Basic strategy
The basic idea of marvin is to automatically run a sequence of computer programs on a number of compounds. The functionality of the entire sequence is called algorithm in the context of marvin. The single programs are addressed by the term application. Therefore, a marvin algorithm is built up by putting together applications and running them on molecules. marvin is only the integration platform that links together all pieces of data and software needed in a chemoinformatics project.
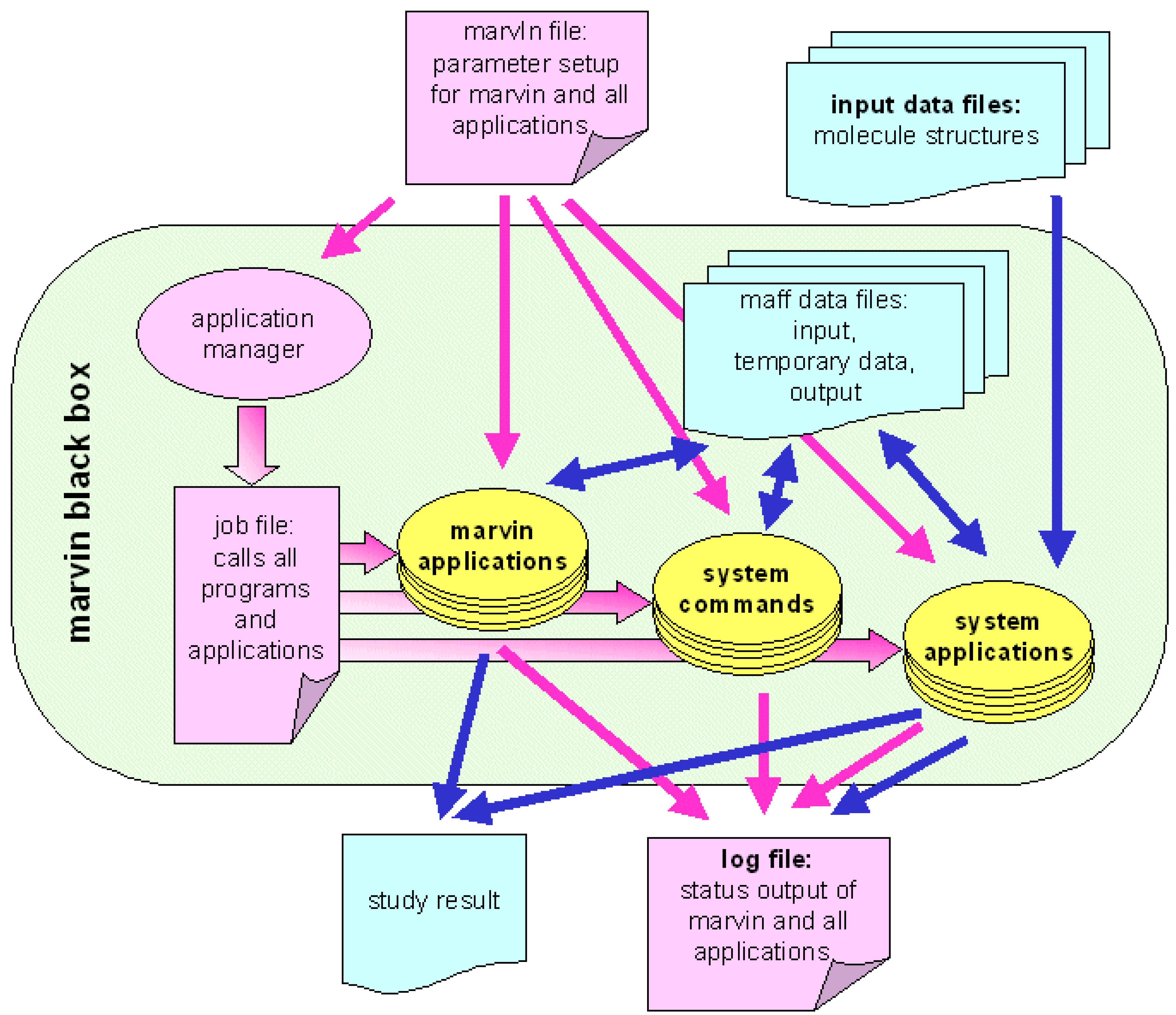
marvin as a black box
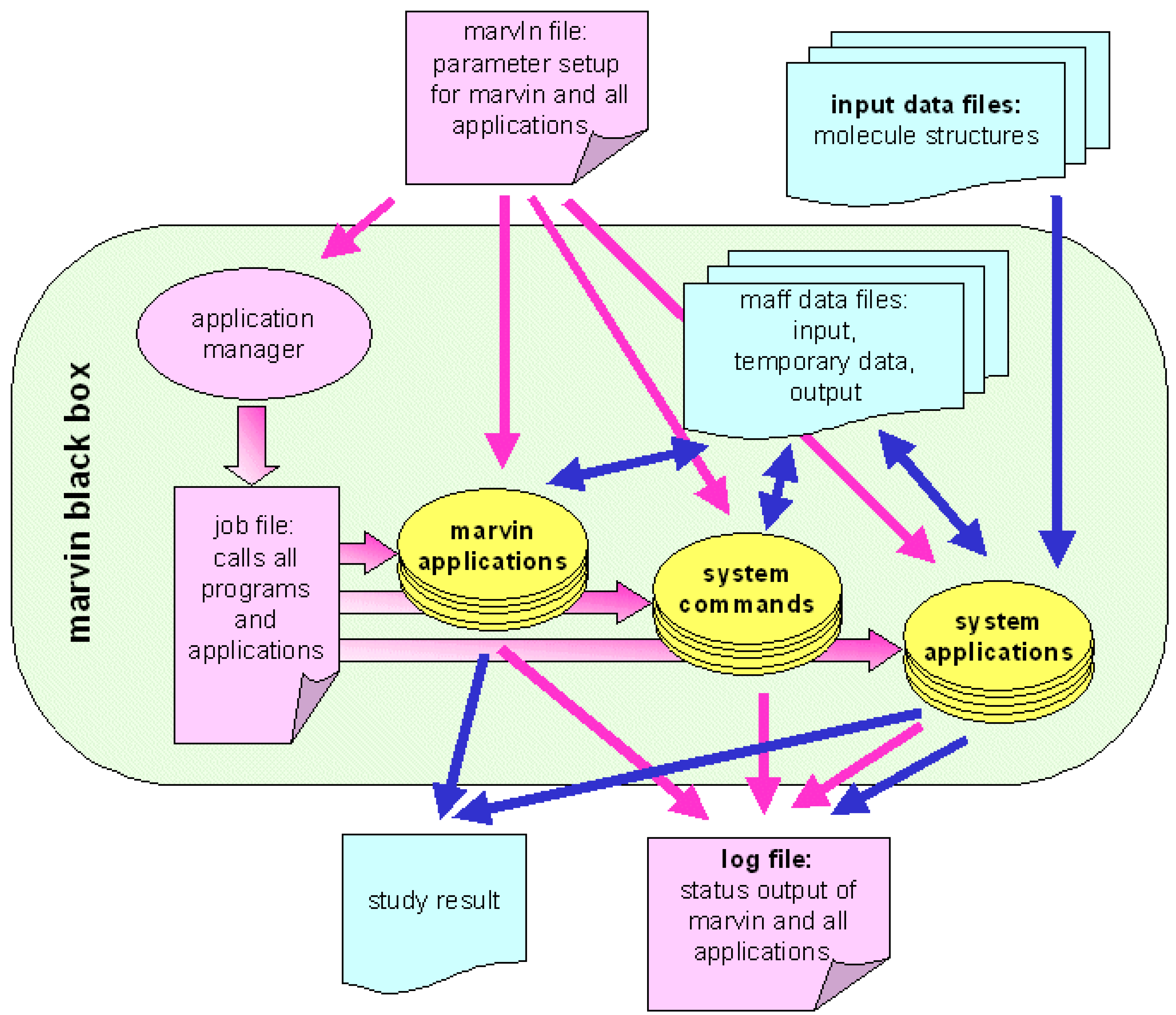
Because of the high level of automation, any marvin algorithm can be applied as a black box: Chemical structures are used as input and results of the entire study are presented as output. Usage as a black box is an important demand on an integrated chemoinformatics software solution, because applying a variety of computational methods, implemented in different software packages on different computational platforms, to millions of compounds would be a very time consuming job. High efficiency and fast application of computational methods however, is one of the major preconditions for chemoinformatics software within the strategy of NT drug discovery.
The concept of marvin allows manual setup and flexible optimization of an algorithm. Once it is tested and validated, even a complex algorithm can be run at the touch of a button without any user interaction until the final result is presented.
marvin as a box of bricks
Looking inside the black box,
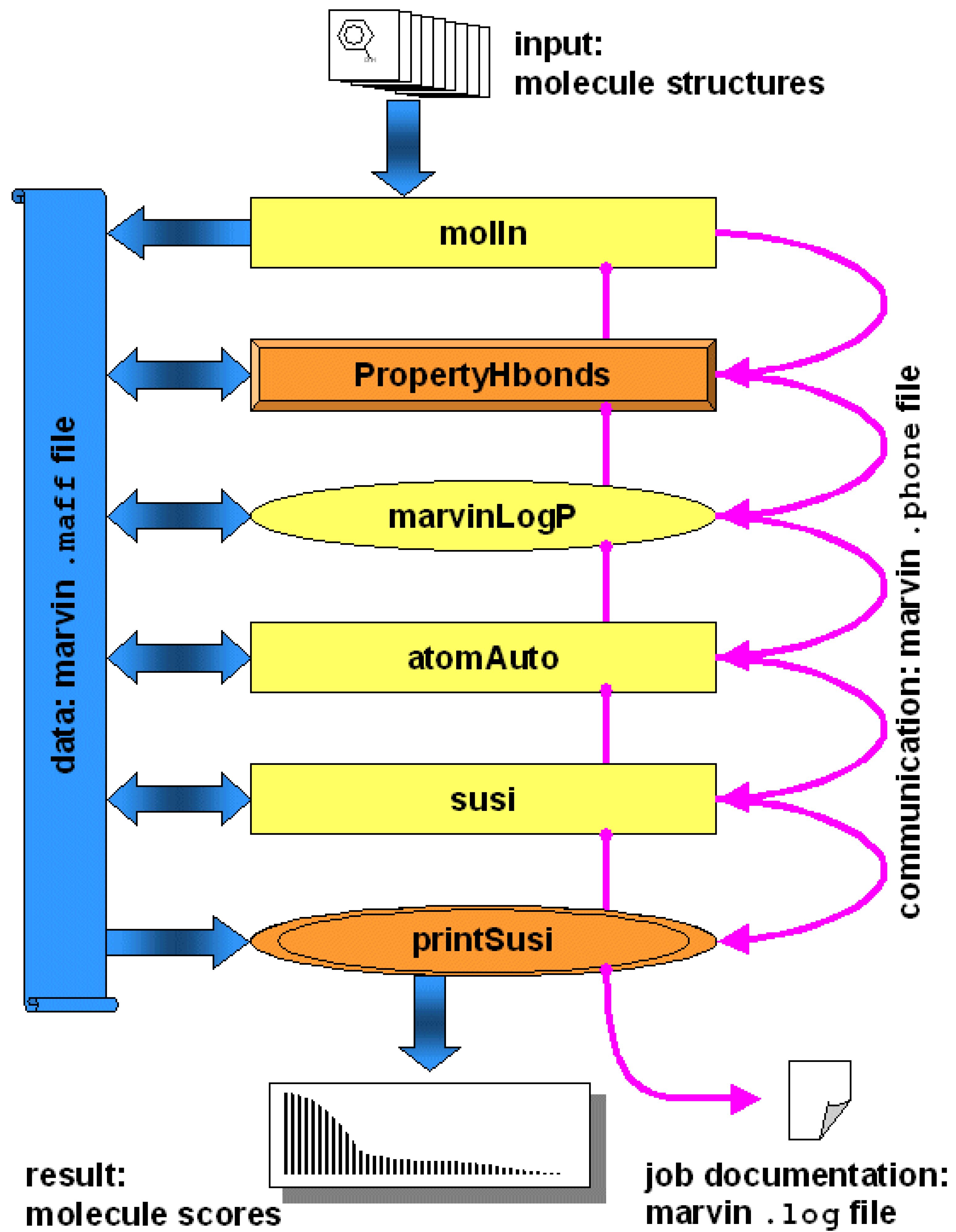
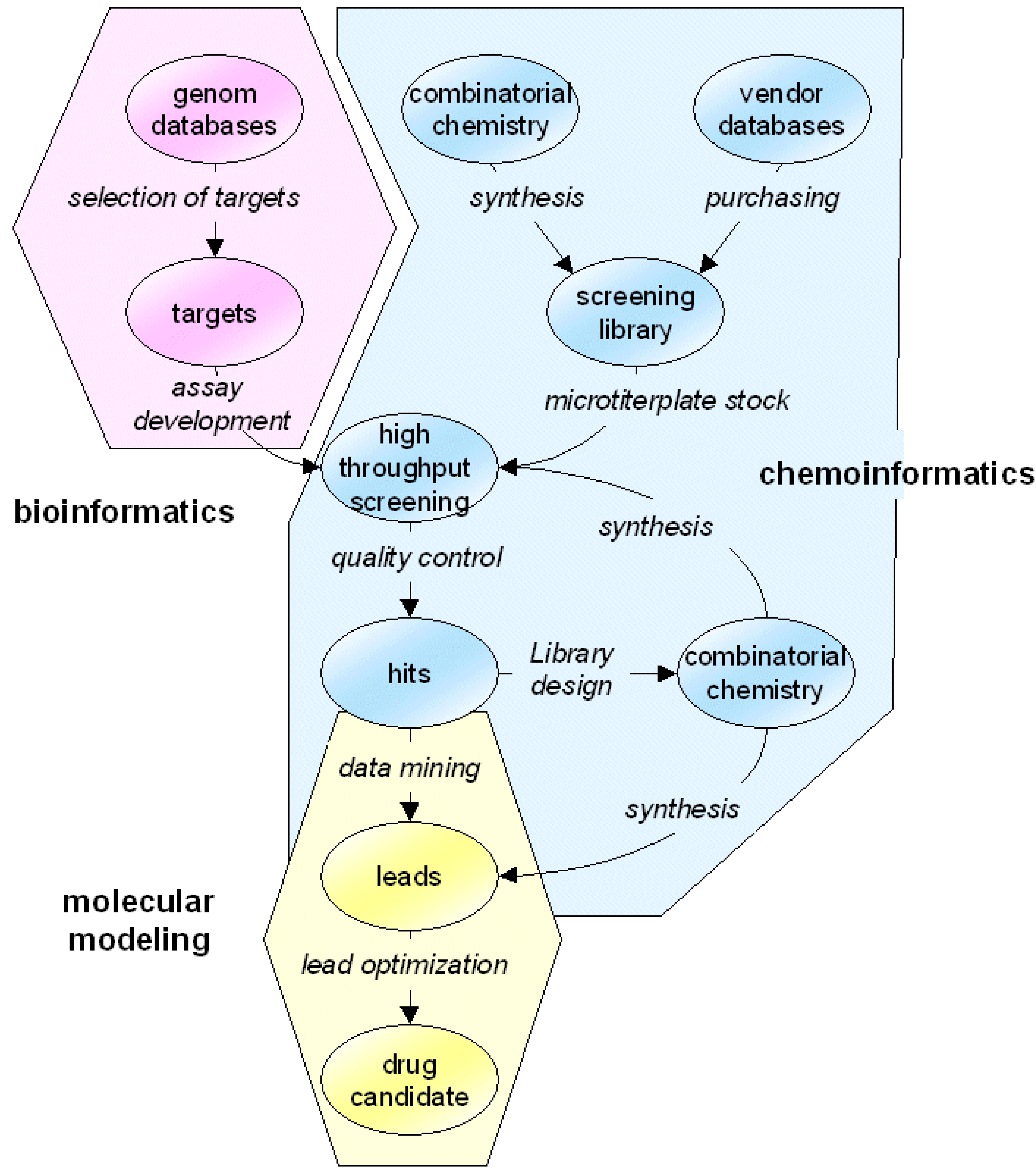
marvin presents itself as a box of bricks (see
Figure 2). All applications - the modules of the algorithm - are built up based on a small number of precisely defined interfaces. These interfaces are data file formats and communication file formats, handled by
marvin library functions. All communication between applications is performed via these files. This is why all applications can be designed individually and every application can be used together with any other one. There is no need to interface modules from different software suppliers - as long as integrating of the single modules into the integration platform is simple.
Before running an algorithm on this file based platform, the global setup file, that holds all runtime parameters for all applications of the algorithm, must be created. The library functions read this setup file and pass the parameters to the corresponding applications. Every application writes its output to data files which are readable by all other marvin applications. Additional inter-application communication is possible via the communication file, which is how error messages and warnings are passed from one application to another.
The status output (such as warnings and error messages) are written to a common output file that comprises a detailed documentation of the entire study. This status file is generated automatically.
Figure 2.
Visualization of the modular structure of a
marvin algorithm (see
Section 4 for the example study). Data sets are transferred between applications by so-called maff data files. Different types of applications, such as generic applications (displayed as yellow rectangles), interfaced applications (ovals) or high-level applications (in orange and 3D representation) are used seamlessly in order to include all functionality needed by the
marvin algorithm. Blue arrows indicate data flow, magenta arrows indicate control flow.
Figure 2.
Visualization of the modular structure of a
marvin algorithm (see
Section 4 for the example study). Data sets are transferred between applications by so-called maff data files. Different types of applications, such as generic applications (displayed as yellow rectangles), interfaced applications (ovals) or high-level applications (in orange and 3D representation) are used seamlessly in order to include all functionality needed by the
marvin algorithm. Blue arrows indicate data flow, magenta arrows indicate control flow.
marvin modules
mavin modules are the application manager (APM), applications, file format definitions and the marvin runtime library. The application manager reads basic parameters from the input files and sets up the entire marvin job.
Applications are computer programs, which address small functional parts of an algorithm (such as generation of 3-dimensional structure from molecular topology, calculate one molecular property, etc.).
The parameter input files contain all run-time parameters for all applications and the marvin specific setup.
The marvin run-time library is used to integrate new software into the marvin system. The library functions covers functionality concerning user interface, data input and output, protocol recording and documentation.
Application manager
The minimum requirement of an application manager is to apply a number of applications to a number of molecules. Beyond this, the marvin APM works as a data management and networking module by controlling host-to-host file transfers, batch job submission, inter-application communication etc. The marvin APM is controlled by keywords given in the input files (see Listing 2 for an example). The functionality of the marvin APM includes:
Multiple run modes (single, all, list): The application manager decides whether an application is to run with all molecules or just once for all molecules or only with a certain class of molecules (e.g. special handling of ionic compounds).
Network support: The APM finds applications in a local area network or the internet and starts them on the remote host. Data files are transferred if necessary, default settings are adapted and output files are concatenated automatically.
Integration of resources: Databases and other local or remote resources are included.
Commercial software packages: Functionality supported by commercial software packages is included. External packages can be run as interfaced applications as an integrated part of any marvin study.
Innovative applications: Novel and innovative applications can be implemented in c/c++ and seamlessly combined with the interfaced applications.
File format conversions: Necessary file-format conversions are recognized and performed automatically.
Runtime parameter organization: Every application is provided with the correct parameter input from the input file.
Checkpointing and restart of studies: Already calculated results are recognized and noticed (e.g. in order to restart stopped studies or rerun with only some of the application parameters changed).
Parallelisation: Time-consuming calculations are optimized by starting multiple instances of an application in parallel. This allows "cluster-like" parallelisation on multi-cpu computer servers. Individual applications need not to be parallelized by compiling special versions for specific host computers. Even software modules of which no parallelized versions are available (e.g. commercial software packages) can be run in parallel using this concept.
Batch processing: If processes are submitted to a batch queue, the APM waits for the completion of the job. The marvin algorithm is continued as soon as all needed results are available.
Documentation and error log: The APM generates a detailed documentation for the run of all executed applications. Errors, warnings, status output and cpu-times are reported to the marvin output file.
Optimized data management: Data files are stored in a compressed format and temporary files are removed automatically.
Fail save concepts: The APM recognizes technical problems and tries to find work-arounds or notifies the user.
Applications
marvin applications are built up as generic, as high-level or as interfaced applications.
Generic applications are programs developed for usage within
marvin algorithms by linking the
marvin runtime library. The functions and data types of the
marvin library can be used to handle all data and parameter input and output (see
Section 3.4 for details on the runtime library). Generic applications are most commonly used for external data interfaces, such as reading external file formats, or for implementing innovative computational methods.
In addition, most of the marvin system functions (such as the application manager) are implemented as generic applications.
High-level applications are defined in one of the marvin input files. High-level applications run any other marvin applications with a different set of default parameters (see Listing 1 for an example).
Interfaced applications are external software packages, integrated by using the generic application cmdLine [
24] or by implementing a generic
marvin interface application.
Example applications are given in
Table 1. All types of applications can be used to build up an algorithm, regardless of their type.
Table 1.
Example applications for use within a marvin algorithm
Table 1.
Example applications for use within a marvin algorithm
| Application | Type | Description |
| Add | Generic | Performs simple arithmetic operations on datasets |
| AutoAtom | Generic | Calculates autocorrelation coefficients based on 3-dimensional molecular structures and atomic properties (e.g. atomic point charges or extended properties) |
| Application | Type | Description |
| AutoScale | Generic | Scaling of all data sets in a study |
| AtomProperty | Generic | Calculates extended atom properies (e.g. lipophilicity, hydrogen bond acceptor/donor, etc.) |
| CmdLine | Generic | Interface to external command processor. cmdLine can be used to run external applications from within a marvin study |
| Derivat | Generic | Calculates the derivative of a multi-dimensional data set |
| ft2 | Generic | Calculates the fast fourier transformation of a multi-dimensional data set (forward real to complex and backward) |
| GnuPlot2 | Interfaced | Interface to gnuplot [30] program for plotting marvin data sets to a terminal, file or printer |
| MarvinBabel | Interfaced | Interface to the molecular structure file format translator babel [25] (babel reads and writes more than 60 file formats) |
| MarvinMOPAC | Interfaced | Interface to the quantum chemical program package MOPAC [23] |
| MarvinSybyl | Intefaced | Interface to the molecular modeling software package Sybyl |
| Mip | Generic | Calculates the molecular interaction potential as sum of the electrostatic potential and the 6-12-Lennard-Johnes potential in a rectangular box around a molecule |
| MkData | Generic | Generates a multidimensional dummy test-dataset (e.g. triangle, ramp, cube...) for software evaluation and testing |
| MolIn | Generic | Imports two- or three-dimensional molecular structures into marvin data files |
| MolOut2 | Generic | Exports molecule structures |
| NLM | Generic | Performs non-linear mapping of high-dimensional data sets into one-, two-, or three-dimensional maps |
| PotSelect | Generic | Applies individual scaling factors to the points of a 3-dimensional potential depending on their distance to atoms of specified types. The application is used to select regions in space around atoms or groups in a molecule molecule for further calculation. |
| QSARtable | Generic | Organizes data from all molecules of a study in a pivot table |
| Application | Type | Description |
| Scale | Generic | Implements several different methods for linear and non-linear scaling of data sets |
| Smooth | Generic | Implements several different methods for smoothing (e.g. integral-smoothing, fourier-filtering, etc.) of data sets |
| Susi | Generic | Calculates the similarity between a molecular structure and a cluster of structures |
marvin files
Communication between the different applications and between applications and APM is based on text files. Formats of these files are fixed and implemented in the marvin runtime library. Most important marvin files are input files (parameter setup), data files, phone files (communication between applications) and output files.
Data files
All
marvin applications output molecular data into a standard data file for every molecule (molecule.
maff =
marvin
file
format). Maff files are text-files that store molecule date and history information in a readable form. Therefore every data file includes a brief documentation displaying the applications, used to generate these data. Listing 6 shows an example maff data file from the example algorithm described in
Section 4. For optimized data management
marvin allows automatic compression and decompression of data files. Maff files may contain different types of information assigned to molecules, such as molecular topology (i.e. structural formula), three-dimensional structure, additional properties of the molecule or the atoms, tables of high-dimensional data (e.g. potentials, surfaces, etc.) and comments.
Communication file
The marvin communication file (study.phone) allows applications of a study to communicate to each other (e.g. an application can exclude some molecules from the data set at run-time, see Listing 9 for an example).
Output file
The marvin output file (study.log) comprises all status information from all applications, such as warnings, error messages, computation times, etc. The thoroughness of this information is adjusted by setting the logfile size: parameter in the %%setup section of the marvin input file (possible values are small, medium, big or debug, see Listings 7 and 8).
marvin run-time library
The marvin run-time library is a compilation of functions and data types that help software developers in implementing novel marvin applications. The library functions are designed to address problems of data handling, file handling, user interface and marvin system management.
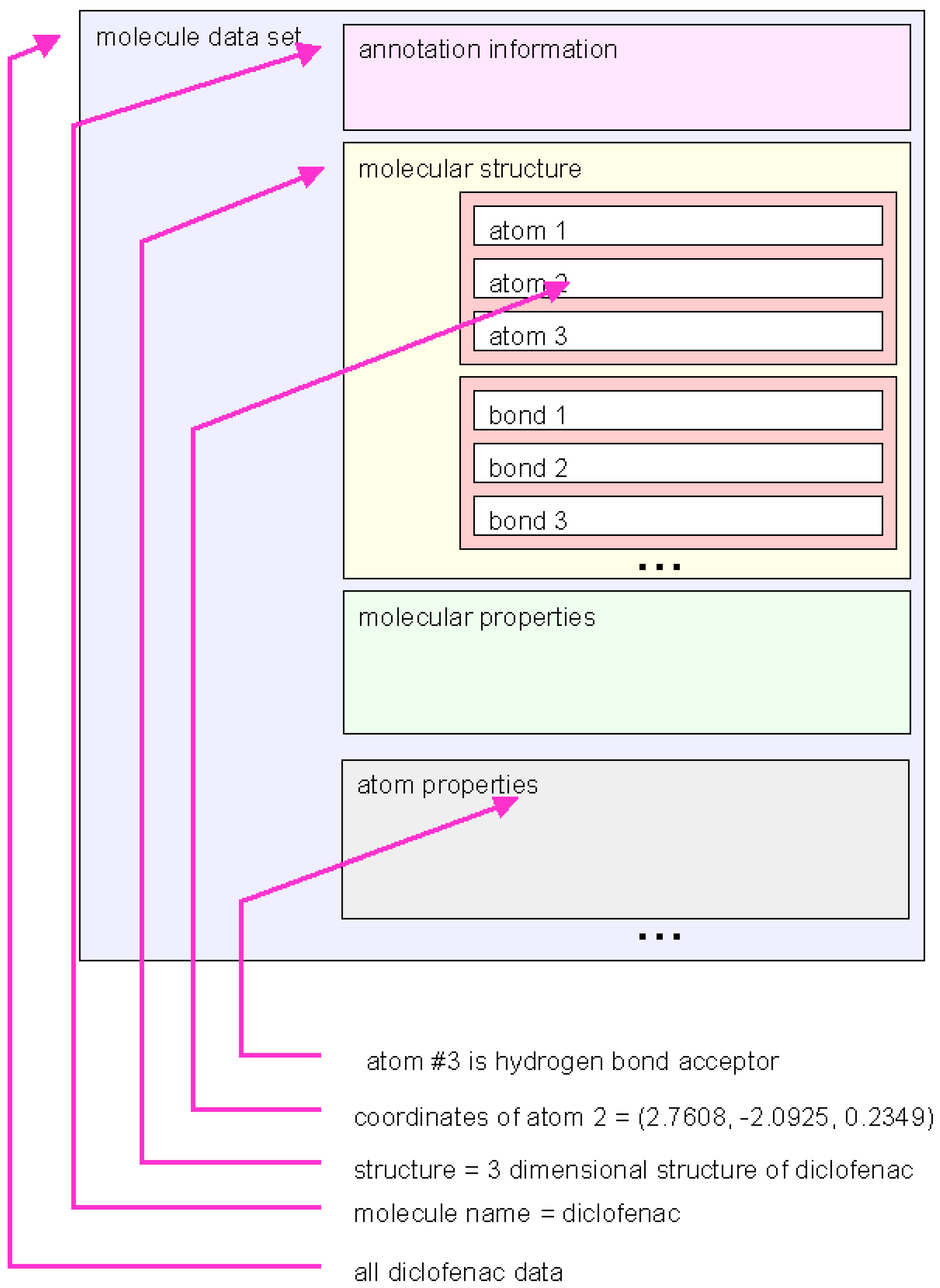
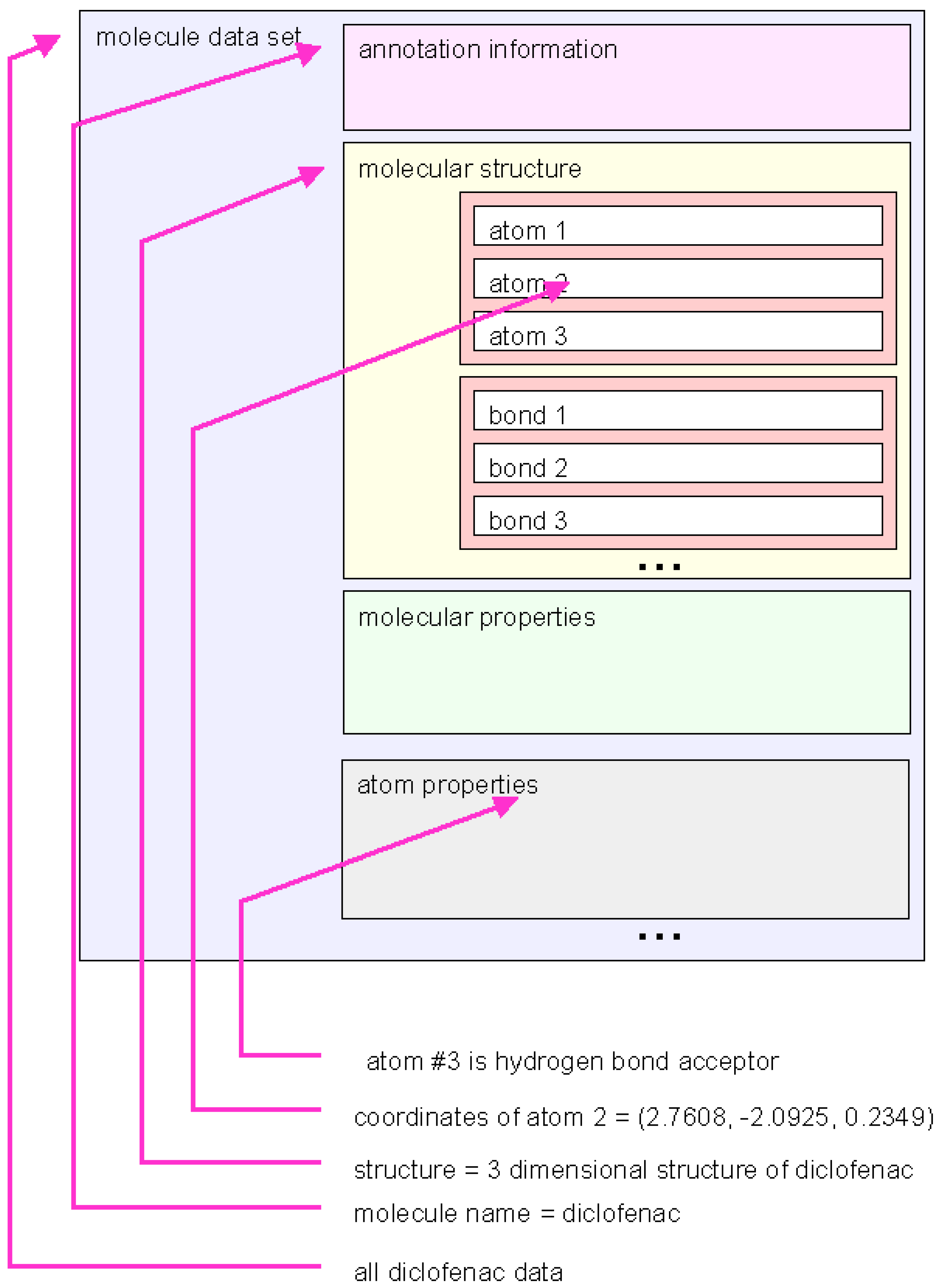
Data structures: marvin library functions are used to accessing molecular data stored in predefined data structures. The data show the same modular structure as the data files (see
Figure 4).
Figure 4.
Schematic representation of a marvin dataset. The molecule data can be handled on different levels of graining. The marvin library function provide access to many types of grouped information.
Figure 4.
Schematic representation of a marvin dataset. The molecule data can be handled on different levels of graining. The marvin library function provide access to many types of grouped information.
Different nesting levels of variables are used to address different levels of detail of information. In the highest hierarchical level entire sets of information about one molecule are handled as one object. But the interface gives handles to more fine-grained information, such as molecular structure, molecular properties or even properties of single atoms.
Functions for data handling: The data-handling library includes functions for writing and reading data from maff data files into predefined data structures and functions for managing these data.
marvin data structures include molecular data (e.g. atom coordinates, atom properties, topologies, molecular properties, etc) and high-dimensional vectors (e.g. potential fields, description vectors, etc).The functions allow accessing data in different ways, such as addressing values by index or by coordinates (see
Table 3 for example functions).
User interface functions: The user interface library includes functions to read run time parameters from one of the input files. The parameters must be specified by section name, parameter name and parameter element number in the parameter list. The routines are searching all
marvin input files for the demanded parameter hierarchically: If the parameter is not found in the setup of the current job (i.e. the
marvIn file), the file applications.defaults and – if necessary – the file local.defaults are searched. This way parameters are set to default settings automatically (see
Table 4 for example functions).
marvin system management: The
marvin system management library includes functions for setting and accessing information in the
marvin environment (see
Table 5 for example functions).
Table 3.
marvin runtime library: Example functions for data handling
Table 3.
marvin runtime library: Example functions for data handling
| Function | Description |
|---|
| ReadData(Name) | reads a molecular data set from maff file
Name |
| MarvinWriteData(Name) | writes a molecular data set into maff file
Name |
| MarvinGridPoint(Index) | returns the value referenced by index vector
Index |
| MarvinGridInterp(Coors) | returns the value of a n-dimensional vector at the point given by the coordinates
Coors by interpolating the grid |
| MarvinGridNumber(Coors) | returns a pointer to the data point next to the point defined by the coordinates
Coors in a n-dimensional vector |
| MarvinAtomProperty(Num) | returns the properties of atom number
Num |
| … | … |
Table 4.
marvin runtime library: Example user interface functions.
Table 4.
marvin runtime library: Example user interface functions.
| Function | Description |
|---|
| MarvinGetFloat( Sect, Para, Num, Val) | Reads the value at position
Num from the input parameter Par in the section Sect into the variable addressed by Val. |
| MarvinGetChar( Sect, Para, Num, Val) | Reads the first character of the word at position
Num from the input parameter Par in the section Sect into the variable addressed by Val (e.g. yes or no) |
MarvinGetInt(), MarvinGetString(),
MarvinGetNum(), MarvinGetList(),
MarvinGetLList(), MarvinGetLine(), … | Different types of run time parameters in the input files are accessible directly from within an application (e.g. words, lines, lists (as vectors), lists (as linked lists), etc. |
| MarvinLog(
size, string) | writes a message into the status output file if size matches the global setting of the current output detail level (one of small, medium, big, debug). E.g. the command MarvinLog( “d”, “The value of Errorlevel is -1”) will write the message into the log file only if the output detail level is set to debug. |
| MarvinError( Num, Mssg) | The function reports the error message corresponding to the error number
Num. The correct error message is assigned to Num and the string Mssg is printed as an additional explanation. |
| MarvinPercent( Actual, Total) | Reports the progress of a computation in percent. |
| ... | ... |
Table 5.
marvin runtime library: Example functions for marvin system management.
Table 5.
marvin runtime library: Example functions for marvin system management.
| Function | Description |
|---|
| MarvinStart() | Initializes all
marvin related settings. The marvin library functions can be used after call of MarvinStart(). |
| MarvinEnd() | Cleans up all data structures previously used by
marvin. |
| MarvinStop(Error) | Stops the execution of the current application or of the entire
marvin job, depending on the error level. |
| MarvinPhone(Recipient, Message, Signal) | Transmits a message to another application. |
| MarvinReadPhone() | Reads messages from other applications. |
| … | … |
Listings
The listings illustrate the way parameters are set in a marvin algorithm. All listings are clippings from the control files and data files of the example study.
Listing 1
Example definitions for nested high-level applications (file: application.defaults): The high-level application “mopacAM1” runs the external program mopac [
23] with the AM1 hamiltonian. The second high-level application “mopac-on-Sun” runs mopac with the same parameters on a remote host named bigsun. Both high-level applications refer to the interfaced application marvinMOPAC.
mopac high-level applications:
%%mopacAM1
default keys: AM1 PRECISE NOINTER \
MMOK GEOOK
geo opt: all
time: 3600 sec
default: marvinMOPAC
%%end of mopacAM1
%%mopac-on-Sun
machine: bigsun target host name
launch: run QUEUE run on normal queue
arguments: no
wait: yes wait for all mopac output files output
default: mopacAM1
%%end of mopac-on-Sun
the mopac interface application:
%%marvinMOPAC
run mode: files
remove: * .dat
MOPAC execution parameters:
mopac execute: $MARVIN_DIR/bin/mopac
batch queue: normal
wait interval: 1 48 h ask for mopac results every hour,
maximum wait time: 48 hours
mopac id: _mopac suffix of all temporary mopac files
result file name: arc
MOPAC default keywords:
hamilton: AM1
time: 172000 maximum mopac run time in seconds
geo opt: yes
default keys: NOINTER XYZ
mopac keys:
mopac comment line: mopac job from marvinMOPAC version 3.1
retrieve options:
retrieve: energy geo charges
%%end of marvinMOPAC
Listing 2
marvin input file for the example algorithm. Text outside a section or outside parameter definitions is ignored by the ReadParameter() routines of the marvin runtime library. Section %%setup contains marvin settings. Other sections (e.g. %%molIn, %%PropertyHbonds, %%marvinLogP, etc.) are used to define application specific setup.
selection of similar compounds from a database
%%setup
owner name: dominik
project: similarity selection
hold temp files: no
compress: yes
handle existing files: new
logfile size: small
system logfile: no
echo to stdout: yes
molecules = reference and
database (in separate file db.list)
molecules: diclofenac \
flufenamic_acid flurbiprofen \
ibuprofen indometacin \
ketoprofen meclofenamic_acid \
mefenamic_acid naproxen \
nabumeton piroxicam \
sulindac_sulfide tenoxicam \
meloxicam 6-mna
inline: db.list
program sequenz: molIn PropertyHbonds marvinLogP \
atomAutoCharge atomAutoHbond susi printSusi
%%end of setup
***** import molecules *****
%%molIn
paralell: 1
input type: mol (mdl mol file)
comment: molecule from db.list
%%end of molIn
***** h-bond property *****
%%PropertyHbonds
default: atomProperty
parallel: 8
machine: local
property: h-bond property to compute
%%end of PropertyHbonds
***** log P *****
%%marvinLogP
parallel: 8
machine: local
%%end of marvinLogP
***** autocorrelation *****
%%atomAutoCharge
default: atomAutoAll
property: charges use atom charges
%%end of autoAtomCharge
%%atomAutoHond
default: atomAutoAll
property: h-bond use extended property h-bond
%%end of autoAtomHbond
%%atomAutoAll
parallel: 1
machine: computeServerSGI
range: 5 20 20 calculate 20 autocorrelation coefficients
for distances between 5 and 20 angstroems)
%%end of autoAtomAll
***** susi *****
%%susi
machine: local
dataset number: 3 (use the 3. data set in maff file)
use the first 15 molecules as reference:
reference: 1 2 3 4 5 6 7 8 9 10 \
11 12 13 14 15
scaling: 0.5
score function: linear
%%end of susi
***** print susi results *****
%%printSusi
machine: computeServerSGI
default: GnuPlot
title string: susi of similarity dataset
set title: yes setname owner name date
printer: ps001
%%end of printSusi
end of marvin input file
ADo.
Listing 3
marvin setup file local.defaults. Pathnames and specific settings of the local marvin installation and remote host information are given in this file.
************************************************************************
* marvin.defaults *
************************************************************************
%%local
local host: myWorkstation
marvin dir: /usr1/local/Soft/marvin
tables dir: /usr1/local/Soft/marvin/tables
compress: compress -f
uncompress: uncompress -f
remove: rm -rf
remote execute: rsh
remote copy: rcp
jobfile shell: /sbin/csh
system applications: setup cpuTime wait
maff version: maff by ADo: Version 4.20
read maff versions: maff by ADo: Version 4.x
%%end of local
%%setup
machine: local
execute: $MARVIN_DIR/bin/setup
launch:
arguments:
parallel: 1
maff status line: marvin application manager
maff history line: setup: marvin application manager, 0.1v4.0
%%end of setup
%%computeServerSGI
work dir: $MARVIN_DIR/tmp/NetWork
login script: .login
network log: computeServerSGI.netlog
login name: sgi1 -l dominik -n
rcp adress: dominik@sgi1
remove: rm -rf
%%end of computeServerSGI
%%molIn
machine: local
execute: $MARVIN_DIR/bin/molIn
launch:
arguments:
parallel: 1
table for Sybyl atom types: $MARVIN_DIR/tables/molIn/AtomTypesSybyl.table
table for Sybyl bond types: $MARVIN_DIR/tables/molIn/BondTypesSybyl.table
maff status line: molecular dataset imported
maff history line: molIn: marvin input interface, 0.11v3.1
%%end of molIn
%%autoAtom
machine: local
execute: $MARVIN_DIR/bin/autoAtom
launch:
arguments:
parallel: 1
maff status line: atom based autocorrelation function
maff history line: autoAtom: autocorrelation, 0.31v1.2
%%end of autoAtom
%%marvinMOPAC
machine: computeServerSGI
execute: $MARVIN_DIR/bin/marvinMOPAC
launch: queue
arguments: -q long
parallel: 8
maff status line: MOPAC V6.0 interface
maff history line: marvinMOPAC: interface to MOPAC V6.0, 0.12v4.1
%%end of marvinMOPAC
...
Listing 4
The marvin setup file application.defaults. Default settings for all marvin applications are defined in this file.
************************************************************************
* application.defaults *
************************************************************************
%%setup
echo to stdout: yes
logfile size: big
system logfile: none
hold temp files: yes
compress: yes
handle existing files: new
owner name: Andreas Dominik
project: unknown
%%end of setup
%%wait
interval: 1 sec
%%end of wait
%%molIn
run mode: files
remove:
format: detect
need all molecules: yes
comment: imported molecule!
%%end of molIn
%%atomAtuo
run mode: files
remove:
range: 5 15 20 range and grid spacing of
autocorrelation (20 coefficients in a
distance of 5 – 20 A)
dataset: 1 number of data set in data file
autocorrelation function: product
%%end of autoAtom
...
Listing 5
marvin job file for the example algorithm. This shell script is automatically gerenated by the application manager. It runs all applications for the molecules of the study. In the example the application molIn is executed on a single CPU of the local host. The application PropertyHbonds runs on eight molecules in parallel and the example application atomAuto is executed on the remote host “sgi1” that is configured as computeServerSGI (see file local.defaults, Listing 3).
#!/sbin/csh
#******************************************************************
# *
# *
# marvin - job - file *
# *
# *
#******************************************************************
#
#
# run molIn:
#
uncompress -f diclofenac.mol2.Z
$MARVIN_DIR/bin/molIn molIn diclofenac similarity 1
compress -f diclofenac.mol2
compress -f diclofenac.molecule
#
# run molIn:
#
uncompress -f flufenamic_acid.mol2.Z
$MARVIN_DIR/bin/molIn molIn flufenamic_acid similarity 1
compress -f flufenamic_acid.mol2
compress -f flufenamic_acid.molecule
#
# run molIn:
#
uncompress -f flurbibrofen.mol2.Z
$MARVIN_DIR/bin/molIn molIn flurbibrofen similarity 1
compress -f flurbibrofen.mol2
compress -f flurbibrofen.molecule
#
...
#
# run PropertyHbonds:
#
uncompress -f diclofenac.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds diclofenac similarity 1 &
#
uncompress -f flufenamic_acid.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds flufenamic_acid similarity 2 &
#
uncompress -f flurbibrofen.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds flurbibrofen similarity 3 &
#
uncompress -f ibuprofen.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds ibuprofen similarity 4 &
#
uncompress -f indometacin.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds indometacin similarity 5 &
#
uncompress -f ketoprofen.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds ketoprofen similarity 6 &
#
uncompress -f meclofenamic_acid.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds meclofenamic_acid similarity 7 &
#
uncompress -f mefenamic_acid.molecule.Z
$MARVIN_DIR/bin/atomProperty PropertyHbonds mefenamic_acid similarity 8 &
#
$MARVIN_DIR/bin/wait wait similarity similarity diclofenac.hb
compress -f diclofenac.hb
rm -f diclofenac.molecule
$MARVIN_DIR/bin/wait wait similarity similarity flufenamic_acid.hb
compress -f flufenamic_acid.hb
rm -f flufenamic_acid.molecule
$MARVIN_DIR/bin/wait wait similarity similarity flurbibrofen.hb
compress -f flurbibrofen.hb
rm -f flurbibrofen.molecule
$MARVIN_DIR/bin/wait wait similarity similarity ibuprofen.hb
compress -f ibuprofen.hb
rm -f ibuprofen.molecule
$MARVIN_DIR/bin/wait wait similarity similarity indometacin.hb
compress -f indometacin.hb
rm -f indometacin.molecule
$MARVIN_DIR/bin/wait wait similarity similarity ketoprofen.hb
compress -f ketoprofen.hb
rm -f ketoprofen.molecule
$MARVIN_DIR/bin/wait wait similarity similarity meclofenamic_acid.hb
compress -f meclofenamic_acid.hb
rm -f meclofenamic_acid.molecule
$MARVIN_DIR/bin/wait wait similarity similarity mefenamic_acid.hb
compress -f mefenamic_acid.hb
rm -f mefenamic_acid.molecule
#
cat similarity.log similarity.log.1 similarity.log.2 similarity.log.3 similarity.log.4 similarity.log.5 similarity.log.6 similarity.log.7 similarity.log.8 > marvin_tmp_logfile.log
#
mv marvin_tmp_logfile.log similarity.log
rm -f similarity.log.1 similarity.log.2 similarity.log.3 similarity.log.4 similarity.log.5 similarity.log.6 similarity.log.7 similarity.log.8
#
...
# run atomAutoCharge:
#
uncompress -f diclofenac.logp.Z
rsh sgi1 -l dominik -n 'source .login ; mkdir $MARVIN_DIR/tmp/NetWork' >>& computeServerSGI.netlog
rsh sgi1 -l dominik -n 'source .login ; rm –rf $MARVIN_DIR/tmp/NetWork/similarity' >>& computServerSGI.netlog
rsh sgi1 -l dominik -n 'source .login ; mkdir $MARVIN_DIR/tmp/NetWork/similarity ' >>& computeServerSGI.netlog
rcp 'diclofenac.logp' 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/'
rcp 'similarity.marvIn' 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/'
rcp 'similarity.log' 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/'
rcp 'similarity.phone' 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/'
rcp '$MARVIN_DIR/tables/marvin/application.defaults' 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/home.defaults'
echo "application is running on remote host computeServerSGI " >> similarity.log
rsh sgi1 -l dominik -n 'source .login ; cd $MARVIN_DIR/tmp/NetWork/similarity; $MARVIN_DIR/bin/atomAuto atomAutoCharge diclofenac similarity 1 ' >>& computeServerSGI.netlog
rsh sgi1 -l dominik -n 'source .login ; cd $MARVIN_DIR/tmp/NetWork/similarity; $MARVIN_DIR/bin/wait wait similarity similarity diclofenac.aac >>& computeServerSGI.netlog
rcp 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/ diclofenac.aac' '.'
rcp 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/similarity.log_1' '.'
rcp 'dominik@sgi1:$MARVIN_DIR/tmp/NetWork/similarity/similarity.phone' '.'
compress -f diclofenac.aac
rm -rf diclofenac.logp
cat similarity.log similarity.log.1 > marvin_tmp_logfile.log
mv marvin_tmp_logfile.log similarity.log
rm -f similarity.log.1
#
...
#
# write end logfile
#
$MARVIN_DIR/bin/cpuTime cpuTime similarity similarity
echo "marvin job similarity finished on host local" |
mail -s "similarity.marvIn" $USER
Listing 6
marvin data file from the example study. The file header includes a brief documentation of status and history of the data. Header information (%%comment and %%history sections) is generated automatically by the library functions. The file holds four data sets in total, including molecular structure (data type mol), clogP value (data type table, one row and one column only) and the autocorrelation coefficients (data type grid) from hydrogen bond properties and atomic charges.
%%maff by ADo: Version 3.x
%%diclofenac.aac2
%%autocorrelation coefficients calculated
%%begin of comment
molecule from db.list
reference molecule for susi similarity scores
%%end of comment
%%begin of history
diclofenac.molecule: molIn: import molecule: 0.11v2.1
Mon May 31 15:00:22 1999
diclofenac.hb: atomProperty: calculate extended atom property: 0.33v1.0
Mon May 31 15:04:13 1999
diclofenac.logp: marvinLogP: calculate clogP value: 0.34v0.9
Mon May 31 15:48:34 1999
diclofenac.aac: atomAuto: atom based autocorrelation: 0.32v1.6
Mon May 31 16:03:41 1999
diclofenac.aac2: atomAuto: atom based autocorrelation: 0.32v1.6
Mon May 31 17:35:13 1999
%%end of history
dataset type: mxgg
number of datasets: 4
%%begin of dataset
dataset number: 1
dataset type: m
dataset name: diclofenac with extended properties
energy type: none
energy: 0
charges type: GASTEIGER
extended properties: 1
number of atoms: 30
number of bonds: 31
%%begin of atoms:
num type X Y Z charge name
1 604 4.0891 -2.4130 0.5652 0.0636 C1
2 604 2.7608 -2.0925 0.2349 0.0002 C2
3 706 5.0994 -1.5224 0.3778 -0.2766 N1
4 604 4.3840 -3.6761 1.0970 -0.0456 C3
5 604 1.7701 -3.0763 0.3662 -0.0678 C4
6 601 2.3982 -0.6806 -0.2673 0.0791 C5
7 604 6.3556 -1.9372 0.0813 0.0852 C6
8 604 3.3920 -4.6493 1.2238 -0.0789 C7
9 604 2.0830 -4.3516 0.8417 -0.0804 C8
10 602 0.9569 -0.5015 -0.6750 0.2409 C9
11 604 7.4428 -1.4508 0.8180 0.0613 C10
...
%%end of atoms
%%begin of bonds
number from to type
1 1 2 16
2 1 3 10
3 1 4 16
4 2 5 16
5 2 6 10
6 3 7 10
7 4 8 16
8 5 9 16
9 6 10 10
10 7 11 16
11 7 12 16
12 10 13 10
13 10 14 20
14 11 15 16
...
%%end of bonds
%%begin of property:
property type: h-bond
property name: h-bond
0
0
-1
0
0
0
0
0
0
0
0
0
-1
-1
0
0
0
0
0
0
0
0
0
0
0
1
0
0
0
%%end of property
%%end of dataset
%%begin of dataset
dataset number: 2
dataset type: x
dataset name: clogP
number of columns: 1
%%X1
min: 4.711
max: 4.711
number of points: 1
%%begin of data
4.711
%%end of data
%%end of dataset
%%begin of dataset
dataset number: 3
dataset type: g
dataset name: autocorrelation coefficients
number of axis: 2
%%X1:
min: 5
max: 20
grid points: 20
%%X2 (values):
min values: -0.350815
max values: 0.837681
number of points: 20
%%begin of data
0.837681
0.767013
0.599279
0.34065
-0.0288451
-0.280594
-0.350815
-0.33142
-0.277938
-0.21335
-0.149994
-0.0906098
-0.0420099
-0.0112209
0.00534179
0.0108576
0.0108935
0.00837755
0.00577135
0.00371506
%%end of data
%%end of dataset
%%begin of dataset
dataset number: 4
dataset type: g
dataset name: autocorrelation coefficients
number of axis: 2
%%X1:
min: 5
max: 20
grid points: 20
%%X2 (values):
min values: -0.0833
max values: 1.0000
number of points: 20
%%begin of data
0.7083
1.0000
0.7500
0.4166
-0.1667
-0.0833
-0.0833
-0.0416
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
0.0000
%%end of data
%%end of dataset
%%end of file
Listing 7
Clippings from the marvin status output file of the example study recorded with log mode small. Log mode small is the default for running studies with a validated and tested algorithm.
filename: similarity.log
*****************************************************************
* *
* marvin - logfile *
* *
*****************************************************************
* job: similarity
* owner: dominik
* project: similarity selection
*****************************************************************
* files in fileset:
* diclofenac
* flufenamic_acid
...
* programs of the job:
* molIn
* PropertyHbonds
* marvinLogP
* atomAutoCharge
* atomAutoHbond
* susi
* printSusi
*****************************************************************
application: molIn
host: myWorkstation
file: diclofenac
at: Mon May 31 15:00:21 1999
molecule diclofenac imported!
CPU-time used by Marvin: 0.01 sec
application: molIn
host: myWorkstation
file: flufenamic_acid
at: Mon May 31 15:00:21 1999
molecule flufenamic_acid imported!
CPU-time used by Marvin: 0.01 sec
...
application: PropertyHbonds
host: myWorkstation
file: diclofenac
at: Mon May 31 15:04:13 1999
extended property hb calculated for molecule diclofenac!
CPU-time used by Marvin: 0.04 sec
...
application: susi
host: myWorkstation
file: similarity
at: Mon May 31 18:09:23 1999
susis calculated for 5015 structures:
rank name susi
1 flufenamic_acid 1.0000 (ref)
2 ibuprofen 1.0000 (ref)
3 naproxen 1.0000 (ref)
4 nabumeton 1.0000 (ref)
5 piroxicam 1.0000 (ref)
6 tenoxicam 1.0000 (ref)
7 meloxicam 1.0000 (ref)
8 6-mna 1.0000 (ref)
9 flurbiprofen 0.9986 (ref)
10 ketoprofen 0.9865 (ref)
11 diclofenac 0.9855 (ref)
12 meclofenamic_acid 0.9531 (ref)
13 mefenamic_acid 0.9145 (ref)
14 indometacin 0.8722 (ref)
15 sulindac_sulfide 0.8548 (ref)
16 db567 1.0000
17 db571 1.0000
18 db572 1.0000
19 db573 1.0000
20 db586 1.0000
...
5013 db4998 0.0000
5014 db4999 0.0000
5015 db5000 0.0000
CPU-time used by Marvin: 32.40 sec
...
**
* Total CPU time used by marvin: 10713.36 sec
* no error in marvin job!
* marvin done.
Listing 8
Clippings from the marvin status output file of the example study recorded with log mode medium. Log mode medium and log mode large are used for validating an algorithm or a single application. All parameters read by the marvin library functions are echoed to the log file.
application: molIn
host: myWorkstation
file: diclofenac
at: Mon May 31 20:10:01 1999
read: input(2) = .mol2
read: output(2) = .molecule
read: format(1) = detect
read: table for Sybyl atom types(1) = $MARVIN_DIR/tables/molIn/AtomTypesSybyl.table
read: table for Sybyl bond types(1) = $MARVIN_DIRmarvin/tables/molIn/BondTypesSybyl.table
Reading molecule from Sybyl-.mol2 file: diclofenac.mol2
writing maff-file diclofenac.molecule
Molecule imported: diclofenac.molecule
CPU-time used by Marvin: 0.090000 sec
...
application: PropertyHbonds
host: myWorkstation
file: diclofenac
at: Mon May 31 20:34:35 1999
read: input(2) = .lipo
read: output(2) = .hbond
read: molecule(1) = 1
read: property(1) = h-bonds
reading maff-file diclofenac.lipo
Property calculated for molecule diclofenac
writing maff-file diclofenac.hbond
CPU-time used by Marvin: 0.080000 sec
...
application: autoAtomHbond
host: computeServerSGI
file: diclofenac
at: Thu Apr 20 16:01:16 2000
read: input(2) = .hbond
read: output(2) = .aac
read: dataset(1) = 1
read: replace(1) = n
read: range(1) = 5
read: range(2) = 20
read: range(3) = 20
read: autocorrelation function(1) = product
read: property(1) = 1
read: scaling(1) = y
read: scaling(2) = 1.0
reading maff-file diclofenac.hbond
Calculate autokorrelation
input name: diclofenac, (30 atoms) based on property hbond profile
output: 5.000000 A - 20.000000 A, (20 points, 20 total)
Default autokorrelation
...
Listing 9
Example marvin phone file for communication between individual applications.
filename: similarity.phone
*****************************************************************
*
* marvin - phone-file
*
*****************************************************************
%%signal
from: marvinInput
to: all
signal: exclude tenoxicam
%%end of signal
%%signal
...


{kind=link}
{kind=link}
{kind=link}
{kind=link}