One-Pot Quinazolin-4-ylidenethiourea Synthesis via N-(2-Cyanophenyl)benzimidoyl isothiocyanate

1
Department of Organic Chemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
2
National Center for Biomolecular Research, Faculty of Science, Masaryk University, Brno, Czech Republic
3
Department of Inorganic Chemistry, Masaryk University, 611 37 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(7), 574-587; https://doi.org/10.3390/60700574
Submission received: 10 November 2000
/
Revised: 22 May 2001
/
Accepted: 23 May 2001
/
Published: 30 June 2001
Abstract
:1,1-Disubstituted-3-(2-phenyl-3H-quinazolin-4-ylidene)thioureas (8) were synthesized in a one pot reaction of N-(2-cyanophenyl)benzimidoyl isothicyanate (3) with secondary amines. The products underwent transamination reactions. Compounds 8a-8g were identified by FTIR, 1H-NMR, 13C-NMR, mass spectroscopy and X-ray crystallography.
Introduction
The benzodiazocines [1] and quinazolines [2,3] represent two classes of heterocyclic compounds with diverse physiological activities, e.g., acting as central nervous system depressants and calcium sensitizing agents. Several quinazoline derivatives have been tested for their biological and pharmacological activities. These derivatives posses hypnotic [4], diuretic [5], antihistaminic [6], anti-inflammatory [7], bronchodilator [8], antihypertensive [9], antituberculousis [10], antimicrobial [11], anticonvulsant [12], sedative [12] and hypoglycemic activities [13].
These derivatives are intermediates for various pharmaceutical agents. Consequently, numerous research groups have attempted to synthesize these compounds. Others have modified their basic structures by changing the size of the rings or by changing the heteroatoms. Stankovsky’s group [14,15,16] has described the synthesis of 2,4-disubstituted-6H-5,1,3-benzothiadiazocines by the intramolecular substitution reaction of the 1,1-di-R-3-[2-(chloromethylphenylimino}phenylmethyl] thioureas.
Results and Discussion
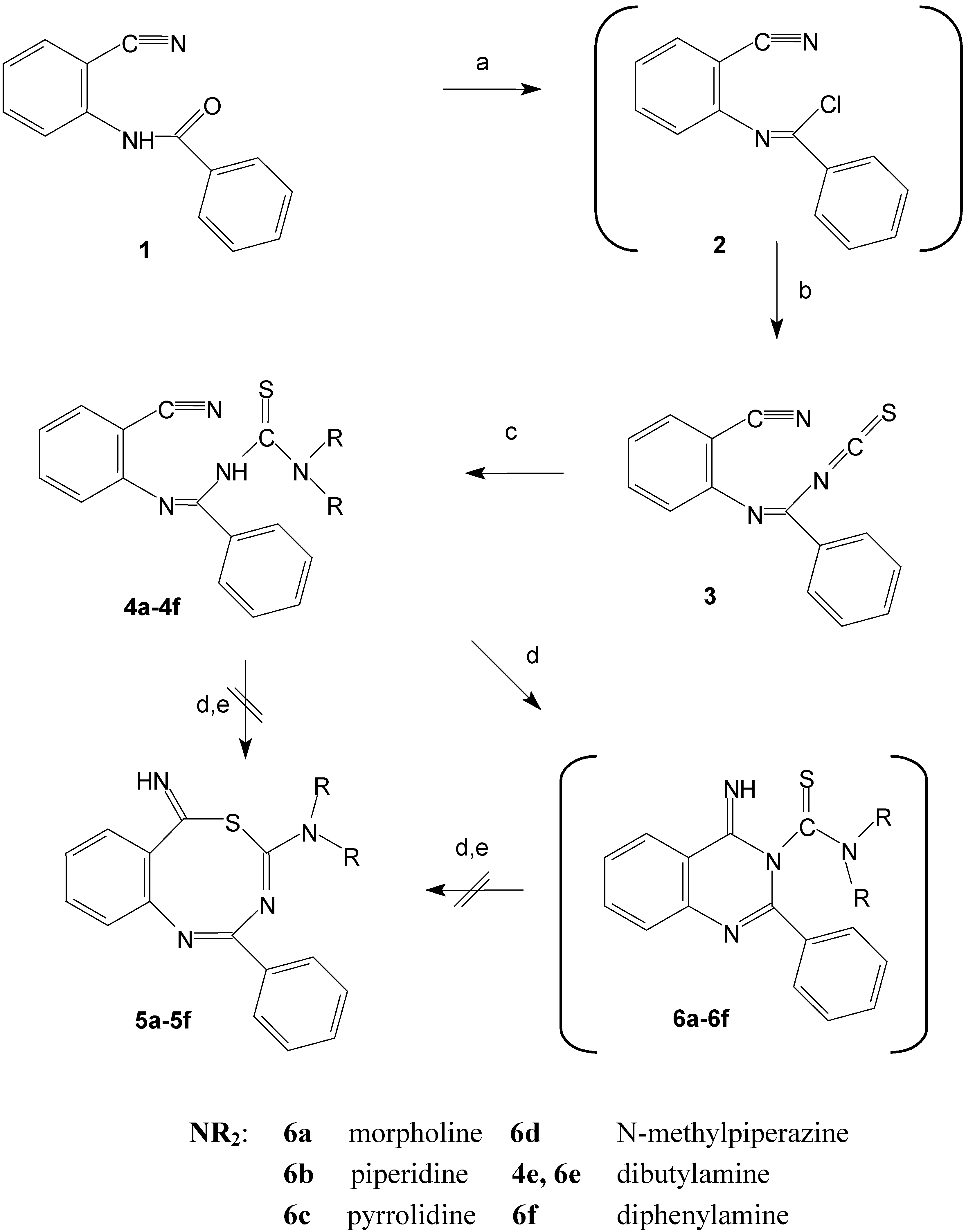
We describe in this paper the intramolecular cycloaddition reactions of the products formed in situ by addition of secondary amines to N-(2-cyanophenyl)benzimidoyl isothicyanate (3). The very reactive starting compound 3 was easily prepared from N-(2-cyanophenyl)benzamide (1). The amide 1 was transformed by the action of phosphorous pentachloride into the benzimidoyl chloride 2 that subsequently reacts with potassium thiocyanate to give the desired isothiocyanate derivative 3. The proposed structure of compound 3 is supported by its IR spectrum that showed the presence of the expected isothiocyanato and cyano moieties. The isothiocyanate 3 reacts with secondary amines (morpholine, piperidine, pyrrolidine, N-methylpiperazine, and diphenylamine) to give the cyclic products.
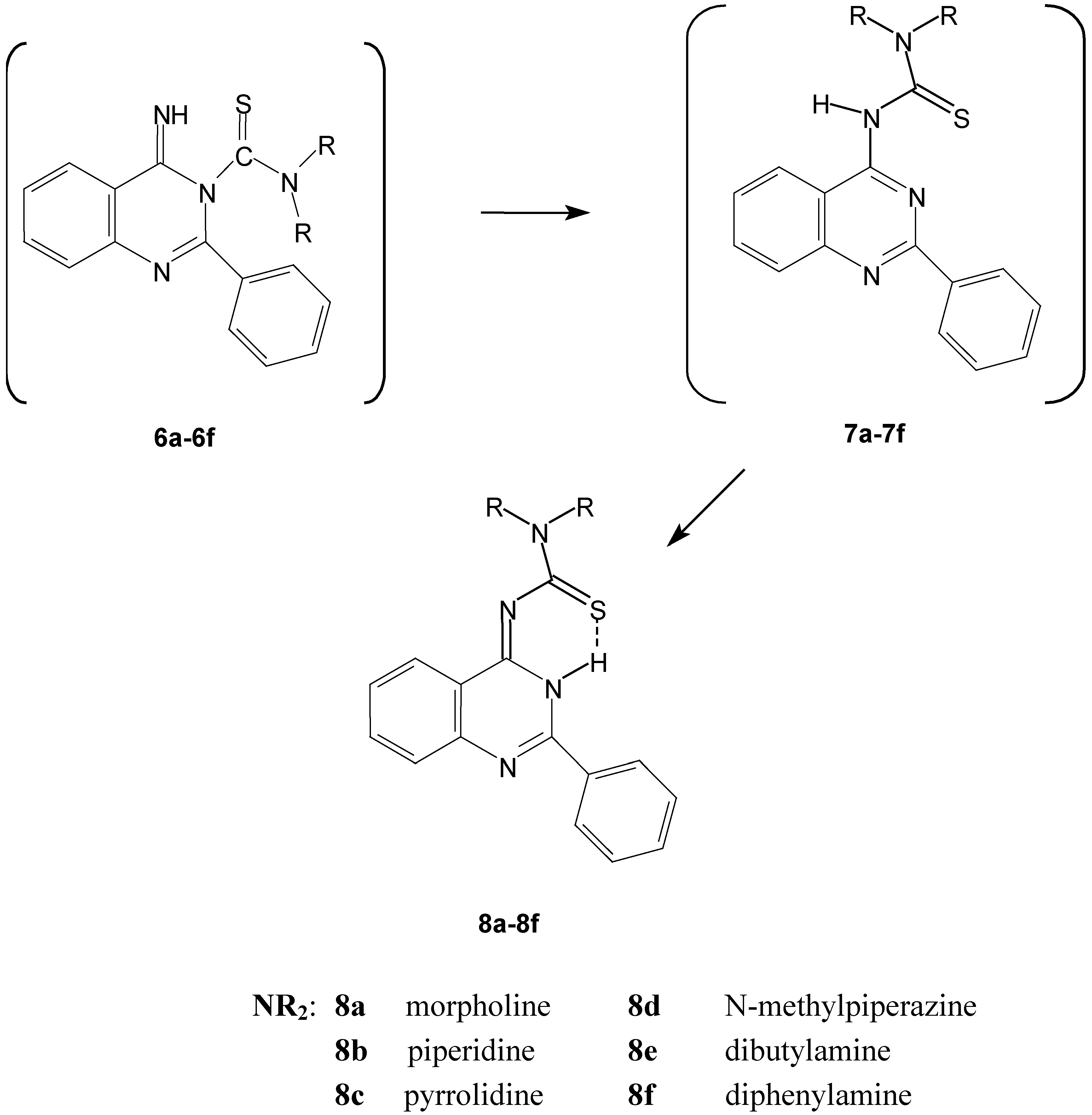
The proposed structures of the cyclisation products can be explained by considering the in situ formation of the thiourea derivatives 4a-4d, 4f (Scheme 1). This fact was confirmed by the isolation and identification of the thiourea derivative 4e obtained when 3 was reacted with dibutylamine. This reaction required basic conditions to accelerate the cyclisation reaction. The intermediates 4a-4f contain two active nucleophilic sites in the molecule, which compete in the attack of the nitrile group and finally give the cyclic product. The reactive sites are the sulfur and nitrogen atoms present in the thioamide group of the thiourea derivatives 4a-4f. The intramolecular reaction would give one or both cyclic products generally: The benzothiadiazocine derivatives 5 via sulfur attack [4,5,6], or the quinazoline derivatives 6 via nitrogen attack. The reaction could be extended to involve the Dimorth rearrangement products 7 or their tautomers 8.
The intramolecular addition reactions of thioureido derivatives containing functionalized carboxy groups under neutral and basic conditions take part on the nitrogen atom [17,18]. Consequently the expected products formed were the quinazoline derivatives 6 giving the more thermodynamically stable six membered rings. The resultant quinazoline derivatives 6 undergo Dimroth rearrangement by the effect of the excess amine to give 7, which were converted spontaneously to finally give the thiourea derivatives 8.
Scheme 1.
Synthesis of the quinazoline derivatives 6 and alternative reaction pathways.
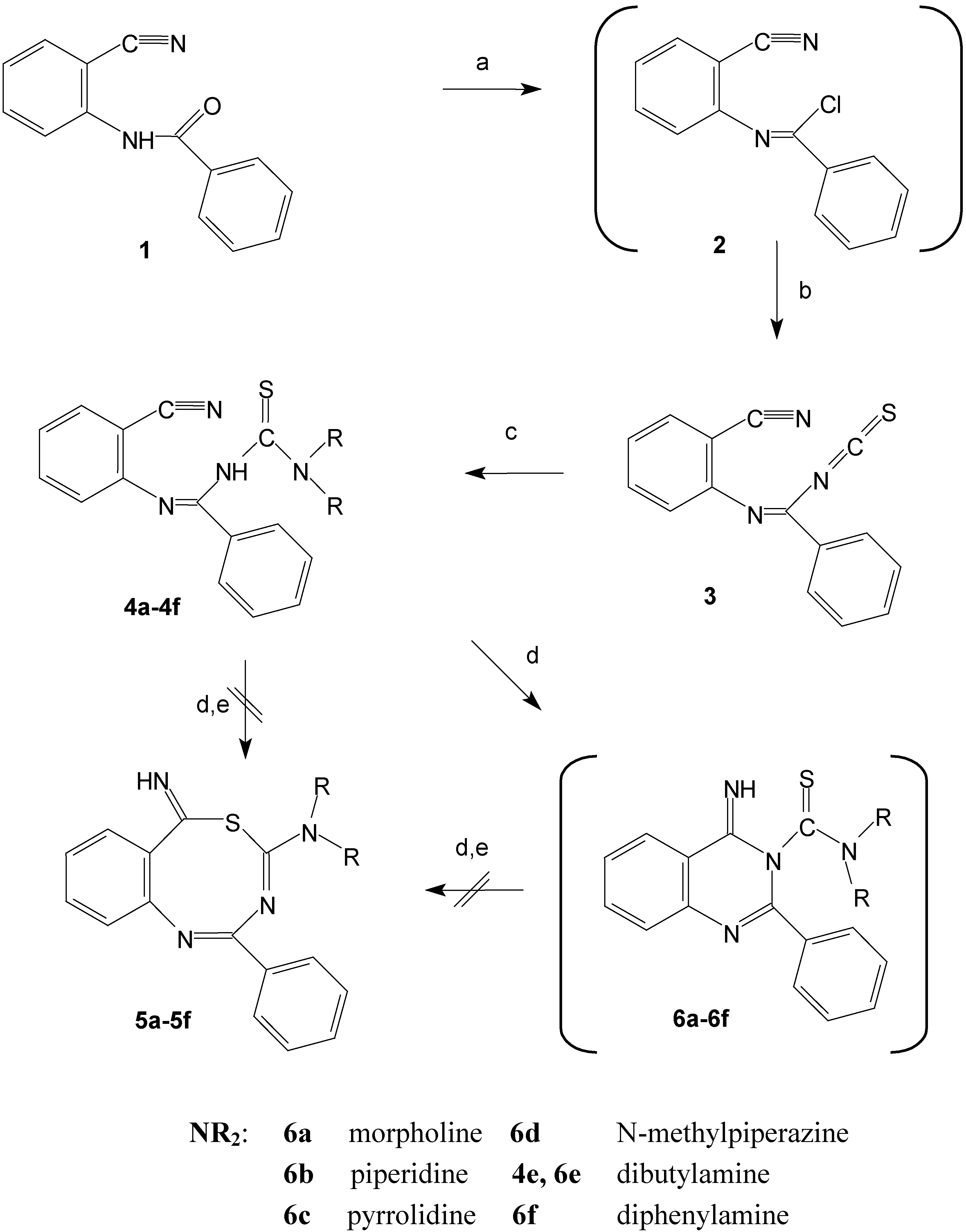
a: PCl5, toluene, 110°C, 8 h; b: KSCN, acetone, -5°C, 2h; c: NHR2, acetone, 25°C, 24 h;
d: CHCl3, N(C2H5)3, 50°C, 4h; e: DMF, reflux, 24h, or MeOH, MeONa, reflux, 24h.
Scheme 2.
The spontaneous conversion of compounds 6 to the quinazoline derivatives 8.
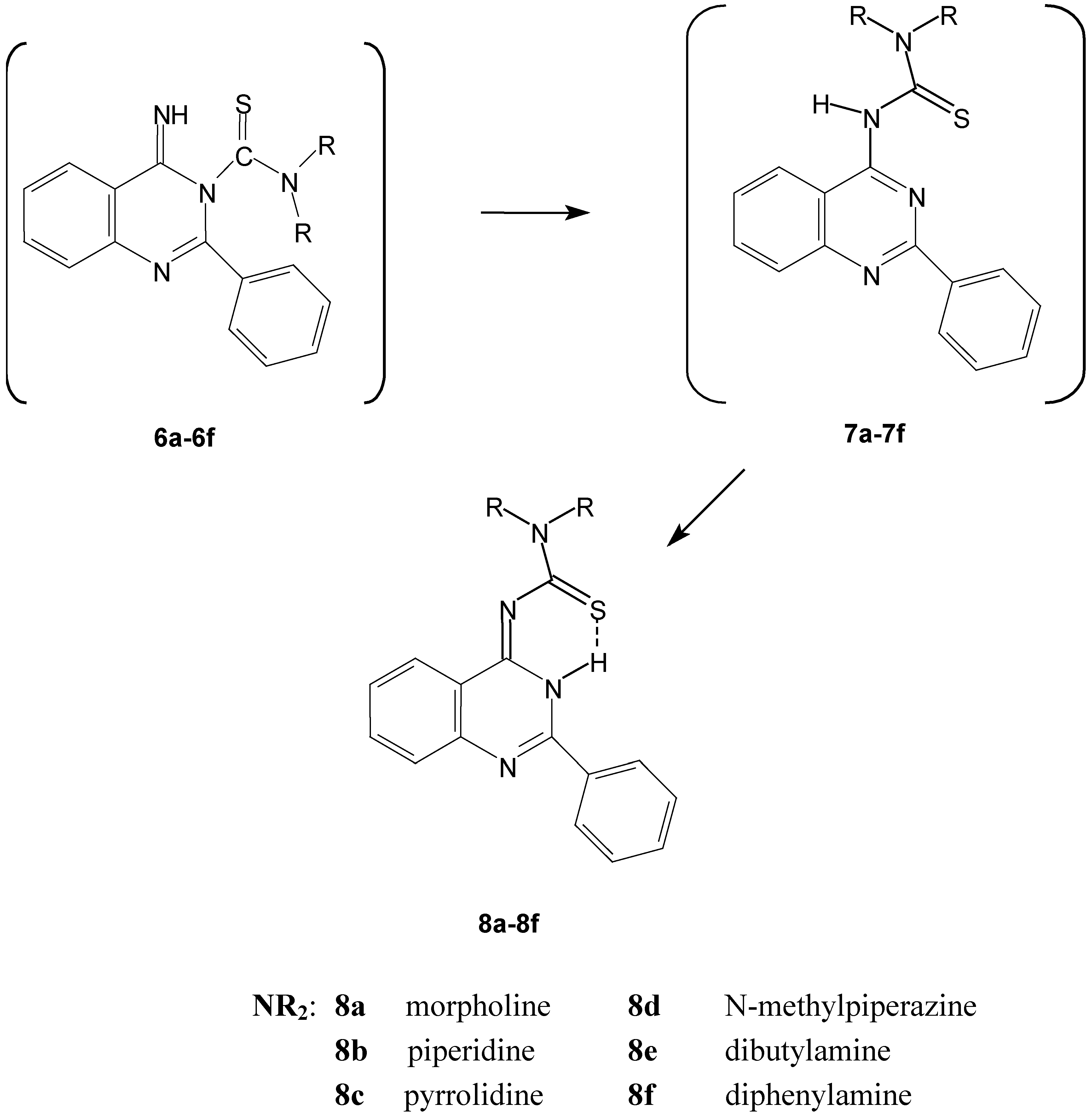
The identities of the synthesized 3, 4, and 8 were supported by the spectral data as follows: the infrared spectra show the disappearance of ν(NCS) and ν(CN) bands present in case of the isothiocyanate 3 and the existence of ν(NH) and ν(C=N) bands at 3220 cm-1, and 1613 cm-1, respectively, for the cyclic forms 8a-8f. The infrared spectra also gave two pieces of evidence in the case of the formation of the open chain thiourea derivative 4e: the first was the ν(CN) band at 2228 cm-1 and the second the ν(NH) band at 3210 cm-1 together with the absence of ν(C=N) band of the quinazoline derivative at 1613 cm-1.
The proton nuclear magnetic resonance spectra show the introduction of the CH2 groups from the amino group when reacting with isothiocyanate 3. It was a surprise to find that the two CH2 groups adjacent to the nitrogen atom of the amines are identical in all cases except for the case of pyrrolidine which gave two distinct peaks with chemical shifts of ca 4.01 and ca 3.87 ppm, respectively. This feature can be readily explained if we examine the geometry of 8 (Figure 2, Figure 3).
The 1H-NMR spectra gave no clear evidence supporting the formation of the open chain intermediate in case of 4e. The important evidence for the formation of the quinazolines 8 is the chemical shift at ca 16.73 ppm due to the NH group. The high chemical shift is attributed to high interactions between the thiocarbonyl group and the NH of the quinazoline ring. The alternative formation of 7 would have the NH signal at a lower chemical shift (ca 12 ppm) according to the simulated NMR spectra.
The most important peak shown by the 13C-NMR is the peak corresponding to C=S with a chemical shift of ca 183-184 ppm, which plays an important role in the structure identification of the cyclic products. Of coarse, it is associated with the quinazoline structures 6-8 not the thiadiazocines 5a-5f. 13C-NMR spectra gave two different peaks corresponding to the -CH2- group of the amines 8a-8e although we expected to observe only one peak. This is obviously due to the geometry of the product, in which one -NCH2- group from the amine (this group has almost the same chemical environment with respect to the other -NCH2- group) is affected by anisotropy of the C=S group. This fact is maximized in the 1H-NMR spectrum when using pyrrolidine as an amine. It is apparent that the two CH2 groups adjacent to the nitrogen atom in the compound 8e are not identical (51.60 ppm, 51.27 ppm) and the other two neighboring NCH2CH2 (30.83 ppm, 28.90 ppm) groups are not equivalent due to the same mentioned reason. The 13C-NMR spectrum of thiourea 4e show that the C=S group appeared at 178 ppm and the nitrile group at 107 ppm.
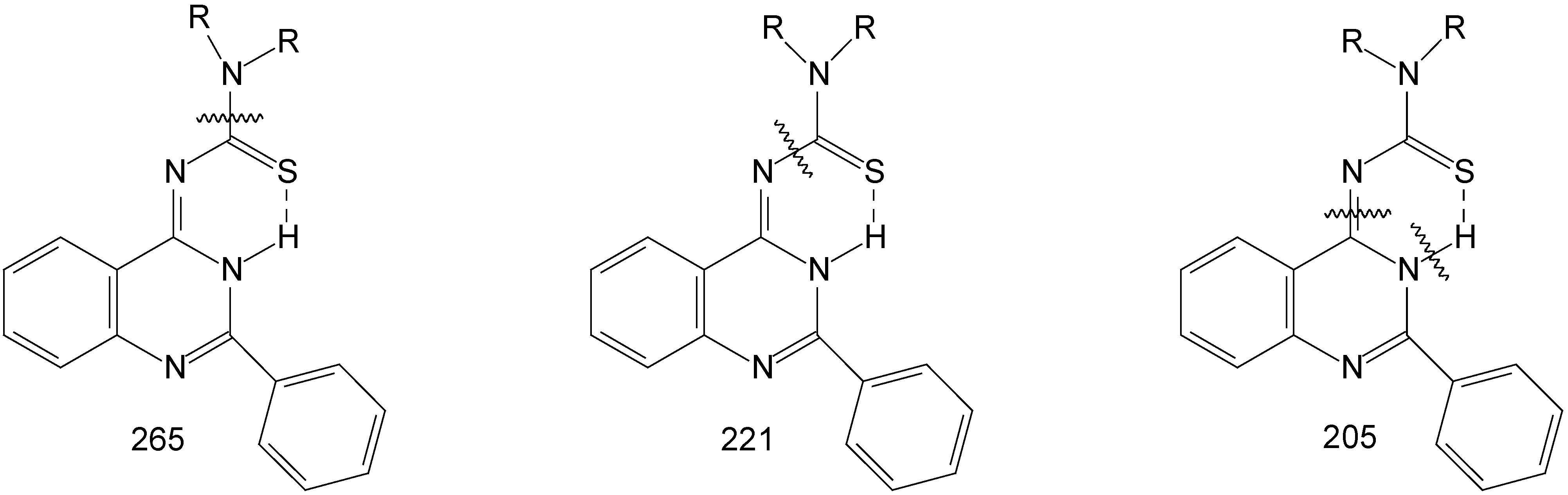
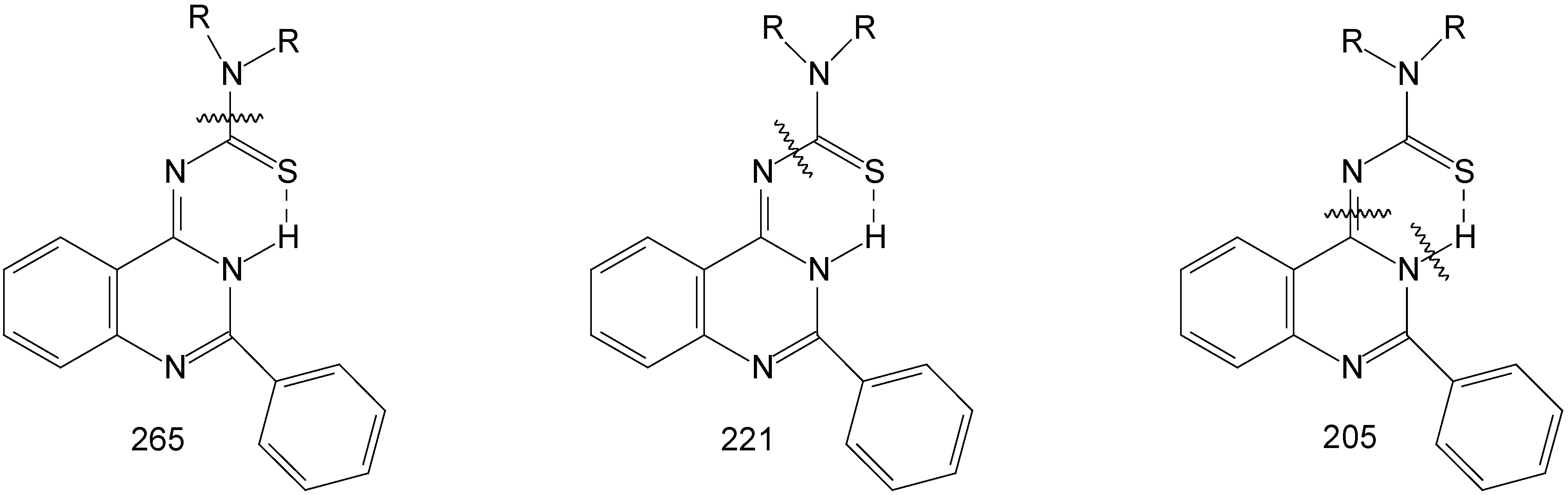
The mass spectra gave the molecular ion for all the products 8a-8d, next it gave molecular fragment which corresponds to the thioamide moiety at m/z 131 (morpholine), 128 (piperidine), 114 (pyrrolidine), and 142 (N-methylpiperazine) and the molecular fragment at 221 corresponding to the residue of the compound (2-phenyl-4-quinazoilinimine moiety). The products of fragmentation are shown on Figure 1.
Figure 1.
Mass spectral fragmentations in compounds 8
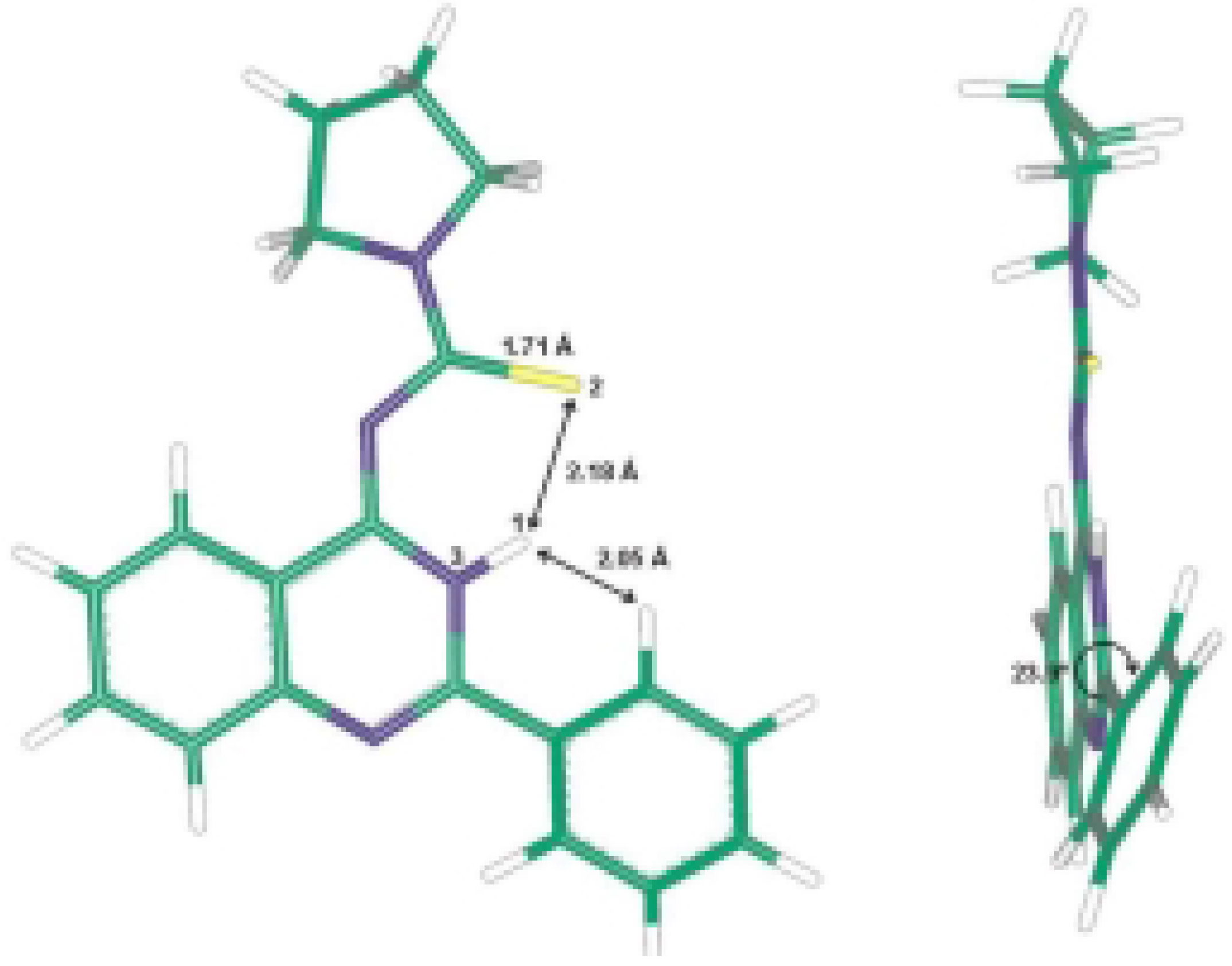
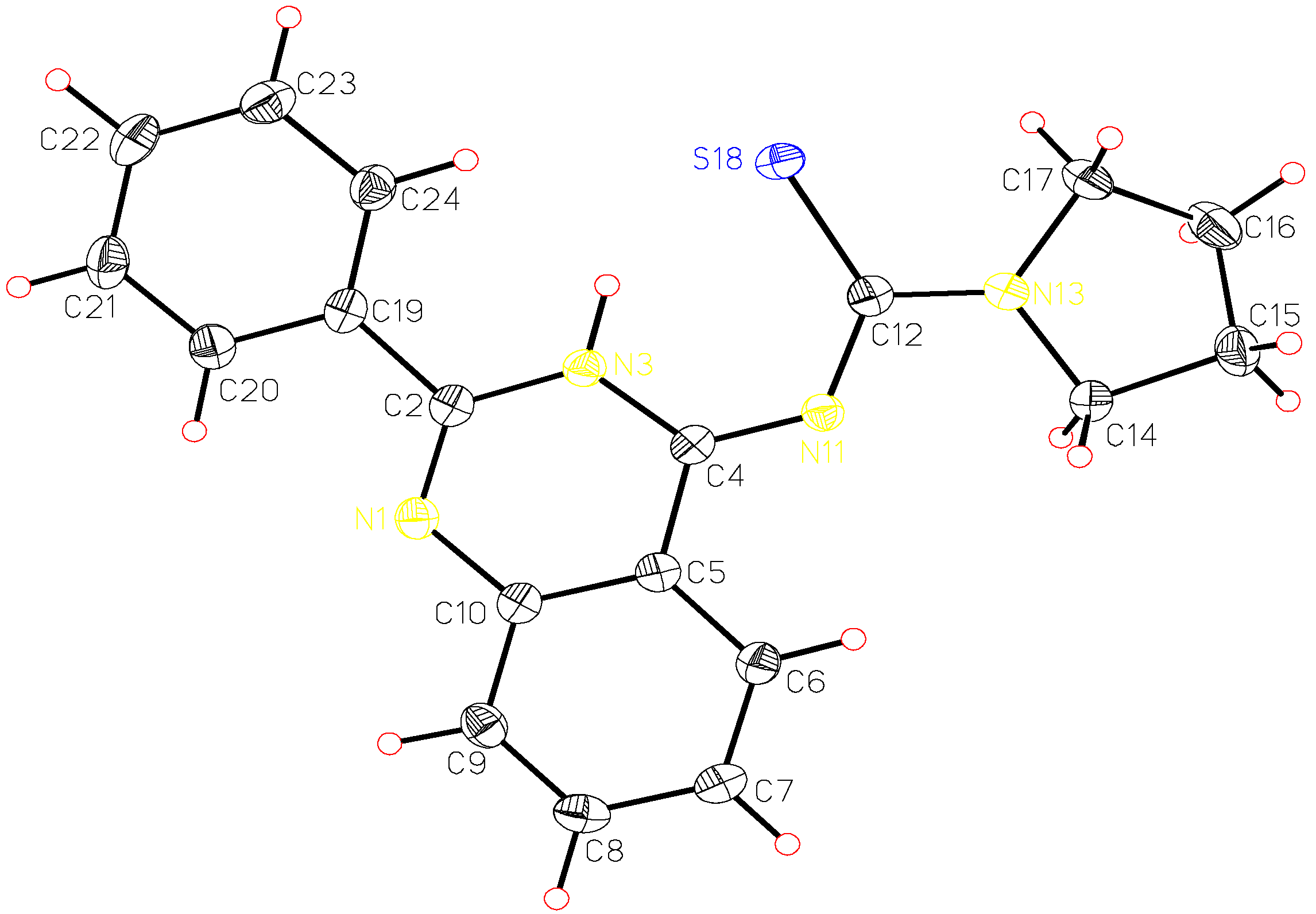
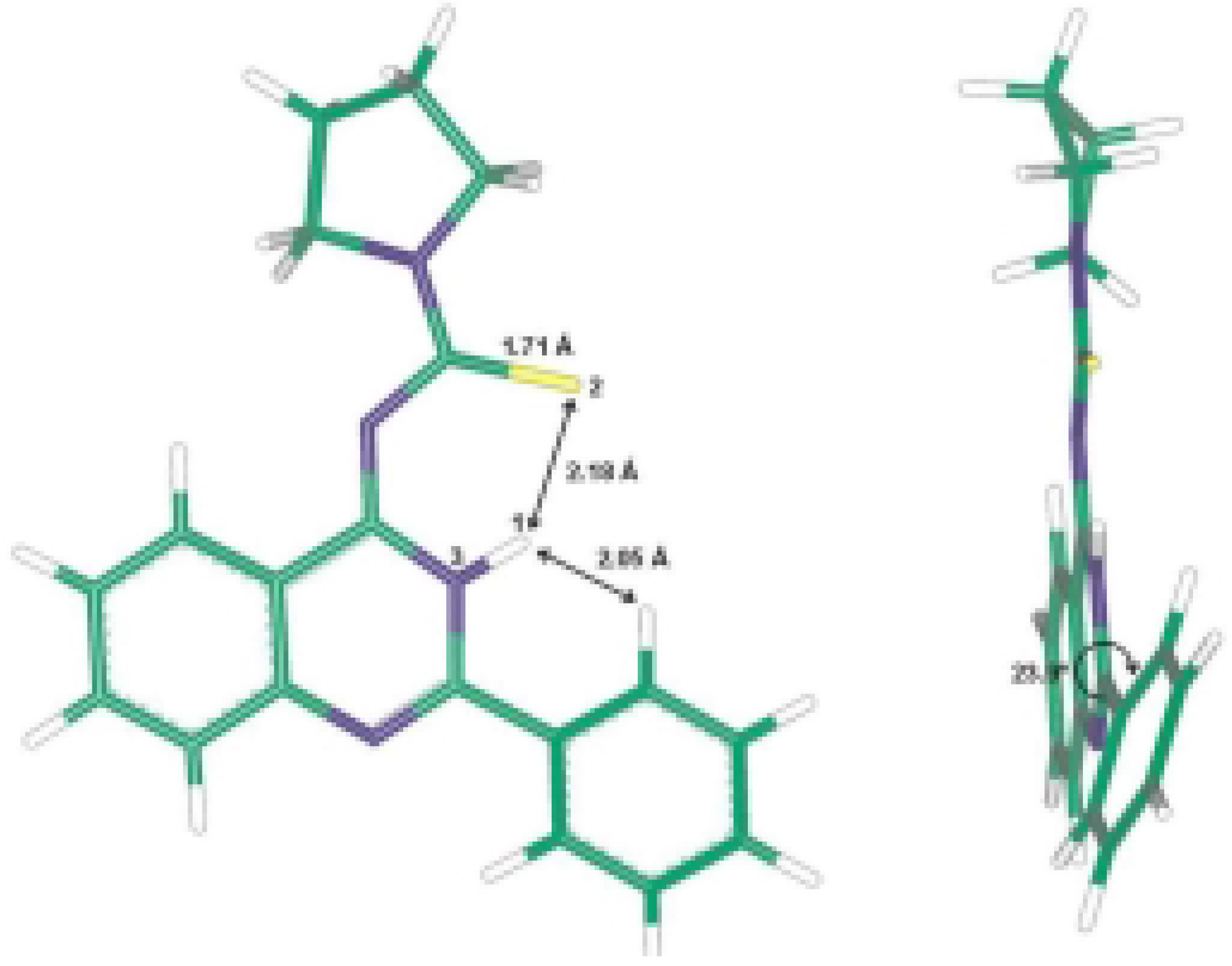
A single crystal of pyrrolidine-1-carbothioic acid (2-phenyl-3H-quinazolin-4-ylidene) amide (8c) was used for X-ray structure analysis. The structural data obtained are presented in Table 1, Table 2 and Table 3 and correspond to a great extent with those calculated by ab initio HF/6-31G** method. The calculated and the X-ray molecular structures of 8c are presented in Figure 2 and Figure 3. The analysis shows that the structure formed is the tautomeric form 8 of the Dimroth rearrangement product 7. The reason for this could be explained if we examine the structures of all possible products 5-8 and the structures reported by Stankovsky [14,15,16,19].
The ab initio studies together with the X-ray analysis show that the compound 8c is almost planar with the phenyl ring at position 2 slightly out of plane and the pyrrolidine methylene groups out of plane. The electrons in π-orbitals are in good conjugation as presented in Figure 3. The hydrogen bond interactions between the thiocarbonyl group and the NH of the quinazoline ring add extra stability to the isolated product (Scheme 2). These hydrogen bond interactions were identified from the X-ray analysis and the computational studies, which show a bond distance of about 2.17 Å and 2.18 Å, respectively.
The intermediary thiourea derivatives 6 have the quinazoline moiety in one plane while the phenyl group, thioamide moiety and the pyrrolidine are each in a different plane. The same results were obtained for above mentioned benzothiadiazocine 5. The calculated (HF/6-31G** method) energy difference between 6 and 8 is 30.69 kcal.mol-1, while the energy difference between 5 and 6 is 36.61 kcal.mol-1. This shows that the more stable compound is the isolated quinazoline derivative 8. The 6-methylene analogues of 5 reported in the literature [14,15,16] have the benzene ring and the N1 together with the CH2 group in one plane, while the [phenyl-C2-N3-C4-S5-NR] is in another plane, indicating that the electrons in the π-orbitals are in good conjugation.
Figure 2.
X-ray analysis of compound 8c.
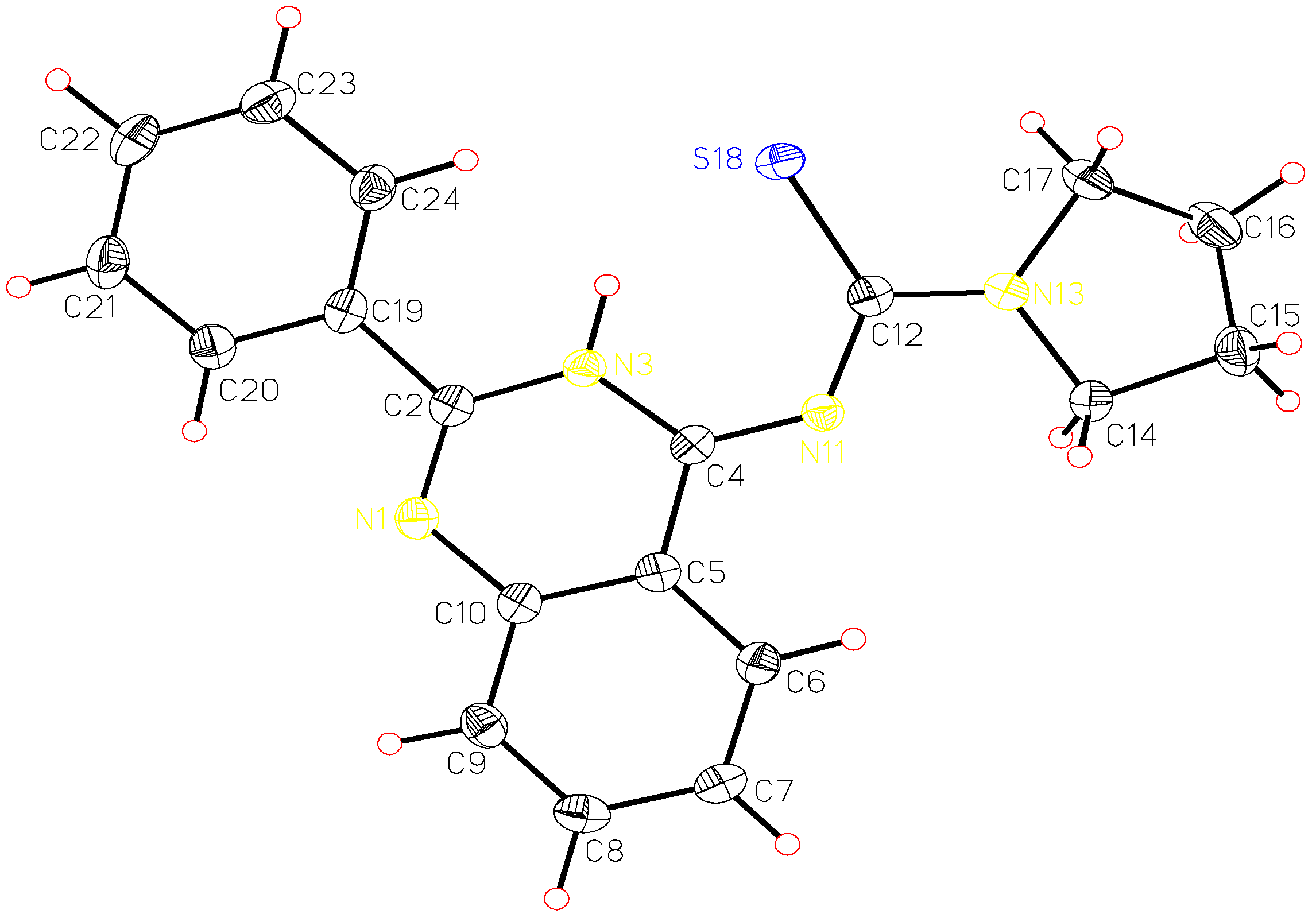
Figure 3.
Molecular design of 8c according to HF/6-31G** method.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Bond Length [Å] | Bond | Bond Length [Å] | ||
|---|---|---|---|---|---|
| X-ray | HF/6-31G** | X-ray | HF/6-31G** | ||
| N1-C2 | 1.30 | 1.27 | C12-N13 | 1.34 | 1.33 |
| N1-C10 | 1.39 | 1.38 | C12-S18 | 1.72 | 1.71 |
| C2-N3 | 1.38 | 1.37 | N13-C14 | 1.48 | 1.47 |
| C2-C19 | 1.49 | 1.49 | N13-C17 | 1.47 | 1.47 |
| N3-C4 | 1.36 | 1.35 | C14-C15 | 1.52 | 1.52 |
| C4-N11 | 1.32 | 1.30 | C15-C16 | 1.52 | 1.52 |
| N11-C12 | 1.37 | 1.36 | C16-C17 | 1.52 | 1.52 |
| Angle [°] | X-ray | HF/6-31G** | Angle [°] | X-ray | HF/6-31G** |
|---|---|---|---|---|---|
| N1-C2-C19 | 119.80 | 120.14 | C14-N13C17 | 111.84 | 112.15 |
| C2-N1-C10 | 117.34 | 117.99 | N13-C17-C16 | 103.17 | 103.47 |
| N1-C2-N3 | 123.17 | 122.99 | N1-C10-C5 | 122.81 | 122.56 |
| C2-N3-C4 | 123.63 | 124.19 | C10-C5-C6 | 120.25 | 120.07 |
| N3-C4-N11 | 125.66 | 126.65 | C5-C6-C7 | 119.86 | 120.10 |
| N11-C12-N13 | 111.85 | 112.38 | C6-C7-C8 | 119.88 | 119.80 |
| N13-C12-S18 | 119.05 | 119.49 | C7-C8-C9 | 120.82 | 120.79 |
| C12-N13-C17 | 125.13 | 124.17 | C2-C19-C24 | 122.91 | 122.15 |
| N13-C14-C15 | 103.49 | 103.46 | C2-N3-H3 | 121.32 | 120.53 |
| Torsion angle [°] | X-ray | HF/6-31G** | Torsion angle [°] | X-ray | HF/6-31G** |
|---|---|---|---|---|---|
| C14-N13-C17-C16 | 15.73 | 12.06 | N11-C4-N3-C2 | -178.35 | -178.96 |
| C17-N13-C14-C15 | 6.92 | 11.68 | C4-N3-C2-N1 | -0.56 | -1.43 |
| C16-C17-N13-C12 | -163.11 | -168.09 | N3-C2-N1-C10 | 0.93 | 023 |
| C17-N13-C12-S18 | -1.31 | -2.84 | C2-N1-C10-C5 | -0.63 | 0.92 |
| C17-N13-C12-C11 | 178.84 | 176.89 | C12-N11-C4-N3 | 2.05 | -0.44 |
| S18-C12-N11-C4 | 7.02 | -3.05 | N1-C2-N3-H3 | 179.60 | 175.29 |
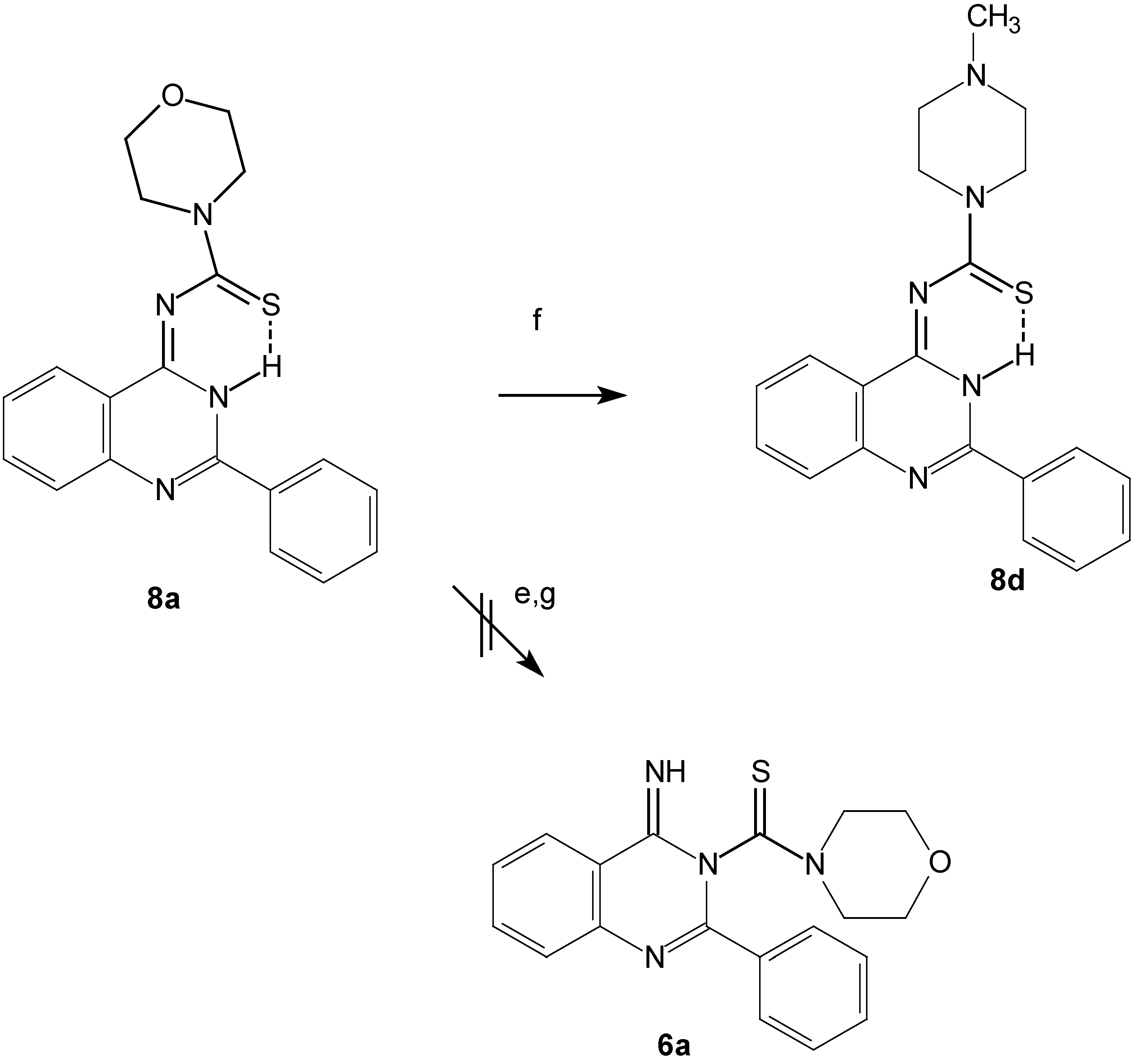
The predicted structure contains a thioamide group, so we can confirm the structure chemically through a transamination reaction of compound 8a by N-methylpiperazine to give 8d (Scheme 4).
Scheme 4.
Transamination reaction for compound 8a.
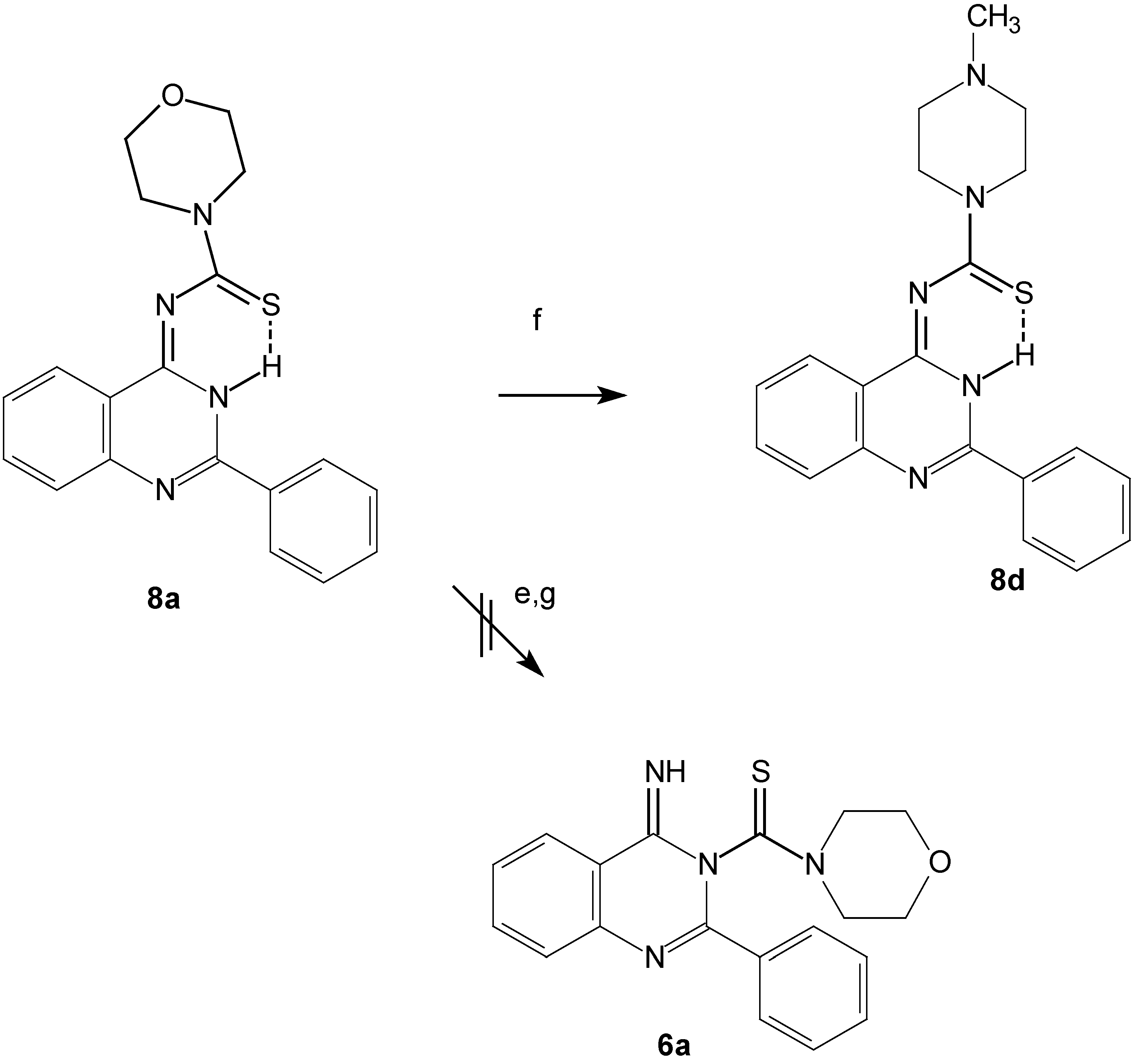
e: DMF, reflux, 24h, or MeOH, MeONa, reflux, 24h.
f: HNR2, DMF, reflux, 2-4h; g: Conc. H2SO4, 2 d.
The Dimroth rearrangement reaction might take place upon refluxing the expected quinazoline derivatives 6a-6f in DMF or in MeOH / MeONa for a long period, but no such reactions were detected, providing further support for the formation of the quinazolin-4-ylidenthioureas 8. A retro-Dimroth rearrangement reaction to give 6a might be expected to take place when 8a was left standing for 2 days in conc. sulfuric acid but formation of this product was not detected. We confirmed the structures of the 1,1-disubstituted-3-(2-phenyl-3,H-quinazolin-4-ylidene) thiourea compounds 8a-8f on the basis of the above spectral data, by comparison of this data with the computer simulated spectra and from knowledge about the chemistry of the thiourea derivatives [17,18].
Conclusions
3-[(2-cyanophenyl-imino)-phenylmethyl]-1,1-disubstituted thioureas undergo a regioselective intramolecular cycloaddition reaction to give intermediate 1,1-disubstituted-4-imino-2-phenyl-4H-quinazolin-3-carbothioic acid amides (6) that are converted spontaneously into 1,1-disubstituted-3-(2-phenyl-3,H-quinazolin-4-ylidene)thioureas 8. These products undergo transamination reactions.
Acknowledgments
This work was supported by the grants of the Ministry of Education of the Czech Republic (Grant No. CEZ: J07/98: 143100011) and Grant Agency of the Czech Republic (Grant No. 203/01/1333). We would like to thank analytical department of Lachema Co., Brno, Czech Republic for elemental analysis. The authors thank the Czech Academic Supercomputer Center in Brno for providing the access to computer facilities.
Experimental
General
Melting points of all the compounds were measured on a Boetius Rapido PHMK 79/2106 (Wägetechnik) instrument. TLC was carried out on Silufol UV 254 plates (Kavalier, Votice). The eluent used was a 20:80 mixture of acetone-benzene. TLC detection was accomplished with a Fluotes universal instrument (Quarzlampen, Hanau) and iodine vapors. The purity of compounds 4 and 8 were proven by their elemental analysis, measured on an Erba 1102 instrument. FTIR spectra (reported as /cm-1) were taken on a Genesis (Unicam) spectrometer in potassium bromide pellets. NMR spectra were measured on a Bruker Avance DRX-500 spectrometer. Unless stated otherwise 1H, 13C-NMR spectra were measured in CDCl3 solutions and tetramethylsilane was used as the internal standard. Results are reported in ppm on the δ scale. The measured 13C and 1H-NMR spectra were correlated with those obtained by on-line simulation (Advanced Chemistry Development, Inc., Toronto, Canada). The X-ray structural data of compound 8c (Table 4) were collected with a KUMA KM-4 kappa fourcircle diffractometer. The structure was solved by direct methods using SHELXS86 [20] and refined on F2 for all reflections using SHELXl93 [21]. Crystals suitable for X-ray determination were obtained as white prisms by crystallization from CHCl3 - petroleum ether at room temperature. The crystallographic data for 8c have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 143663. Geometry optimization of structure 8c was performed at ab initio level of quantum chemical calculation by HF/6-31G**. Mass spectrometry was determined (electron impact, 70 eV) with a Fisons TRIO 1000 and GC 8000 series instrument.
| Empirical formula | C19H18N4S |
| Molecular weight | 334.43 |
| Temperature | 120(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | orthorhombic, Pbca |
| Unit cell dimensions | |
| a, α | 9.51557(6) Å, α= 90 ° |
| b, β | 15.8703(10) Å, β= 90 ° |
| c, γ | 21.6351(12) Å, γ= 90 ° |
| Volume | 3267.3(3) Å3 |
| Z; density calculated | 8 mg.m-3 |
| Absorption coefficient | 0.206 mm-1 |
| F(000) | 1408 |
| Crystal size | 0.65 X 0.25 X 0.10 mm |
| θ , Range for data collection | 3.47- 25.00° |
| Range of h, k, l | -8< = h< =12, -20< = k< = 20, -28< = l< = 27, |
| Reflections collected | 15684 |
| Independent reflections | 2865 |
| Refinement method | full-matrix least-squares on F2 |
| Data; restraints; parameters | 2818 / 0 / 289 |
| Goodness-of-fit on F2 | 1.020 |
| Final R indices [I > 2σ(I)] | R1= 0.0354, wR2 = 0.0827 |
| R indices (all data) | R1= 0.0426, wR2 = 0.0856 |
| Largest diff. Peak and hole | 0.240 and –0.262 e. Å-3 |
N-(2-Cyanophenyl)benzimidoyl isothicyanate - acetone solution (3).
A mixture of N-(2-cyanophenyl)benzamide (1) [22] (0.5 g, 2.25 mmol), and phosphorous pentachloride (0.5 g, 2.4 mmol) was refluxed in dry toluene for 8 h. The solvent was removed under reduced pressure to give a brownish colored oil of N-(2-cyanophenyl)benzimidoyl chloride (2) [23] which was not further purified. To a solution of this crude oil in dry acetone a solution of potassium thiocyanate (0.22 g, 2.25 mmol) in dry acetone was added portionwise with stirring and cooling at - 5°C for 2 h. The precipitated potassium chloride was filtered off , thus leaving an acetone solution of N-(2-cyanophenyl)benzimidoyl isothicyanate (3). The IR spectrum of this solution displayed a strong peak at 2059 cm-1 corresponding to the νNCS band. The isothiocyanate was used without further isolation and purification (to avoid polymerization and destruction of this very reactive compound).
1,1-Disubstituted-3-(2-phenyl-3H-quinazolin-4-ylidene)thioureas (8).
Procedure A
The appropriate secondary amine (2.25 mmol) was added in portions with stirring at room temperature to the acetone solution of isothiocyanate 3 over a period of 1h. The reaction mixture was then stirred for 24h. The precipitated quinazolines 8a-8d and 8f were filtered off and recrystallized from ethyl alcohol. Spectral data are given below.
Procedure B
The reaction mixture consisting of the chloroform solution of thiourea derivative 4e (0.28 g) and triethylamine (0.2 ml) was refluxed for 4h. The reaction mixture was evaporated under reduced pressure to give the desired product 8e, which was recrystallized from ethyl alcohol.
Procedure C
Quinazoline 8a (0.2 g, 0.6 mmol) was dissolved in DMF (3mL) and 1.8 mmol of the corresponding amine (N-methylpiperazine or 40% aqueous methylamine) was added to this solution. The reaction mixture was heated under reflux at 150°C for 2-4 h. Afterwards the DMF was evaporated under reduced pressure (80°C/ 2.67 kPa.) and the resultant product was recrystallized from ethyl alcohol.
Morpholine-1-carbothioic acid (2-phenyl-3H-quinazolin-4-ylidene) amide (8a).
From 3 (procedure A); Yield: 0.17 g (22%); M.p. 185-186°C; Calc. for C19H18N4OS (350.44): 65.12% C, 5.18% H, 15.99% N, 9.15% S; found: 64.98% C, 5.05% H, 16.02% N, 9.00% S; FTIR: 3220 (NH), 2961,2923,2859 (CH), 1614 (C=N); 1H-NMR: 8.41– 7.39 (9H, m, ArH), 4.29-4.25 (4H, m, OCH2), 3.84 - 3.80 (4H, m, NCH2); 13C-NMR: 184.70 (C=S), 154.98 (C2), 149.60 (C4), 148.76 (Cq), 134.78 (CHAr), 132.43 (Cq), 132.09 (CHAr), 129.21 (CHAr), 128.21 (CHAr), 127.42 (CHAr), 127.26 (CHAr), 125.49 (CHAr), 121.17 (Cq), 66.82 (OCH2), 47.95 (NCH2), 47.79 (NCH2); Mass spectrum, m/z (Ir/%): 350 (5), 265 (16), 263 (82), 206 (9), 205 (100), 132 (11), 103 (15), 102 (39), 87 (25), 86(14), 77 (42), 76 (16), 57 (27) , 51 (14).
Piperidine-1-carbothioic acid (2-phenyl-3H-quinazolin-4-ylidene) amide (8b).
From 3 (procedure A); Yield: 0.23 g (29%); M.p. 135-136°C; Calc.for C20H20N4S (348.47): 68.94% C, 5.78% H, 16.08% N, 9.20% S; found: 68.79% C, 5.70% H, 16.10% N, 9.09% S; FTIR: 3180 (NH), 2926,2852 (CH), 1610 (C=N); 1H-NMR: 8.42– 7.39 (9H, m, ArH), 4.25-4.29 (4H, m, NCH2), 1.70 – 1.74 (6H, m, CH2); 13C-NMR: 183.35 (C=S), 154.28 (C2), 149.70 (C4), 148.76 (Cq), 134.29 (CHAr), 132.65 (Cq), 131.62 (CHAr), 129.34 (CHAr), 128.14 (CHAr), 127.61 (CHAr), 127.15 (CHAr), 125.56 (CHAr), 121.17 (Cq), 49.06 (NCH2), 48.55 (NCH2), 26.53 (CH2), 25.92 (CH2), 24.82 (CH2); Mass spectrum, m/z (Ir/%): 348 (3), 264 (18), 263 (75), 206 (19), 205 (100), 132 (8), 103(11), 102 (28), 85 (22), 84 (53), 77 (47) , 76 (19), 75 (15), 56 (12).
Pyrrolidine-1-carbothioic acid (2-phenyl-3H-quinazolin-4-ylidene) amide (8c).
From 3 (procedure A); Yield: 0.19 g (25%); M.p. 205-206°C; Calc. for C19H18N4S (334.44): 68.24% C, 5.42% H, 16.75% N, 9.59% S; found: 68.07% C, 5.33% H, 16.60% N, 9.43% S; FTIR: 3200 (NH), 2968,2867 (CH), 1611 (C=N); 1H-NMR: 8.45– 7.44 (9H, m, ArH), 4.01 (2H, t, NCH2) (JA,B= 6.90 Hz), 3.87 (2H, t, NCH2) (JA,B= 6.90 Hz), 1.99-2.07 (4H, m, CH2); 13C-NMR: 183.19 (C=S), 155.66 (C2), 151.04 (C4), 149.18 (Cq), 134.72 (CHAr), 133.17 (CHAr), 132.11 (CHAr), 129.68 (CHAr), 128.59 (CHAr), 128.02 (CHAr), 127.34 (CHAr), 125.96 (CHAr), 121.84 (Cq), 52.21 (NCH2), 51.69 (NCH2), 27.05 (CH2), 26.31 (CH2); Mass spectrum, m/z (Ir/%): 334 (22), 301 (12), 265 (21), 264 (39), 263 (48), 232 (35),206 (25), 205 (100), 103 (12), 102 (24), 77 (38), 70 (30).
4-Methylpiperazine-1-carbothioic acid (2-phenyl-3H-quinazolin-4-ylidene) amide (8d).
From 3 (procedure A); Yield: 0.13 g (16%); M.p. 244-245°C; From 8a (procedure C); Yield: 0.1g (48%); M.p. 244-245°C; Calc. for C20H21N5S (363.48): 66.09% C, 5.82% H, 19.27% N, 8.82% S; found: 65.90% C, 5.70% H, 19.07% N, 8.69% S; FTIR: 3240 (NH), 2934, (CH), 1610 (C=N); 1H-NMR: 8.43–7.48 (9H, m, ArH), 4.42-4.37 (4H, m, NCH2), 2.60-2.56 (4H, m, NCH2), 2.39 (3H, s, CH3); 13C-NMR: 184.38 (C=S), 154.85 (C2), 149.51 (C4), 149.13 (Cq), 134.73 (CHAr), 132.86 (CHAr), 132.00 (CHAr), 129.37 (CHAr), 128.43 (CHAr), 127.60 (CHAr), 127.34 (CHAr), 125.65 (CHAr), 121.51 (Cq), 55.06 (NCH2), 54.71(NCH2), 47.21 (CH3NCH2), 46.78 (CH3NCH2), 45.53 (NCH3); Mass spectrum, m/z (Ir/%): 363 (8), 264 (17), 263 (72), 206 (10), 205 (100), 131 (6), 103 (9), 102 (34), 100 (29), 99 (22), 83 (47), 77 (33), 75 (10), 74 (10), 70 (11), 58 (38) , 56 (10).
1,1--Dibutyl-3-(2-phenyl-3H-quinazolin-4-yliden) thiourea (8e).
From 4e (procedure B); Yield: 0.18 g (64%) (B, from 4e); M.p. 184-185°C; Calc. for C23H28N4S (392.56): 70.37% C, 7.19% H, 14.27% N, 8.17% S; found: 70.21% C, 7.15% H, 14.11% N, 8.07% S; FTIR: 3210 (NH), 2955, 2927, 2869 (CH), 1614 (C=N); 1H-NMR: 8.41– 7.08 (9H, m, ArH), 3.97 (4H, t, NCH2) (JA,B= 7.12 Hz), 1.84-1.70 (4H, m, NCH2CH2), 1.51-1.34 (4H, m, CH2CH3), 1.04 (6H, t, CH2CH3) (JA,B= 7.31 Hz); 13C-NMR: 184.21 (C=S), 154.23(C2), 149.83 (C4), 148.99 (Cq), 134.29 (CHAr), 132.96 (CHAr), 131.62 (CHAr), 129.34 (CHAr), 128.14 (CHAr), 127.61 (CHAr), 127.15 (CHAr), 125.56 (CHAr), 121.85 (Cq), 51.60 (NCH2), 51.27 (NCH2), 30.83 (NCH2CH2), 28.90 (NCH2CH2), 20.68 (CH2CH3), 20.59 (CH2CH3), 14.19 (CH2CH3), 14.11 (CH2CH3)
1,1-Diphenyl-3-(2-phenyl-3H-quinazolin-4-ylidene) thiourea (8f)
From 3 (procedure A); Yield: 0.31 g (32%) M.p. 155 °C; Calc. for C27H20N4S (432.54): 74.97% C, 4.66% H, 12.95% N, 7.41% S; found: 74.95% C, 4.45% H, 12.92% N, 7.39% S; FTIR: 3284 (NH), 3059 (CH), 1612 (C=N); 1H-NMR: 16.73 (1H, bs, NH), 8.32– 7.07 (19H, m, ArH); 13C-NMR: 188.30 (C=S), 158.71 (C2), 155.77 (CHAr), 151.53 (C4), 149.13 (Cq), 134.93 (CHAr), 134.29 (CHAr), 132.52 (Cq), 132.17 (CHAr), 131.62 (CHAr), 129.37 (CHAr), 127.57 (CHAr), 128.14 (CHAr), 127.53 (CHAr), 127.50 (CHAr), 126.09 (CHAr), 121.17 (Cq).
1,1-Dibutyl-3-[(2-cyanophenylimino)phenylmethyl] thiourea (4e).
Compound 4e was prepared via procedure A by the reaction of 3 with dibutylamine. Yield: 0.28 g (32%); M.p. 134-135°C (DMF); Calc. for C23H28N4S (392.56): 70.37% C, 7.19% H, 14.27% N, 8.82% S; found: 70.21% C, 7.01% H, 13.95% N, 8.63% S; FTIR: 3210 (NH), 2955, 2927, 2869 (CH), 1597 (C=C); 1H-NMR (CF3COOD): 8.56– 7.78 (9H, m, ArH), 4.09 (4H, t, NCH2), 2.14-1.94 (4H, m, NCH2CH2), 1.70-1.55 (4H, m, CH2CH3), 1.11 (6H, t, CH2CH3); 13C-NMR (CF3COOD): 178.12 (C=S), 163.81 (N=C) 160.14 (Cq), 140.65 (CHAr), 139.64 (Cq), 138.50 (CHAr), 133.25 (CHAr), 132.47 (CHAr), 130.94 (CHAr), 128.07 (CHAr), 122.20 (CHAr), 117.80 (Cq), 107 (CN), 56.56 (NCH2), 56.10 (NCH2), 31.94 (NCH2CH2), 30.47 (NCH2CH2), 21.68 (CH2CH3), 14.23 (CH2CH3).
References
- Grasso, S.; Zappalá, M.; Chimirri, A. Heterocycles 1987, 26(9), 2477.
- Brown, D. J. Quinazolines, Supplement I (The Chemistry of Heterocyclic Compounds vol. 55); John Wiley & Sons: Chichester (U.K.), 1996. [Google Scholar]
- Abdel-Megeed, M.; Teniou, A. Rev. Roum. Chim. 1988, 33, 981.
- Ravina, A. J. Organic Preparations and Procedures Int. 1973, 5, 174.
- Stevens, G. Diuretics; Academic Press Inc.: New York, 1963; p. 12. [Google Scholar]
- Hayo, S.; Harera, H. J.; Strycher, W. G.; Honge, E. J. Med. Chem. 1969, 12, 936.
- Maillard, J.; Benard, M.; Vinent, M.; Von-Van-Tri, J. R.; Morin, R.; Benharkate, C.; Mennilet, C. Chim. Ther. 1967, 2, 231.
- Malhotra, S.; Kouls, K.; Sharma, R. L.; Anand, K. K.; Gupta, O. P.; Dhar, K. L. Ind. J. Chem. 1988, 27b, 937.
- Hess, H. J.; Cronin, T. H.; Sciabine, A. J. Med. Chem. 1968, 11, 130. [PubMed]
- Waisser, K.; Dostál, H.; Kubicová, L.; Kolář, K. Čes. a Slov. Farm. 2000, 49, 113.
- Murav’eva, K. M.; Akhangle, S. N. V.; Schukina, M. N.; Zykova, T. N.; Perchin, T. N. Khim. Farm. Zh. 1967, 1, 29.
- Leszkuvszky, C.; Erdely, I.; Tardos, L. Acta. Physiol. Acad. Sci. Hung. 1965, 27, 81.
- Gupta, C. M.; Hussain, S. T.; Bhaduri, A. P.; Khana, N. M.; Mukherjee, S. K. Nature 1969, 223, 524.
- Bodajla, M.; Stankovský, Š.; Špirková, K. Collect. Czech. Chem. Commun. 1995, 60, 1415. [CrossRef]
- Bodajla, M.; Stankovský, Š.; Špirková, K.; Jantová, S. Collect. Czech. Chem. Commun. 1996, 61, 1681. [CrossRef]
- Bodajla, M.; Stankovský, Š.; Jantová, S.; Hudecová, D.; Špirková, K. Chem. Papers 1996, 50(1), 28.
- Pazdera, P.; Nováček, E.; Kalviňš, I.; Trapencieris, P.; Pugovics, O. Chem. Papers 1991, 45, 527.
- Pazdera, P.; Ondráček, D.; Nováček, E. Chem. Papers 1989, 43(6), 771.
- The complete computational analysis of all the predicted structures and the similar structures reported by Stankovsky [14,15,16] are available from the authors.
- Sheldrick, G. M. Acta Crystallogr. Sect. A. 1990, 46, 467.
- Sheldrick, G. M. SHELXL93: Program for Structure Refinement; University of Göttingen: Göttingen, 1993. [Google Scholar]
- Osamu, K.; Shigeo, Y.; Toshiro, K. Agric. Biol. Chem. 1980, 44, 2143.
- Houghton, P. G.; Pipe, D. F.; Rees, C. W. J. Chem. Soc. Perkin Trans. I 1985, 7, 1471. [CrossRef]
- Sample Availability: Available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Fathalla, W.M.; Čajan, M.; Marek, J.; Pazdera, P. One-Pot Quinazolin-4-ylidenethiourea Synthesis via N-(2-Cyanophenyl)benzimidoyl isothiocyanate. Molecules 2001, 6, 574-587. https://doi.org/10.3390/60700574
AMA Style
Fathalla WM, Čajan M, Marek J, Pazdera P. One-Pot Quinazolin-4-ylidenethiourea Synthesis via N-(2-Cyanophenyl)benzimidoyl isothiocyanate. Molecules. 2001; 6(7):574-587. https://doi.org/10.3390/60700574
Chicago/Turabian StyleFathalla, Walid M., Michal Čajan, Jaromír Marek, and Pavel Pazdera. 2001. "One-Pot Quinazolin-4-ylidenethiourea Synthesis via N-(2-Cyanophenyl)benzimidoyl isothiocyanate" Molecules 6, no. 7: 574-587. https://doi.org/10.3390/60700574