An Efficient Preparation of 1,2-Diamino-1-phenylheptane

Organic Chemistry Laboratory, Central Leather Research Institute, Adyar, Chennai – 600 020. Tamil Nadu, India
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(6), 487-493; https://doi.org/10.3390/70600487
Submission received: 12 December 2001
/
Revised: 24 June 2002
/
Accepted: 25 June 2002
/
Published: 30 June 2002
{kind=link}
{kind=link}
Abstract
:A convenient four-step method for the preparation of the lipophilic vicinal diamine 1,2-diamino-1-phenylheptane is described. Condensation involving octan-2-one, benzaldehyde and ammonia is reported. Regioselective Schmidt rearrangement of 2,6-diphenyl-3-pentyl-piperidin-4-one (1) to 2,7-diphenyl-3-pentyl hexahydrodiazapin-5-one (3) is presented. Hydrolysis of 2,7-diphenyl-3-pentyl hexahydrodiazapin-5-one to 1,2-diamino-1-phenylheptane (4) is also reported for the first time.
Introduction
Vicinal diamines have assumed a significant importance in Organic Synthesis [1] especially in drug design [2] and in the development of salen complexes for cancer chemotherapy [3,4]. Several lipophilic amines and diamines have been shown to exhibit antiinflamatory [5] and antimicrobial activities [6]. They are also useful intermediates in the production of surfactants, resins, drugs, pesticides and heterocyclic compunds [7]. Previously available methods for the preparation of vicinal diamines involve (i) reduction of diazides in the presence of Mn (III) salt [8], (ii) catalytic reduction of dioximes [9], (iii) nucleophilic ring opening of aziridines [10,11], (iv) irradiation of imines [12], (v) reductive amination of α-amino ketones [13], (vi) reduction of α-amino nitriles [14], (vii) reductive coupling of aldimines [15], and (viii) reduction of aliphatic α,β-dinitro compounds [16]. The applicability of these methods is limited by several disadvantages such as the explosive nature of diazides, requirement for careful selection of reducing agents and the formation of by-products. As a part of our investigation to identify the biophysical properties governing the antimicrobial activity of lipophilic vicinal diamines and diamides, this paper describes an efficient and straightforward four-step synthetic sequence leading in excellent yields to a relatively inaccessible lipophilic vicinal diamine, 1,2-diamino-1-phenylheptane from easily available starting materials, with cinnamic acid being the only side product. Using this protocol we have prepared a series of vicinal diamines starting from appropriate methyl ketones. Preparation of some of these vicinal diamines via Beckmann rearrangement has been reported in the literature [22].
Results and Discussion
Three component condensation involving benzaldehyde, octan-2-one and ammonia proceeded smoothly in ethanol at higher temperatures (60-70°C) [17,18,19]. The resultant 2,6-diphenyl-3-pentyl-piperidin-4-one (1) was stable and obtained in quantitative yield (Scheme 1).
Scheme 1.

The reaction could be carried out on a large scale in a very simple procedure. Piperidin-4-ones of type 1 derived from octanone or methyl ketones with a long aliphatic chain have not been reported before. The structure of compound 1 was identified by comparing its spectral data with those of other piperidinones of type 1, and confirmed by elemental analysis. The appearance of the strong carbonyl stretch at 1714 cm-1 is typical of a six member cyclic ketone. The Bohlmann bands [19] at 2700–2850 cm-1 are further evidence in favour of structure 1. Bohlmann bands have been observed earlier in both piperidinones of type1 and hexahydrodiazapinones of type 2. Analysis of the 1H-and 13C-NMR spectra also provide evidence in favour of piperidinone 1. The 13C-NMR spectrum of 1 is of particular interest. The carbonyl carbon resonates at 209 ppm. The quaternary carbons on the two phenyl rings appear at 143.0 and 141.8 ppm indicating the asymmetric nature of the molecule.
Use of the Schmidt rearrangement [20,21] usually results in regioselective formation of lactams. However, the yields may vary depending on the nature of addition of reactants, temperature and the mode of neutralization of the reaction mixture. Of the two possible isomers resulting from the Schmidt rearrangement of the piperidinone hydrochloride 2, only one isomer, 3, was obtained as expected. The IR spectrum of the diazapinone 3 displays two bands at 3206 cm-1 and 3084 cm-1, corresponding to amine NH and amide NH, respectively. The strong band at 1674 cm-1 is indicative of the lactam 3. Analysis of 1H and 13C-NMR spectra of the product revealed that it is the alkyl substituted C3 carbon which migrates during Schmidt rearrangement producing the hexahydrodiazapinone 3. Whereas the multiplet corresponding to H3 of 1 appears at 2.6 ppm, the multiplet signal for H3 of 3 resonates at 3.7 ppm. This marked downfield shift of the H3 multiplet provides strong evidence for the regioselective formation of lactam 3 during the Schmidt rearrangement. The disappearance of the singlets at 5.8 ppm and 2.05 ppm upon exchange with D2O is attributed to the labile nature of the amide and amine protons, respectively. The appearance of amide carbonyl peak at 176 ppm and the downfield shift in the resonance position of C3 (70 ppm) in the 13C-NMR spectrum of 3, provide complementary evidence suggesting that C3 is adjacent to amide NH and not to the carbonyl group.
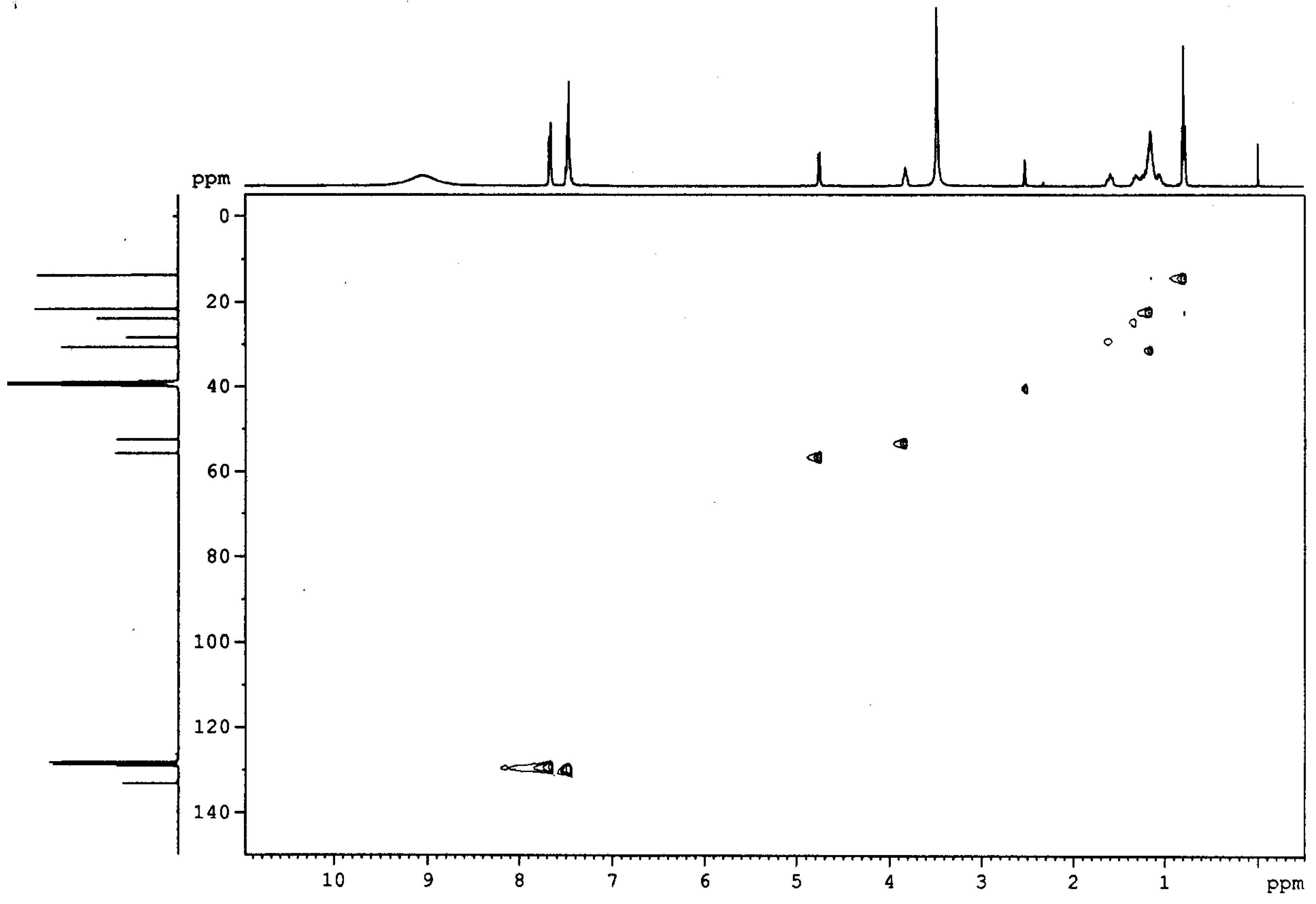
Hydrolysis [22] of the 2,7-diphenyl-3-pentyl-hexahydrodiazapin-5-one 3 under reflux for 12h using 6N hydrochloric acid resulted in the quantitative formation of water soluble vicinal diamine 4 and cinnamic acid (5). The cinnamic acid produced during hydrolysis crystallized readily at room temperature and was easily separated by filtration, and the residual acid was removed by extraction with ether, making the procedure very simple. Compound 5 was identified as cinnamic acid by comparing its spectra with those obtained from the authentic sample. The identity of vicinal diamine 4 was established using physical and spectroscopic data. Unambiguous assignment of the proton and 13CNMR spectra of the diamine 4 was accomplished via the corresponding HETCOR spectrum (See Figure 1). The broad peak at 9.05 ppm in the 1H-nmr of 4 is attributed to the two amino groups in their hydrochloride form. The singlet at 3.35 ppm is from water impurity. All other assignments are given in the experimental section. The formation of diamine was confirmed by elemental analysis and by converting the diamine to lipophilic diamides for further studies.
Figure 1.

Conclusions
This paper describes a convenient method for the preparation of lipophilic vicinal diamine 4. The condensation involving octan-2-one, benzaldehyde and ammonia is reported for the first time. The regioselective Schmidt rearrangement of piperidin-4-one 2 resulting in the formation of lactam 3 is presented. Acid hydrolysis of the lactam yields easily separable vicinal diamine.
Experimental Section
General
Melting points were recorded on a Ragaa hot stage capillary melting point apparatus and are uncorrected. The 1H- (300-MHz) and 13C-NMR (75-MHz) spectra were obtained on a Bruker MSL 300P (FT mode) Spectrometer. The 400 MHz 1H-NMR spectrum for compound 3 was recorded on a Jeol 400 GSX Spectrometer. The 1H- (400 MHz), 13C- (100 MHz) and HETCOR spectra for compound 4 were obtained on a 400 MHz Avance Bruker spectrometer. Tetramethylsilane was used as an internal standard. The mass and IR spectra were recorded on a Perkin-Elmer GC-Mass and Perkin Elmer Spectrum RX I spectrometers, respectively.
3-Pentyl-2,6-diphenylpiperidin-4-one (1)
Benzaldehyde (21.2g, 0.2 mol), octan-2-one (16.0 mL, 0.1 mol; Fluka) and ammonium acetate (7.7 g, 0.1 mol) were added to dehydrated ethanol (100 mL) placed in a conical flask. The contents of the flask were warmed over a water-bath or hot-plate for about 30-45 minutes with occasional shaking during which time the solution turned amber coloured. The solution was left standing overnight and the precipitate formed was separated by filtration. The solid was purified further by crystallization from ethanol or methanol. Yield 16.4g, (51%); mp: 97-98 °C; IR (KBr): 3063, 3033, 2923, 2854, 1713 (C=O), 1457, 759, 700, 653 cm-1; 1H-NMR (CDCl3) δ 7.45- 7.26 (m, 10H, Ar.); 4.08 (d, 1H, H6, J= 8.8 Hz); 3.71 (d, 1H, H2, J=10.2 Hz); 2.72-2.62 (dd, 2H, H5 merged with m, 1H, H3); 2.08 (s, 1H, NH); 1.61 (m, 2H); 1.25 (m, 2H); 1.09 (m, 2H); 0.98 (m, 2H); 0.77 (t, J=7.6 Hz, 3H). 13C-NMR (CDCl3) δ 209.0, 143.8, 141.8, 128.6, 128.5, 128.0, 127.9, 126.5, 67.2, 61.8, 57.0, 51.5, 31.9, 27.3, 24.6, 22.3, 13.9.; MS (m/z, EI+) 322 (M+1), 321 (M+), 264, 208, 194, 165, 145, 131, 116, 103, 91, 77, 55.; Elemental Analysis [Calculated (Found)]: C: 82.13 (82.42); H: 8.40 (8.51); N: 4.36 (4.35).
3-Pentyl-2,6-diphenylpiperidin-4-one Hydrochloride (2).
Dry powdered compound 1 (16.0 g, 49.8 mmol) was dissolved in ether (300 mL) in a conical flask and HCl gas was passed through the solution till the precipitation of the white salt was complete. The precipitated solid was separated by filtration, washed with ether, dried and crystallized from ethanol to afford colourless flakes of 2. Yield 16.50g (92%); mp: 168-169 °C.
3-Pentyl-2,7-diphenylhexahydro-1,4-diazepin-5-one (3).
Powdered 2 (10.0g, 27.9 mmol ) was added, in small portions, to ice-cold concentrated H2SO4 (50 mL) in a conical flask equipped with a magnetic stirrer. After the addition was complete, the solution was allowed to equilibrate to room temperature. Sodium azide (3.0 g, 46.15 mmol) was added in portions over a period of one hour. After the addition was over, the solution was poured into crushed ice, the pH was adjusted to ~8.0 using 2N NaOH solution and the white solid separated was isolated by filtration. The solid was further purified by crystallization from benzene. Yield 6.75g (72%); mp: 149-150 °C; IR (KBr) 3206, 3084, 2958, 1674 (NHC=O), 1428, 1327, 758, 697, 538 cm-1; 1H-NMR (CDCl3 ; 400 MHz) δ 7.41-7.29 (m, 10 H, Ar); 5.88 (s, 1H, CONH); 4.13 (d, 1H, H7, J=10.74 Hz); 3.77-3.67 (merged d, 1H, H2, J=7.81 Hz and m, 1H, H3); 3.14 (dd, 1H, axial H6, 3J=10.74 Hz, 2J=14.21 Hz); 2.64 (dd, 1H, equatorial H6, 3J=1.71 Hz, 2J=14.02 Hz); 2.03 (s, 1H, NH); 1.35 (m, 2H); 1.16 (m, 2H); 1.08 (m,2H); 1.04 (m,2H); 0.80 (t, 3J=7.08 Hz, 3H). 13C-NMR (CDCl3) δ 176.0 (CONH), 144.8, 142.0, 128.6,127.9,127.7,127.6,126.4, 70.3, 59.7, 58.9, 47.4, 32.5, 31.1, 24.9, 22.3, 13.8.; MS (m/z, EI+) 338 (M+2), 314, 292, 118, 100, 91, 11, 55.; Elemental Analysis [Calculated (Found)]: C: 78.46 (78.86); H: 8.32 (8.45); N: 8.32 (8.31).
1,2-Diamino-1-phenylheptane dihydrochloride (4).
Dry solid 3 (5.0g, 14.9 mmol ) was added to 6N HCl (150 mL) in a 250 mL RB Flask and heated under reflux for 24 h. Upon cooling to RT, colourless crystals separated. The solution was filtered through a filter paper and washed with ether (3 x 50 mL). The aqueous layer was separated and evaporated to dryness over a water-bath. The white solid obtained was washed with ether, dried and crystallized from ethanol. Yield 3.30g, (79%); decomposition point: >230 °C; IR (KBr) 3129, 3041, 2954, 1584, 1488, 1404, 1051, 757, 696, 538 cm-1; 1H-NMR (DMSO-d6; 400 MHz) δ 9.05 (s, broad, 6H, NH2·HCl); 7.67 (m, 2H, Ar); 7.48 m, 3H, Ar); 4.75 (d, 1H, H1, J=6.38 Hz); 3.82 (m, 1H, H2); 1.58 (m, 2H); 1.32 (m, 2H); 1.18 (m, 2H); 1.07 (m, 2H); 0.80 (t, 3H, J= 6.92 Hz); 13C-NMR (DMSO-d6; 100 MHz) δ 133.12 (Ar), 129.12 (Ar), 128.77 (2C, Ar), 128.29 (2C, Ar3), 55.73 (C1), 52.45 (C2), 30.58 (C6), 28.26 (C3), 23.86 (C4), 21.53 (C5), 13.66 (C7); MS (m/z, EI+) 281 (M+2), 277 (M-2), 207, 149, 128, 118, 106, 100, 91, 77, 55.; Elemental Analysis [Calculated (Found)]: C: 48.92 (49.13); H: 7.90 (7.98); N: 10.06 (10.22).
References
- Le Gall, T.; Mioskowski, C.; Lucet, D. Angew. Chem. Int. Ed. 1998, 37, 2580.
- Kasina, S.; Fritzberg, A. R.; Johnson, D. L.; Eshima, D. J. J. Med. Chem. 1986, 29, 1933. [PubMed]
- Gust, R.; Burgemeister, T.; Mannschreck, A.; Schonenberger, H. J. Med. Chem. 1990, 33, 2535. [PubMed]
- Khokhar, A. R.; Al-Baker, S.; Shamsuddin, S.; Siddik, Z. H. Bioorg. Med. Chem. Lett. 1997, 40, 112.
- Kokotos, G.; Constantinou-kokotu, V.; Naoula, C.; Hadjiparlou-litina, D. Lipids 1999, 34, 307. [PubMed]
- Telang, A. J.; Ebert, S.; Foght, J. M.; Westlake, D. M. S.; Voordouw, G. Can J. Microbiol. 1998, 44, 1061.
- Mukoyama, M.; Iwagawa, K.; Hata, E.; Yamada, T. Jpn. Kokai Tokkyo Koho JP 1095753. 1998. [Chem. Abstr. 1998, 128, 243824n].
- Jones, D. S.; Srinivasan, A.; Kasina, F.; Fritzbery, A. R.; Wilkening, D. W. J. Org.Chem. 1989, 54, 1940.
- Nagareda, K.; Susuki, S. Jpn. Kokai Tokkyo Koho JP 10 87573. 1998. [Chem. Abstr.,1998, 128, 257174n].
- de Sousa, S. E.; O’Briew, P. Tetrahedron Lett. 1997, 38, 4885.
- Wu, J.; Hou, X-L.; Dai, L-X. J. Org. Chem. 2000, 65, 1344. [PubMed]
- Katrizky, A. R.; Fan, W. Q.; Fu, C. J. Org. Chem. 1990, 55, 3209.
- Reetz, M. T.; Kaysar, F.; Harms, K. Tetrahedron Lett. 1994, 35, 8764.
- Gutsche, C. D.; Mei, G. C. J. Am. Chem. Soc. 1985, 107, 7964.
- Dutta, M. P.; Baruah, B.; Boruah, A.; Dipak, P.; Sandhu, J. S. Synlett. 1998, 8, 857.
- Ji, B.; Chen, H. Huaxue Yanjin Yu Yingyong 1999, 11, 673, [Chem. Abstr., 2000, 132, 264928x].
- Noller, C. R.; Baliah, V. J. Am. Chem. Soc. 1948, 70, 3853. [PubMed]
- Baliah, V.; Ekambaram, A.; Govidarajan, T. S. Curr. Sci. 1954, 23, 264.
- Ravindran, T.; Jeyaraman, R.; Murray, R. W.; Singh, M. J. Org. Chem. 1991, 56, 4833.
- Smith, P. A. S. Molecular Rearrangements; de Mayo, P., Ed.; Interscience: New York, 1963; Vol. 1, p. 507. [Google Scholar]
- Senthilkumar, U. P.; Jeyaraman, R.; Murray, R. W.; Singh, M. J. Org. Chem. 1992, 57, 6006.
- Baliah, V.; Lakshmanan, M. R.; Pandiarajan, K. Indian J. Chem. Section B. 1978, 16B, 72.
- Sample availability: Samples of compounds 1, 3 and 4 are available from MDPI.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Thennarasu, S.; Perumal, P.T. An Efficient Preparation of 1,2-Diamino-1-phenylheptane. Molecules 2002, 7, 487-493. https://doi.org/10.3390/70600487
AMA Style
Thennarasu S, Perumal PT. An Efficient Preparation of 1,2-Diamino-1-phenylheptane. Molecules. 2002; 7(6):487-493. https://doi.org/10.3390/70600487
Chicago/Turabian StyleThennarasu, S., and P. T. Perumal. 2002. "An Efficient Preparation of 1,2-Diamino-1-phenylheptane" Molecules 7, no. 6: 487-493. https://doi.org/10.3390/70600487