Result and Discussion
Photodimerization of compounds
5a-d was carried out by the action of sunlight in 0.1M methanol solution and the results are given in
Table 1. In order to explore the influence of the experimental conditions on the reaction products, photodimerization of the bromo derivative
5c was also performed in solvents with different polarities, (Methods A-D), and the corresponding results are given in
Table 2.
Table 1.
Photochemical dimerization of 3-substituted 2-oxo-2H-1,2-benzoxaphosphorines 5a-d
Table 1.
Photochemical dimerization of 3-substituted 2-oxo-2H-1,2-benzoxaphosphorines 5a-d
| 5 | R | X | Reaction time | Yields, % | 6:7 |
|---|
| Unreacted starting compd. | Overall | 6 | 7 |
|---|
| a | CH3 | H | 40 days
90 days | 24
20 | 45
44 | 56
68 | 44
32 | 1.3:1
2.1:1 |
| b | C2H5 | H | 90 days | 7 | 71 | 72 | 28 | 2.6:1 |
| c | C2H5 | 6-Br | 42 days | - | 89 | 56 | 44 | 1.3:1 |
| d | C2H5 | 6-Cl | 20 days
50 days | 51
- | 26
92 | 68
60 | 32
40 | 2.1 :1
1.5 :1 |
| e | C2H5 | 7-N(C2H5)2 | 90 days | 72 | * | - | - | - |
Table 2.
Photochemical dimerization of ethyl 6-bromo-2-ethoxy-2-oxo-2H-1,2-benzoxaphosphorine-3-carboxylate 5c in various solvents.
Table 2.
Photochemical dimerization of ethyl 6-bromo-2-ethoxy-2-oxo-2H-1,2-benzoxaphosphorine-3-carboxylate 5c in various solvents.
| Method | Reaction conditions | Yields, % | 6c : 7c |
|---|
| Overall | 6c | 7c |
|---|
| A | 5 mL spectr. grade CH3OH, 42 days | 89 | 56 | 44 | 1.3:1 |
| B | 2 mL dry benzene, 55 days | 83 | 64 | 36 | 1.8:1 |
| C | 1.5 mL glacial CH3COOH, 4 days | 94 | 76 | 24 | 3.2:1 |
| D | suspension in 1.5 mL water, 14 days | 83 | 72 | 28 | 2.6:1 |
As it is seen from
Table 1 and
Table 2, in all cases the photodimerization reaction proceeds with complete regio- and stereoselectivity, giving only the
anti head-to-tail isomers
6 or
7. However, with regards to the phosphorous stereogenic centre, both diastereomers
6 and
7 were isolated. The symmetric
exo,exo-6,12-diethoxyderivatives
6 prevailed in all cases over the non symmetric
exo,endo-6,12-diethoxyderivatives
7, with the
6/
7 ratio being between 1.3:1 to 3.2:1 (
Table 1 and
Table 2). The separation of the diastereomers and their cleanup was carried out by multi-stage column chromatography. Here it should be noted that the alternative centrosymmetric diastereomer, i.e.
endo,endo-6,12-diethoxyderivative
8 was not detected among the reaction products.
In almost all experiments some amount of the starting material was isolated (see
Table 1). The higher overall yields of dimers were obtained from the reaction of the halogenated 6-bromo and 6-chloro derivatives
5c and
5d (89% and 92%), respectively (
Table 1), whereas the higher yields and reaction rates of dimerization for the 6-bromoderivative
5c were observed in the polar solvents, e.g. in acetic acid (94% of dimer in 4 days, Method C), versus those in benzene (83% of dimer in 55 days, Method B) (
Table 2).
It was also found that ethyl 7-N,N-dietylamino-2-ethoxy-2-oxo-2
H-1,2-benzoxaphosphorine-3-carboxylate
5e is very stable towards sunlight irradiation in methanol solution. In this case the starting compound
5e (72%) and a small amount (9%) of the 6-ethylaminoderivative
5f (as a result of dealkylation of the starting compound
5e) were isolated from the reaction mixture after 90 days. Such dealkylations have been described earlier [
23,
24] in irradiation of 7-N,N-diethylaminocoumarins.
From the experimental data given above it is obvious that the studied photodimerization of the esters of 2-alkoxy-2-oxo-2
H-1,2-benzoxaphosphorine-3-carboxylic acid
5a-d, contrary to the corresponding coumarin reactions, proceeds with high regioselectivity to the formation of the head-to-tail dimers. Moreover, the [2+2]-photodimerization of
5a-d is also a stereoselective reaction and leads to the formation of the
anti head-to-tail, but not
syn head-to-tail, dimer. The stereoselectivity of the above photodimerization is also apparent in respect to the stereogenic P2-atom of compounds
5. Of the corresponding three possible diastereomers, i.e.
6,
7 and
8, the formation of the symmetric
6 predominated (
Table 1 and
Table 2) whereas the symmetric
8, with the two alkoxy groups directed over the cyclobutane ring, were not detected in the reaction products.
It is not possible to give any strong evidence supporting a particular mechanism for the above [2+2] photodimerization. According to the classical mechanism [
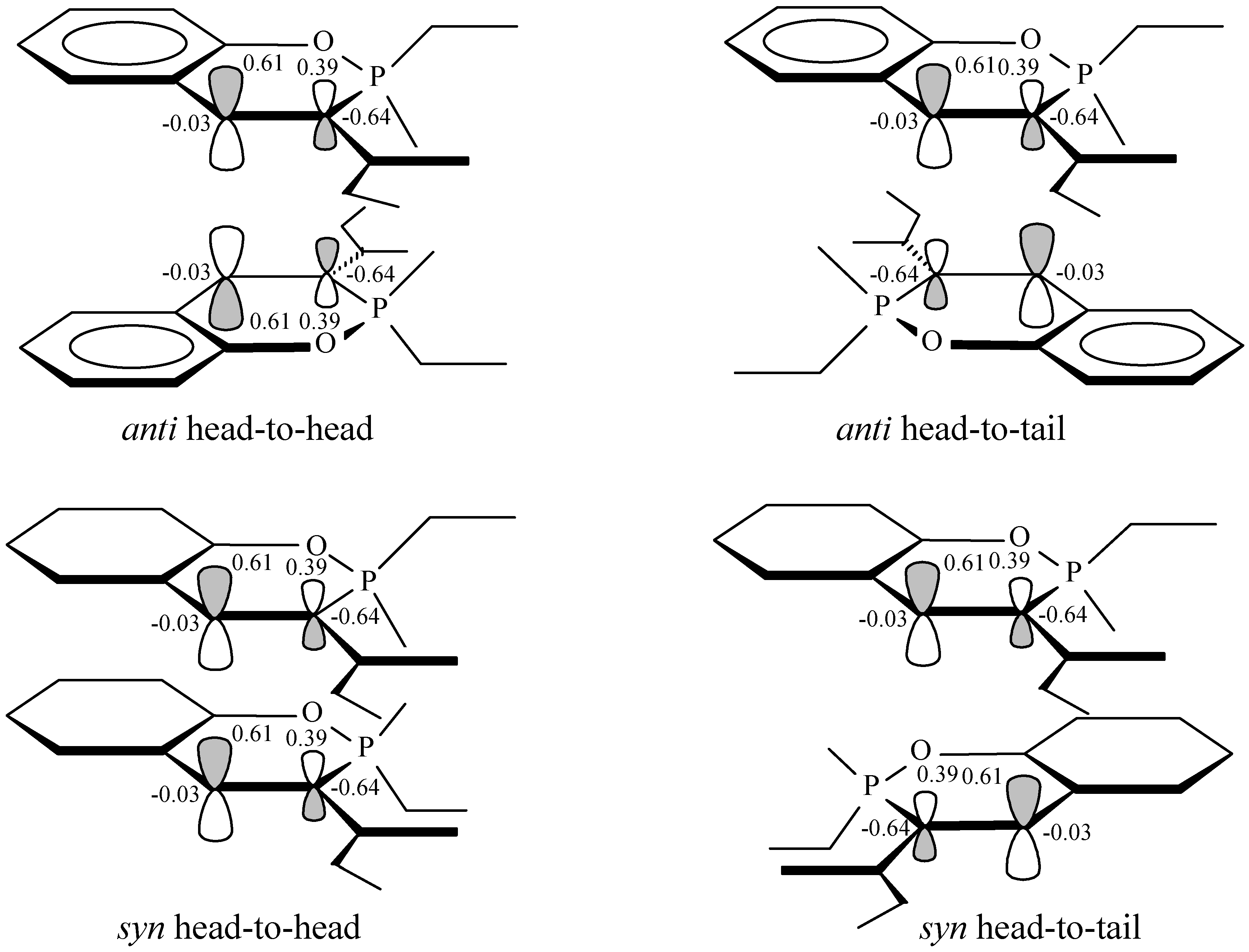
25], the synchronous [2+2] photodimerization takes place through interaction of the empty LUMO orbital of a molecule in the ground state and a HOMO orbital of a molecule in excited state (that originates from the same LUMO of the ground state). MO calculations performed for the methyl benzoxaphosphorin-3-carboxylate
5a, have shown that the p
z-orbital coefficients of the C3-C4 double bond have values of 0.39 and 0.61 respectively and the same values show also the corresponding coefficients in the HOMO orbital of the excited state (
Figure 1). Therefore, it would be expected that if a synchronous reaction takes place, by interaction of the lobes with the same sign and according to the maximum overlapping principle (see
Figure 1), then the head-to-head regioisomers (with structures analogous to
1 or
2) would predominate in the reaction products of compounds
5. In the studied reactions however only the head-to-tail regioisomers,
6 or
7, have been isolated and this fact implies that a synchronous mechanism of this reaction should be excluded. It is therefore obvious that the [2+2] photodimerization of compounds
5 is a multistep reaction that proceeds through a triplet excited state of the molecule of oxaphosphorine
5 or through the formation of a bimolecular triplet transition state as it is shown for coumarin photodimerization [
17,
18,
19,
26]. This assumption is in accordance also with the known results [
26,
27], that in the presence of a "heavy" atom in the molecule of the starting compound or in the solvent the transition from a basic singlet into excited triplet condition is taking place directly.
Figure 1.
Orbital interactions for synchronous anti-and syn-HH and anti- and syn- HT photo-dimerization of methyl ester 2-methoxy-2-oxo-2H-1,2-benzoxaphosphorine-3-carboxylic acid. The calculated AO coefficients and charges of C3 and C4 carbon atoms obtained by NBO analysis are shown
Figure 1.
Orbital interactions for synchronous anti-and syn-HH and anti- and syn- HT photo-dimerization of methyl ester 2-methoxy-2-oxo-2H-1,2-benzoxaphosphorine-3-carboxylic acid. The calculated AO coefficients and charges of C3 and C4 carbon atoms obtained by NBO analysis are shown
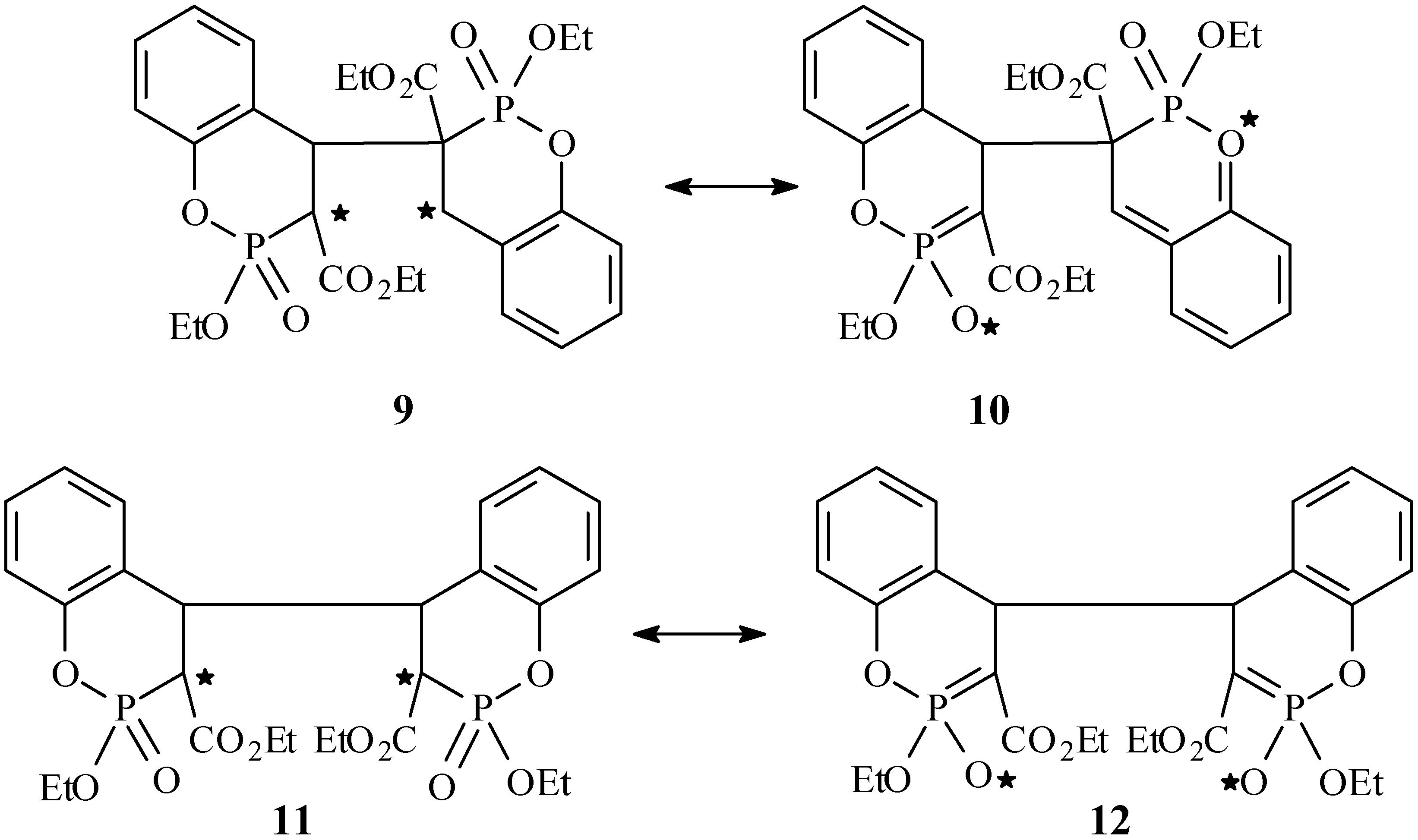
The regioselectivity of the reaction could be explained by a diradical or dipolar intermediate, 9 and 10, which is formed by a C3, C4' interaction of two molecules of 5. This one would be much more stabilized by electron or charge delocalisation than the alternatives, e.g. that would be formed by a C3,C3' or C4,C4' attachment, 11 and 12, where such a delocalisation is much more restricted. A C3, C4' interaction could be also considered as favoured by the charges (obtained by NBO analysis) of the C3 and C4 atoms, where C3 bears a negative charge with a high value of –0.64 e, whereas C4 appears with essentially no charge. Hence, while formation of a head-to-head product through e.g. a C3, C3' interaction is highly disfavoured, a C3, C4' interaction leading to head-to-tail products appears more likely to occur.
Scheme 1.
Possible C3-C4' and C4-C4' diradical or dipolar intermediates leading in HH or HT stereoisomers respectively, of the cyclophotodimerization of compounds 5. * Denotes radical or ion.
Scheme 1.
Possible C3-C4' and C4-C4' diradical or dipolar intermediates leading in HH or HT stereoisomers respectively, of the cyclophotodimerization of compounds 5. * Denotes radical or ion.
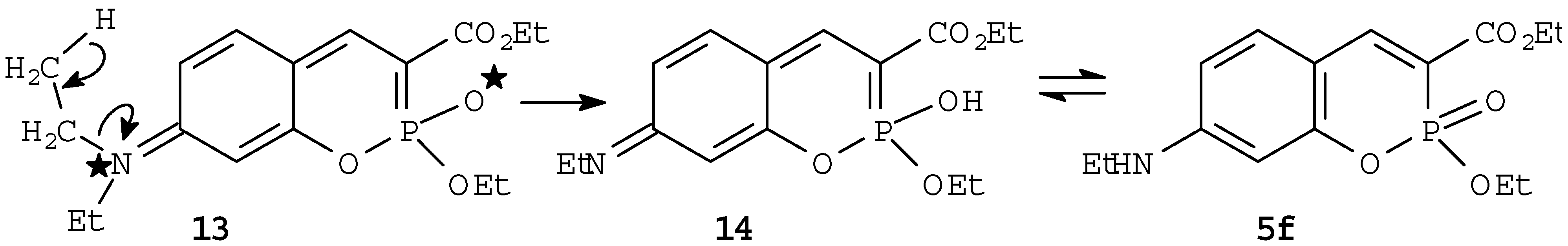
The dipolar or diradical mechanism is supported by the above mentioned behaviour of the 7-dimethylamino-derivative
5e. Thus the formation of the dealkylation product
5f could be explained by assuming a dipolar or diradical intermediate,
13, stabilized by the localization of the charge or radical on the 7-nitrogen atom, giving finally instead of dimerization the dealkylation product 5c, as depicted in
Scheme 2.
Scheme 2.
Possible diradical or dipolar intermediate leading to the dealkylation product 5f.
Scheme 2.
Possible diradical or dipolar intermediate leading to the dealkylation product 5f.
The formation of a dipolar intermediate is also supported by the higher dimerization rates of
5c observed in polar solvents [
25], e.g. 4 days of sunlight irradiation in acetic acid versus 55 days in benzene (
Table 2). The stereoselectivity of the reaction, i.e. the formation of only the
anti isomer as well as the predominance of the centrosymmetric stereoisomer
6 over the non symmetric
7 and the absence of
8 in the reaction products, is most probably the result of steric interactions caused by the oxo and alkoxy groups at P2 atom of the oxaphosphorine ring.
Experimental
General
Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. IR spectra were recorded with a Specord IR 71 or IR 75 spectrophotometers. 1H-NMR and 13C-NMR spectra were obtained with a Bruker WM 250 (at 250 MHz and 62,9 MHz respectively) or a Bruker AM 300 (at 300 MHz and 75,4 MHz respectively) instruments. All NMR spectra were obtained by using TMS as internal standard in CDCl3 and are reported in δ units. E.I. mass spectra were obtained at 70 eV a VG TS-250 spectrometer. Elemental analyses of C, H, P, N and Cl were carried out in the Laboratory of Elemental Analysis at the Department of Organic Chemistry of University of Sofia. Column chromatography was carried out on silica gel (Merck or Fluka 0.063-0.2 mm) using n-hexane/EtOAc or n-hexane/chloroform mixtures of increasing polarity as eluents.
Preparation of the Starting Materials.
The 2-oxo-2
H-1-benzoxaphosphorines
5a-e were prepared by means of the Knoevenagel reaction as described in the literature [
22]. The UV absorption spectra of the starting oxaphosphorins
5a-e were as follows (MeOH), nm (log ε):
5a (MeOH): nm (log ε) = 285 (3.92), 322 (3.38);
5b (MeOH): nm (log ε) = 287 (4.10), 322 (3.56);
5c (MeOH): nm (log ε) = 285 (4.03), 333 (3.34);
5d (MeOH): nm (log ε) = 282 (4.02), 328 (3.36);
5e (MeOH): nm (log ε) = 263 (3.25), 403 (4.11).
Photochemical dimerization of the 3-substituted 2-oxo-2H-1-benzoxa-phosphorines 5. General Procedure:
Depending of the reaction conditions (solvent and reaction time) the following methods are distinguished:
Method A. The solution of corresponding benzoxaphosphorines 1 (0,5 mmoles) in methanol (spectroscopy grade, 5 mL) was left in direct sunlight and monitored by tlc. After evaporation of the solvent, the residue was chromatographed on a silica gel column with n-hexane - chloroform of increasing polarity as eluent. In the case the reaction was not finished, the unreacted starting compound was removed on a silica gel column with n-hexane - ethyl acetate (of increasing polarity) as eluent.
Method B. The same as in method A, but with dry benzene (2 mL) as solvent.
Method C. As in method A, but glacial acetic acid (1.5 mL) was used as solvent.
Method D. As in A, but the reaction was performed in a water suspension (1.5 mL).
Diethyl l6a,6b,12a,12b-tetrahydro-exo,exo-6,12-dimethoxy-endo,endo-6,12-di-oxo-6Н,12Н-cyclobuta-[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (6a).
Yield: 0.04g (30%), when the reaction was carried out for 90 days and 0.034g (25%) when the reaction was carried out for 40 days, m.p. = 235-237
oC (ether). Lit. [
22] m.p. 235-237
oC.
Diethyl 6a,6b,12a,12b-tetrahydro-exo,endo-6,12-dimethoxy-endo,exo-6,12-di-oxo-6Н,12Н-cyclobuta-[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (7a).
Yield: 0.02g (14%), when the reaction was carried out for 90 days and 0.028g (20%), when the reaction was carried out for 40 days, M.p. = 211-213 oС (ether); IR (CHCl3): ν = 1735, 1620, 1580, 1485, 1260, 1190, 1040 cm-1; 1H-NMR (CDCl3) (300 MHz): δ = 0.85 (t, J=7.1 Hz; 3H, CH3), 0.913 (t, J=7.1 Hz; 3H, CH3), 3.65 (dd, J=7.1 and 10.7 Hz; 1H, CHHAO), 3.71 (d, J=12.2 Hz; 3H, OCH3), 3,72-3.88 (m, 2H , CH2), 3.96 (dd, J=7.1 and 10.7 Hz; 1H, CHBHO), 4.08 (d, J=11.1 Hz; 3H, OCH3), 5.32 (dd as t, J=21.5 Hz; 1H) and 5.46 (dd, J=16.8 and 22.3 Hz; 1H) (6b/12b-H), 7.09 (two d as t, 2H), 7.17 (dd as t, J=7.6 Hz; 1H), 7.20 (dd as t, J=7.8 Hz; 1H), 7.28-7.36 (m, 3H), 7.48 (dd, J=1.4 and 7.6 Hz; 1H); 13C-NMR (CDCl3) (75.4 MHz): δ = 13.51, 13.54 (CH3), 42.75 (t, 2JCCP=4.7 Hz) and 43.85 (t, 2JCCP=6.3 Hz) (C-6b/C-12b), 51.24 (dd, J1CP=136.8 HZ; 3JCCCP=6.6 Hz) and 51.35 (dd, J1CP=134.2 HZ; 3JCCCP=4.2 Hz), (C-6a/C-12a), 53.53 (d, 2JCOP=7.7 Hz) and 55.40 (d, 2JCOP=5.1 Hz) (CH3OP), 63.43/62.54 (CH2O), 119.65 (d, 3JCCCP=4.8 Hz) and 120.29 (d, 3JCCCP=4.7 Hz) (C-4/C-10), 122.77 (dd, 3JCCCP=13.9 and 5.7 Hz) and 123.57 (dd, 3JCCCP=13.6 and 3.9 Hz) (C-6c/C-12c), 124.16, 125.26 (C-2/C-8), 130.38 (C-3/C-9), 131.09 (d, 4JCCCCP=1.7 Hz) and 132.10 (d, 4JCCCCP=1.5 Hz) (C-1/C-7), 151.64 (d, 2JCOP=5.2 Hz) and 151.69 (d, 2JCOP=8.7 Hz) (C-4a/C-10a), 166.14 (d, 3JCCCP=1.9 Hz) and 166.64 (d, 3JCCCP≈1.0 Hz) (C=O). – MS: m/z (%) = 537 (8), 536 (M+, 16), 463 (25), 462 (17), 418 (9), 417 (24), 391 (11), 390 (22), 389 (31), 312 (13), 269 (42), 268 (38), 267 (38), 254 (15), 240 (37), 234 (18), 224 (33), 223 (74), 222 (15), 209 (59), 196 (73), 182 (56), 166 (49), 146 (62), 118 (75), 115 (73), 101 (43), 89 (72), 77 (62), 29 (100); Analysis, calcd. for C24H26O10P2 (536.40): C, 53.74; H, 4.89; found: C, 53.46; H, 5.09.
Diethyl 6a,6b,12a,12b-Tetrahydro-exo,exo-6,12-diethoxy-endo,endo-6,12-di-oxo-6Н,12Н-cyclobuta-[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (6b).
Yield: 0.03g (20%), M.p. = 251-253
oС (ether). Lit. [
22] m.p. 251-253
oС.
Diethyl 6а,6b,12а,12b-tetrahydro-exo,endo-6,12-diethoxy-endo,exo-6,12-di-oxo-6Н,12Н-cyclobuta-[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (7b).
Yield: 0.07g (51%), M.p. = 217-219
oС (ether). Lit. [
22] m.p. 217-219
oС.
Diethyl 6а,6b,12а,12b-tetrahydro-2,9-dibromo-exo,exo-6,12-diethoxy-endo, endo-6,12-dioxo-6Н,12Н-cyclobuta[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (6c).
Yields: Method
A: 0.09g (50%), Method
B: 0,1g (53%), Method
C: 0.13g (72%), Method
D: 0.11g (60%), M.p. = 272-274
oC (ether). Lit. [
22] m.p. 272-274
oC; UV (CH
3OH):
nm (logε) = 270 (s, 3.32), 278 (3.53), 287 (3.60), 303 (s, 3.20).
Diethyl 6а,6b,12а,12b-tetrahydro-2,9-dibromo-exo,endo-6,12-diethoxy-endo,exo-6,12-dioxo-6Н,12Н-cyclobuta[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylateC (7c).
Yields: Method A: 0.07g (39%), Method B: 0.06g (30%), Method C: 0.04g (22%), Method D: 0.04g (23%), M.p. = 238-240 oC (methanol); UV (CH3OH): nm (logε) = 270 (s, 3.55), 279 (3.78), 287 (3.79); IR (CHCl3): ν = 1740, 1610, 1560, 1480, 1260, 1230, 1120, 1020 cm-1; 1H-NMR (CDCl3) (300 MHz): δ = 0.952 (t, J=7.2 Hz; 3H) / 0.981 (t, J=7.2 Hz; 3H) (CH3CH2OCO), 1.185 (t, J=7.1 Hz; 3H) / 1.55 (t, J=7.1 Hz; 3H) (CH3CH2OP), 3.75 (dd, J=7.1 and 10.7 Hz; 1H) / 3.83 (dd, J=7.2 and 10.5 Hz; 1H) / 3.87 (dd, J=7.2 and 10.7 Hz; 1H) / 3.95 (dd, J=7.2 and 10.8 Hz; 1H) (CH3CH2OCO), 4.09-4.26 (m, 2H) / 4.30-4.41 (m, 2H) (CH3CH2OP), 5.21 (dd as t, J=21.5 Hz; 1H) and 5.34 (dd, J=15.9 and 22.7 Hz; 1H) (6b/12b-H), 6.95 (d, J=8.4 Hz; 1H) / 7.04 (d, J=8.6 Hz; 1H) (H-4/H-10), 7.40-7.48 (m, 3H), 7.61 (d, J=2.2 Hz; 1H); 13C-NMR (CDCl3) (75.4 MHz): δ = 13.54 / 13.59 (CH3), 16.19 (d, 3JCCOP=5.1 Hz) and 16.50 (d, 3JCCOP=6.5 Hz) (CH3CH2OP), 42.49 (t, 2JCCP=4.7 Hz) and 43.47 (t, 2JCCP=6.3 Hz) (C-6b/C-12b), 50.63 (dd, J1CP=134.4 HZ; 3JCCCP=3.7 Hz) and 52.09 (dd, J1CP=136.7 HZ; 3JCCCP=6.7 Hz), (C-6a/C-12a), 62.71, 62.79 (CH2O), 64.10 (d, 2JCOP=7.5 Hz) and 65.59 (d, 2JCOP=5.5 Hz) (CH2OP), 117.61 (d, 5JCCCCCP=1.3 Hz) and 117.93 (d, 5JCCCCCP≈1 Hz) (C-2/C-8), 121.24 (d, 3JCCOP=4.6 Hz) and 122.06 (d, 3JCCOP=4.7 Hz) (C-4/C-10), 124.63 (dd, 2JCCP / 3JCCCP=13.9 / 6.0 Hz) and 125.59 (dd, 2JCCP / 3JCCCP=12.2 / 2.6 Hz) (C-6c/C-12c), 124.16, 125.26 (C-2/C-8), 133.18, 133.32 (C-3/C-9), 133.79 (d, 4JCCCCP=1.7 Hz) and 135.59 (d, 4JCCCCP=1.8 Hz) (C-1/C-7), 149.80 (d, 2JCOP=5.4 Hz) and 150.85 (d, 2JCOP=8.9 Hz) (C-4a/C-10a), 165.54 (d, 2JCCP=2.1 Hz) and 166.30 (d, 2JCCP≈1.0 Hz) (C=O); MS: m/z (%) = 722 (19), 721 (13), 720 (M+, 32), 649/647 (26), 576/574 (36), 362/360 (37) 334/332 (38), 317/315 (41), 260 (62), 244/242 (53), 235 (45), 232 (65), 214 (40), 212/210 (37), 195 (29), 179 (30), 154 (20), 145 (46), 79 (100), 77 (62); Analysis, calcd. for C26H28O10Br2P2 (722.28): C, 43.24; H, 3.91; found: C, 43.43; H, 4.11.
Diethyl 6a,6b,12a,12b-tetrahydro-2,9-dichloro-exo,exo-6,12-diethoxy-endo,endo-6,12-dioxo-6Н,12Н-cyclobuta[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (6d).
Yield: 0.09g (55%) when the reaction was carried out for 50 days and 0.04g (18%) when the reaction was carried out for 40 days, M.p. = 256-258
oC (ether). Lit. [
22] m.p. 238-240
oC.
Diethyl 6a,6b,12a,12b-tetrahydro-2,9-dichloro-exo,endo-6,12-diethoxy-endo,exo-6,12-dioxo-6Н,12Н-cyclobuta[1,2-c:3,4-c']bis[1,2]benzoxaphosphorine-trans-6а,12а-dicarboxylate (7d).
Yield: 0.06g (37%) when the reaction was carried out for 50 days and 0.02g (8%) when the reaction was carried out for 20 days, M.p. = 229-230 oС (ether); IR (CHCl3): ν = 1740, 1600, 1475, 1270, 1240, 1090, 1020 cm-1; 1H-NMR (CDCl3) (300 MHz): δ = 0.93 (t, J=7.1 Hz; 3H) / 0.97 (t, J=7.1 Hz; 3H) (CH3CH2OCO), 1.18 (t, J=7.1 Hz; 3H) and 1.55 (dt, J=0.7 and 7.1 Hz; 3H) (CH3CH2OP), 3.77 (dq, J=10.7 and 7.1 Hz; 1H) / 3.80 (dq, J=10.6 and 7.1 Hz; 1H) / 3.86 (dd, J=10.6 and 7.1 Hz; 1H) / 4.00 (dd, J=10.7 and 7.1 Hz; 1H) (CH3CH2OCO), 4.06-4.27 (m, 2H) / 4.20-4.40 (m, 2H) (CH3CH2OP), 5.13 (dd as t, J=21.5 Hz; 1H) and 5.44 (dd, J=16.0 and 22.7 Hz; 1H) (6b/12b-H), 7.01 (d, J=9.1 Hz; 1H) / 7.05 (d, J=8.7 Hz; 1H) (H-4/H-10), 7.25-7.32 (m, 3H), 7.48 (d, J=2.5 Hz; 1H); 13C-NMR (CDCl3) (75.4 MHz): δ = 13.50 / 13.52 (CH3), 16.19 (d, 3JCCOP=5.2 Hz) and 16.51 (d, 3JCCOP=6.4 Hz) (CH3CH2OP), 42.54 (t, 2JCCP=4.6Hz) and 43.53 (t, 2JCCP=6.3 Hz) (C-6b/C-12b), 50.51 (dd, J1CP=134.8 HZ; 3JCCCP=3.7 Hz) and 50.95 (dd, J1CP=136.8 HZ; 3JCCCP=6.6 Hz), (C-6a/C-12a), 62.69, 62.78 (CH2O), 64.10 (d, 2JCOP=8.4 Hz) and 65.58 (d, 2JCOP=5.6 Hz) (CH2OP), 120.88 (d, 3JCCOP=4.6 Hz) and 121.69 (d, 3JCCOP=4.6 Hz) (C-4/C-10), 124.22 (dd, 2JCCP / 3JCCCP=14.1 / 6.0 Hz) and 125.13 (dd, 2JCCP / 3JCCCP=13.4 / 3.8 Hz) (C-6c/C-12c), 130.24, 130.35 (C-3/C-9), 130.22 (d, 5JCCCCCP=~1 Hz) and 130.52 (d, 5JCCCCCP=1.0 Hz) (C-2/C-8), 130.84 (d, 4JCCCCP=1.7 Hz) and 131.71 (d, 4JCCCCP=1.7 Hz) (C-1/C-7), 149.22 (d, 2JCOP=5.4 Hz) and 150.27 (d, 2JCOP=8.7 Hz) (C-4a/C-10a), 165.77 (d, 2JCCP=2.3 Hz) and 166.32 (dd, 2JCCP≈2 and 1.0 Hz) (C=O). – MS: m/z (%) = 636/634/632 (M+, 25), 588/586 (5), 562/561/560/559 (12), 514/512 (10) 318/316 (78), 290/2988 (92), 271 (74), 245 (82), 218/216 (74), 207 (81), 180 (82), 154/152 (78), 115 (21), 111 (19), 89 (78), 75 (54), 63 (83), 39 (100); Analysis, calcd. for C26H28O10Cl2P2 (633.36): C, 49.31; H, 4.46; found: C, 49.33; H, 4.39.
Diethyl 7-N-ethylamino-2-oxo-2Н-1-benzoxaphosphorine-3-carboxylate (5f).
Yield: 0.02g (9%), M.p. = 88-89 oC (n-hexane/ether); IR (CHCl3): ν = 3700, 3480, 1710, 1630, 1595, 15450, 1525, 1250, 1200, 1075, 1040 cm-1; 1H-NMR (CDCl3) (300 MHz): δ = 1.27 (t, J=7.2 Hz; 3H, CH3), 1.37 (t, J=7.1 Hz; 3H)/ 1.38 (t, J=7.0 Hz; 3H) (CH3CH2OP), 3.20 (m; 2H, CH2NH), 4.21-4.46 (m; 4H, CH3CH2OP), 4.64 (bs; 1H, NH), 6.28 (d, J=2.2 Hz; 1H, 8-H), 6.35 (dd, J=2.2 and 8.5 Hz; 1H, 6-H), 7.17 (d, J=8.5 Hz; 1H, 5-H), 8.13 (d, J=37.0 Hz; 1H, 4-H); 13C-NMR (CDCl3) (75.4 MHz): δ = 14.33 / 14.38 (CH3), 16.42 (d, 3JCCOP=6.6 Hz), 37.75 (CH2NH), (t, 2JCCP=4.6Hz) and 43.53 () (C-6b/C-12b), 50.51 () and 50.95 (dd, J1CP=136.8 HZ; 3JCCCP=6.6 Hz), (C-6a/C-12a), 61.29 (CH2O), 64.10 (d, 2JCOP=6.3 Hz) (CH2OP), 100.15 (d, 3JCCOP=7.7 Hz) (C-8), 109.10 (d, J1CP=180.7 HZ) (C-3), 109.20 (C-6), 109.4 (d, 3JCCCP=15.3 Hz) (C-4a), 133.17 (d, 4JCCCCP=1.4 Hz) (C-5), 151.27 (d, 2JCCP=4.9 Hz) (C-4), 153.41 (d, 4JCCCOP=1.9 Hz) (C-7), 155.48 (d, 2JCCP=8.2) (C=O), 164.84 (d, 2JCOP=13.7 Hz) (C-8a); Analysis, calcd. for C17H24O5NP (353.36): C, 57.79; H, 6.85; N, 3.96; found: C, 57.68; H, 6.84; N, 3.77.
Computational details
The quantum chemical calculations were performed with the NWChem 4.0 package [
28]. The geometry of the molecules was optimised at the B3LYP [
29] level with TZP basis sets of all atoms. The atomic charges and orbital coefficients were obtained by natural bond orbital analysis using the NBO 4.M program [
30] at HF-MP2 level with 6-31+G* basis sets.




{kind=link}
{kind=link}
{kind=link}



