Synthesis of “Acetylene-Expanded” Tridentate Ligands

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
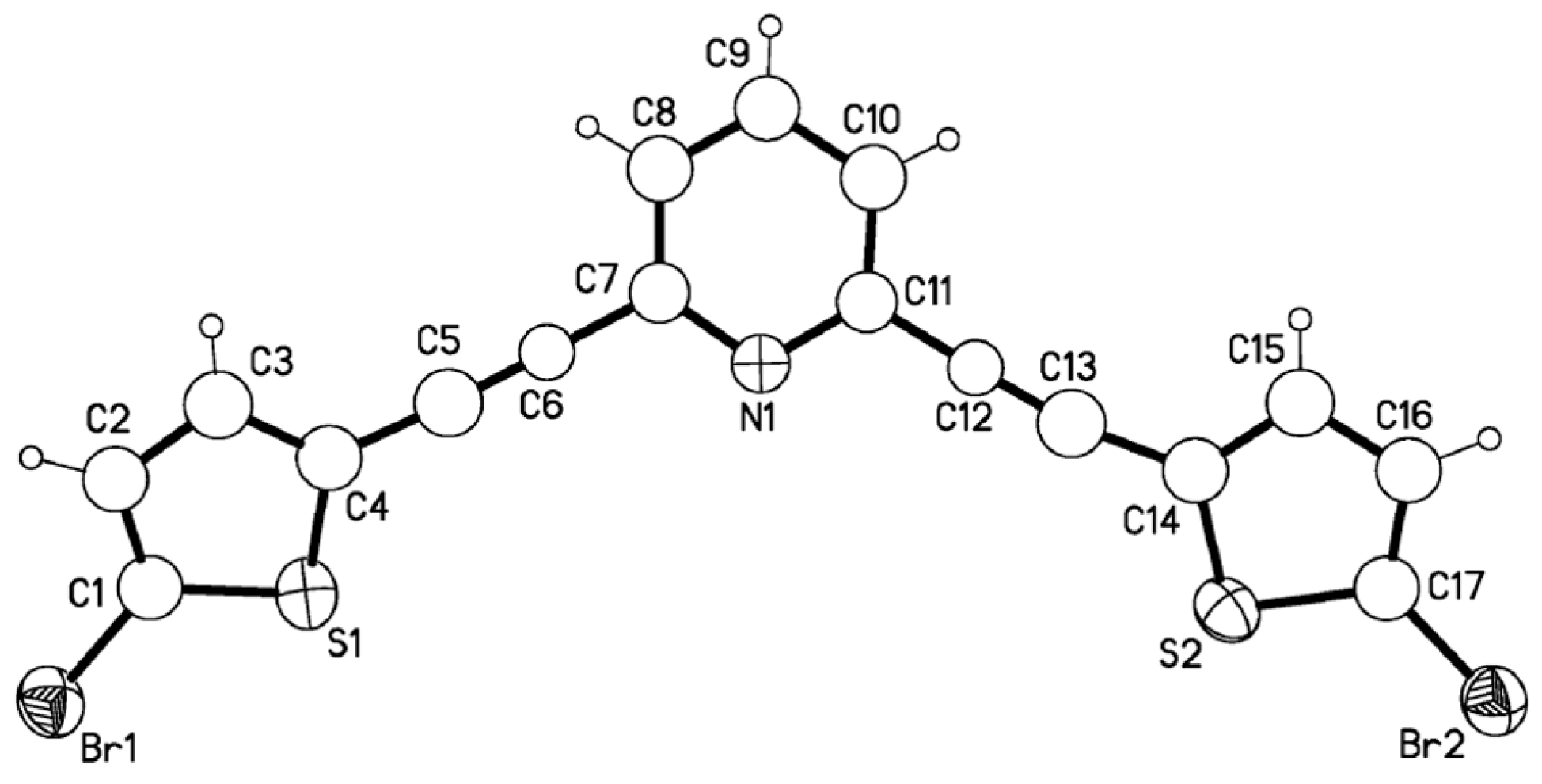
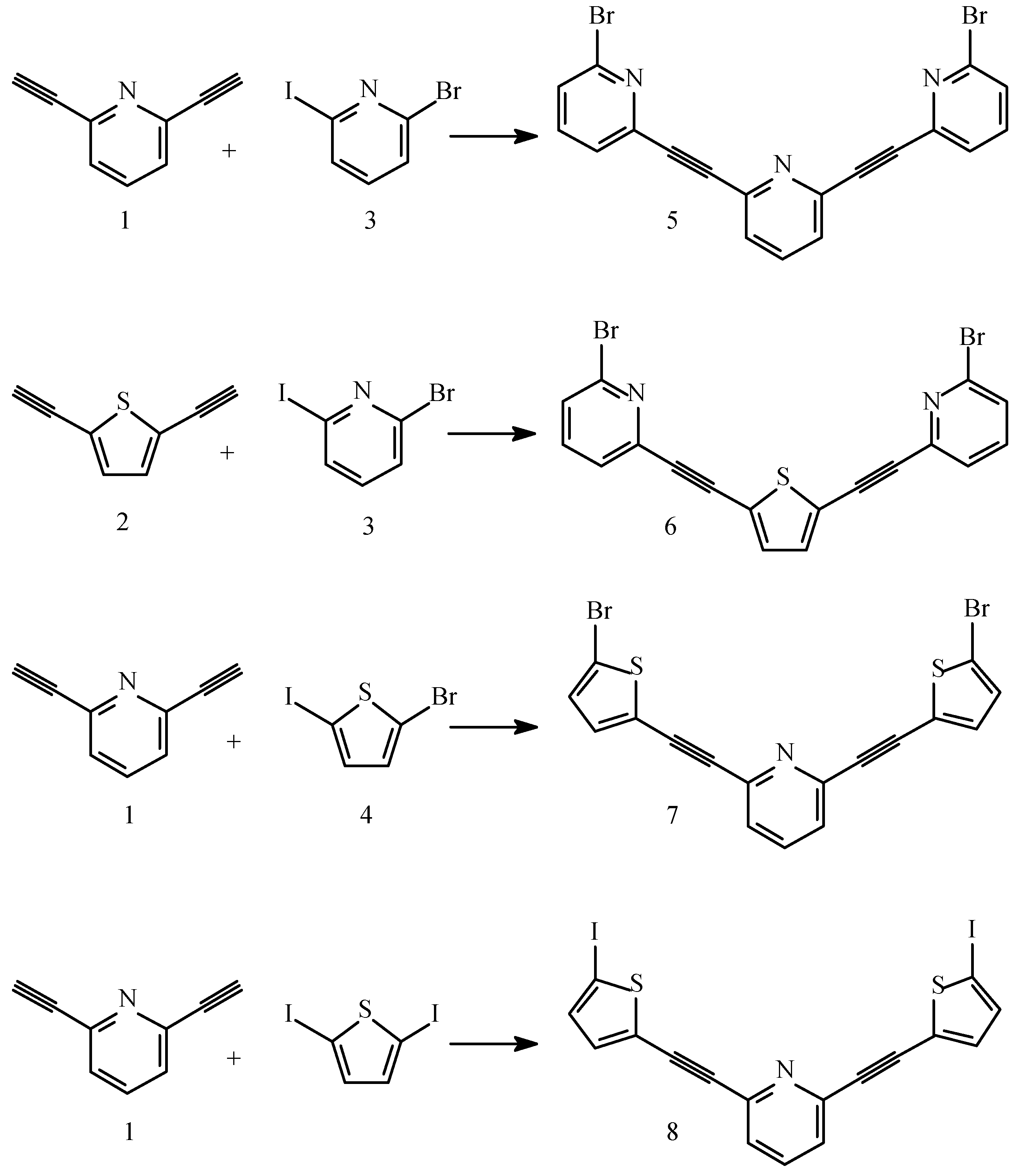
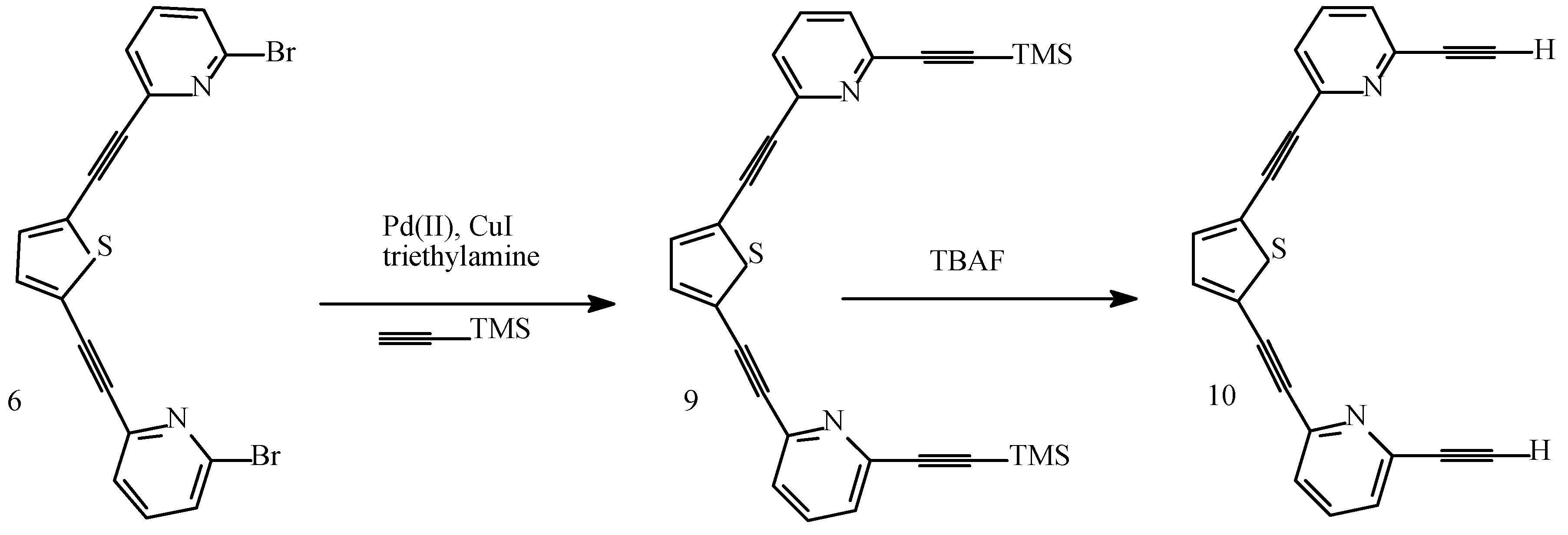
Results and Discussion




Conclusions
Experimental
General
X-ray crystallography
Acknowledgements
References and Notes
- Lehn, J. -M. Perspectives in Supramolecular Chemistry- From Molecular Recognition towards Molecular Information Processing and Self-Organization. Angew. Chem. Int. Ed. Engl. 1990, 29, 1304–1319. [Google Scholar] [CrossRef]
- Zaworotoko, M. J. Crystal Engineering of Diamondoid Networks. Chem. Soc. Rev. 1994, 23, 283–288. [Google Scholar] [CrossRef]
- Rimmer, E. L.; Bailey, R. D.; Hanks, T. W.; Pennington, W. T. Complexes of Acridine and 9-Chloroacridine with I2: Formation of Unusual I6 Chains through Charge-Transfer Interactions Involving Amphoteric I2. Chem. Eur. J. 2000, 6, 4071–4081. [Google Scholar] [CrossRef]
- Hanks, T. W.; Pennington, W. T.; Bailey, R. D. Iodine and Organoiodide Templates in Supramolecular Synthesis. In Anisotropic Organic Materials Approaches to Polar Order, ACS Symp. Ser.; Glaser, R., Kaszynski, P., Eds.; American Chemical Society: Washington, DC, 2001; Vol. 798, pp. 83–96. [Google Scholar]
- Bailey, R. D.; Hook, L. L.; Watson, R. P.; Hanks, T. W.; Pennington, W. T. Tetraiodoethylene: A Supramolecular Host for Lewis Base Donors. Crystal Eng. 2000, 3, 155–171. [Google Scholar] [CrossRef]
- Walsh, R. B.; Padgett, C. W.; Metrangolo, P.; Resnati, G.; Hanks, T. W.; Pennington, W. T. Crystal engineering through halogen-bonding: Complexes of nitrogen heterocycles with organic iodides. Cryst. Growth & Des. 2001, 1, 165–175. [Google Scholar]
- Tour, J.M. Molecular Electronics. Synthesis and Testing of Components. Acc. Chem. Res. 2000, 33, 791–804. [Google Scholar] [CrossRef]
- Diederich, F. Carbon-rich Acetylenic Scaffolding: Rods, Rings, and Switches. Chem. Commun. 2001, 219–227. [Google Scholar] [CrossRef]
- Organic Materials for Nonlinear Optics III; Ashwell, G.J.; Bloor, D. (Eds.) Royal Society of Chemistry: Cambridge, 1993.
- Sukwattanasinitt, M.; Lee, D. -C.; Kim, M.; Wang, X.; Li, L.; Yang, K.; Kumar, J.; Tripathy, S. K.; Sandman, D.J. New Processable, Functionalizable Polydiacetylenes. Macromolecules 1999, 32, 7361–7369. [Google Scholar] [CrossRef]
- Xiao, J.; Yang, M.; Lauher, J. W.; Fowler, F. W. A Supramolecular Solution to a Long-Standing Problem: The 1,6-Polymerization of a Triacetylene. Angew. Chem. Int. Ed. 2000, 39, 2132–2135. [Google Scholar] [CrossRef]
- Phelps, D.; Crihfield, A.; Hartwell, J.; Hanks, T. W.; Pennington, W. T.; Bailey, R. D. Synthesis of Polydiacetylene Charge-Transfer Complexes. Mol. Cryst. Liquid Cryst. 2000, 354, 523–530. [Google Scholar] [CrossRef]
- Bunten, K.; Kakkar, A. K. Synthesis, Optical Absorption, Fluorescence, Quantum Efficiency, and Electrical Conductivity Studies of Pyridine/Pyridinium Dialkynyl Organic and Pt(II)-σ-Acetylide Monomers and Polymers. Macromolecules 1996, 29, 2885–2893. [Google Scholar] [CrossRef]
- Lewis, J.; Long, N. J.; Raithby, P. R.; Shields, G. P.; Wong, W.; Younus, M. Synthesis and characterization of New Acetylide-Functionalized Oligothiophenes and their Dinuclear Platinum Complexes. J. Chem. Soc., Dalton Trans. 1997, 22, 4283–4288. [Google Scholar] [CrossRef]
- Barker, J. L.; Huddleston, P. R.; Wood, J. L. Mannich Reactions of Methoxy Thiophenes. Synth. Commun. 1975, 5, 59–64. [Google Scholar] [CrossRef]
- Trécourt, F.; Breton, G.; Bonnet, V.; Mongin, F.; Marsais, F.; Quéguiner, G. Pyridylmagnesium Chlorides from Bromo and Dibromopyridiens by Bromine-Magnesium Exchange: A Convenient Access to Functionalized Pyridines. Tetrahedron Lett. 1999, 40, 4339–4342. [Google Scholar]
- Johnson, F.; Panella, J. P.; Carlson, A. A. Polyfunctional Aliphatic Compounds. I. The Preparation of 3-Hydroxyglutaronitriles. J. Org. Chem. 1962, 27, 2241–2243. [Google Scholar] [CrossRef]
- Johnson, F.; Panella, J. P.; Carlson, A. A.; Hunneman, D. H. Polyfunctional Aliphatic Compounds. II. The Cylization of Dinitriles by Anhydrous Halogen Acids. Pyridines. J. Org. Chem. 1962, 27, 2473–2478. [Google Scholar] [CrossRef]
- Smith, W. B.; Ho, O. C. Application of the Isoamyl Nitrite-Diiodomethane Route to Aryl Iodides. J. Org. Chem. 1990, 55, 2543–2545. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohada, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975, 4467–4470. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Holmes, B.T.; Pennington, W.T.; Hanks, T.W. Synthesis of “Acetylene-Expanded” Tridentate Ligands. Molecules 2002, 7, 447-455. https://doi.org/10.3390/70500447
Holmes BT, Pennington WT, Hanks TW. Synthesis of “Acetylene-Expanded” Tridentate Ligands. Molecules. 2002; 7(5):447-455. https://doi.org/10.3390/70500447
Chicago/Turabian StyleHolmes, Brian T., William T. Pennington, and T. W. Hanks. 2002. "Synthesis of “Acetylene-Expanded” Tridentate Ligands" Molecules 7, no. 5: 447-455. https://doi.org/10.3390/70500447
APA StyleHolmes, B. T., Pennington, W. T., & Hanks, T. W. (2002). Synthesis of “Acetylene-Expanded” Tridentate Ligands. Molecules, 7(5), 447-455. https://doi.org/10.3390/70500447