-H), 1.91 (quintet, J=7.5 Hz, 2 H, 2’-H), 3.45 (m, 2 H, 1’-H), 6.25 (s, 1 H, 11-H), 7.63 (br s, 1 H, NH), 7.87 (t, J=8.0 Hz, 1 H, 3-H), 7.87 (t, J=8.0 Hz, 1 H, 6-H), 7.96 (t, J=8.0 Hz, 1 H, 2-H), 7.98 (t, J=8.0 Hz, 1 H, 7-H), 8.35 (d, J=8.0 Hz, 1 H, 1-H), 8.68 (d, J=8.0 Hz, 1 H, 8-H), 9.06 (d, J=8.0 Hz, 1 H, 5-H), 9.09 (d, J=8.0 Hz, 1 H, 4-H). 31b: MS (EI); m/z: 630 (100) [(M- 2)+, C43H58N4], 475 (33) [(M- 2)+- (CH2)10CH3]; IR: = 2922, 2851, 1640, 1611, 1563, 1467, 1442, 1408 cm-1; 1H-NMR (CDCl3): δ = 0.85 (t, J=7.0 Hz, 6 H, 12’-H), 1.2 (br m, 28 H, 5’-H - 11’-H), 1.38 (m, 4 H, 4’-H), 1.55 (m, 4 H, 3’-H), 1.96 (m, 4 H, 2’-H), 3.91 (m, 4 H, 1’-H), 6.22 (s, 1 H, 11-H), 7.92 (t, J=8.0 Hz, 2 H, 3-, 6-H), 8.00 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.55 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.99 (d, J=8.0 Hz, 2 H, 4-, 5-H), 9.83 (br s, 1 H, NH).
12-Amino-10H-quino[2,3,4-kl]acridin-10-one (32a):
The procedure of Couladouros [
17] was adopted. To a solution of
24 (10 mg, 0.036 mmol) in methanol (2 mL), under nitrogen, was added a solution of sodium azide (14 mg, 0.22 mmol) in water (0.5 mL) and the solution was acidified to pH 4 with 1N HCl. After stirring at room temperture for 15 h. the reaction mixture was extracted with chloroform (2 x 10 mL) and the combined organic layer was washed with water (20 ml) and evaporated to afford
32a (8 mg, 76%); HRMS calcd. for C
19H
11N
3O [M
+] 297.0902, found 297.0901; IR:
= 3425, 1644, 1616, 1546, 1515 cm
-1;
1H-NMR (CDCl
3/CD
3OD, 20:1): δ = 6.23 (s, 1 H, 11-H), 7.77 (t,
J=8.0 Hz, 1 H, 3-H), 7.80 (t,
J=8.0 Hz, 1 H, 6-H), 7.86 (t,
J=8.0 Hz, 1 H, 2-H), 7.89 (t,
J=8.0 Hz, 1 H, 7-H), 8.20 (d,
J=8.0 Hz, 1 H, 1-H), 8.49 (d,
J=8.0 Hz, 1 H, 8-H), 8.94 (d,
J=8.0 Hz, 1 H, 5-H), 8.96 (d,
J=8.0 Hz, 1 H, 4-H).
10,12(N,N)-Di(2-amino-1,3-propanediol)quino[2,3,4-kl]acridine (33b):
24 (10 mg, 0.036 mmol) and 2-amino-1,3-propanediol (serinol) (10 mg, 0.11 mmol) were reacted in ethanol (5 mL) to afford 33b (8mg, 50%), by the procedure described for the synthesis and purification of 30b. MS (FAB); m/z: 445 (100) [(M+1)+, C25H25N4O4]; IR: = 3332, 1613, 1559, 1415, 1390, 1050 cm-1; 1H-NMR (CD3SOCD3): δ = 3.84 (m, 8 H, 2’-H), 4.45 (quintet, J=5.0 Hz, 2 H, 1’-H), 5.37 (t, J=5.0 Hz, 4 H, OH), 6.91 (s, 1 H, 11-H), 8.08 (t, J=7.5 Hz, 2 H, 3-, 6-H), 8.16 (t, J=7.5 Hz, 2 H, 2, 7-H), 8.47 (d, J=7.5 Hz, 2 H, 1-, 8-H), 9.28 (d, J=7.5 Hz, 2 H, 4-, 5-H).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




